Relationships between molecular structure and kinetic and thermodynamic controls in lipid systems. Part I: propensity for oil loss of saturated triacylglycerols
Laziz
Bouzidi
a,
Tolibjon S.
Omonov
b,
Nissim
Garti
c and
Suresh S.
Narine
*a
aTrent Centre for Biomaterials Research, Physics & Astronomy and Chemistry Departments, Trent University, Peterborough, Ontario K9J 7B8, Canada. E-mail: sureshnarine@trentu.ca; Fax: +1-705-748-1652; Tel: +1-705-748-1011
bAlberta Lipid Utilization Program, Department of Agricultural Food and Nutritional Science, University of Alberta, Edmonton, Alberta T6G 2P5, Canada
cCasali Institute of Applied Chemistry, Faculty of Science, The Hebrew University of Jerusalem, Givat Ram, Jerusalem 91904, Israel
First published on 26th September 2012
Abstract
Pure saturated triacylglycerols (TAGs) in canola oil were used as model systems to analyse oil loss in structured oil both from thermodynamic and kinetic perspectives. Two important parameters which effectively and predictively measure the relative propensity of a solid network to lose/hold oil were defined: (1) the rate of oil loss, K, which is a quantified representation of the kinetics of oil loss and (2) the initial amount of oil susceptible to be lost, i.e., the propensity for oil loss (POL), which is a representation of the thermodynamics of oil binding. It was found that the POL and K values do not always trend in the same fashion, suggesting that the mechanism of oil binding is complex, depending on the structurant's crystalline form locked within the oil network. The two parameters were, however, correlated to the melting and thermal behavior of the structurants, to the polymorphic structures that are obtained during the cooling process and to the habit (shape, size and morphology) of the crystalline phase in the oil. Both POL and K had a strong correlation to the oil loss.
1. Introduction
The structuring of liquid organic phases is a topic of increasing attention due to broad scientific and practical significance in a wide variety of industries, such as food, pharmaceutical, cosmetic and petrochemical.1,2 Structuring of binary and multi component mixtures dispersed in oil-continuous phases (including water-in-oil emulsions) is particularly important in lipid-based food systems, due to requirements for the elimination of trans-fats, reduction of the amount of saturated fat and incorporation of healthy oils into our diet.3–7 The generally quasi-stable nature of structured mixtures of oils-based systems results in component migration and phase transitions, which can lead to serious quality problems and therefore product shelf-life limitations. “Oiling off” in low-trans spreads, margarines and shortenings, oil separation in products like peanut butter, and oil migration in confectionary products are common associated problems.8–10Oil transport is the result of the competing effects of driving and retarding forces. The driving force for oil loss is gravitational and the forces opposing oil loss are capillary, viscous drag and surface interactions.11 It has been predominantly characterized as a diffusion process described by simplified solutions to Fick's Second Law, which stress a kinetic approach.12–14 It is important to note that Fickian diffusion alone fails to model oil migration.15 Many findings have suggested that a capillary rise mechanism plays an important role in the migration of oil.16,17 The capillary forces are expected to be important for oil migration in structures which are similar to those of porous media, and is relevant to some crystallized fat networks.18 As network structures establish and firm, the capillary forces, as well as surface forces which oppose oil loss, increase whilst viscous drag continues to act.16 When large amounts of unbound liquid are present, morphological details of the solid matrix do not significantly affect transport phenomena and the flow is mainly driven by gravitational pressure. Even then, viscous drag is still acting and the weak van der Waals interactions (crystal–crystal, liquid–liquid and liquid–crystal) cannot be ignored. Recently, predictive models based on an explicit formulation of the diffusion problem in terms of the molecular diffusivity and the internal microstructure of a fat, which add some thermodynamic aspects, has been devised to account for structural parameters such as fat crystal microstructure and tortuosity, polymorphic form and phase volume.19
Usually, the propensity for oil loss of a structurant made of aggregates of fat crystals is explained phenomenologically by considering microstructure, solubility and polymorphic transitions.20 The rate and extent of oil migration/loss is linked to the molecular ensemble in the solid network as well as the habit (shape, size and morphology) of the solid network and storage conditions. The spatial distribution of the oil in the mixture, distribution and morphologies of fat crystals on the surface or change in the surface porosity of stored samples are also considered in modeling oil migration and loss. Furthermore, specific regions, such as those where imperfections (e.g., cracks, pores) are dominant, promote undesired growth, migration and oil loss.15 This is justifiable as the major factors determining the rheological and textural properties of crystallized TAG systems are related to the crystal network organization, partly reflected by the polymorphic state of the TAG crystals and the distribution of crystallites in the microstructure.21 The dynamic processes occurring in a network made of aggregates of fat crystals with liquid oil dispersed between the crystals directly impact long-timescale physical functionality.
Our research group focuses on better understanding the fundamentals of the structuring process. Our goal is to identify the mechanisms through which structuring is achieved, and eventually formulate optimal solutions for existing structurants (such as saturated TAGs), design specialized materials, and help propose novel structuring materials. We have first devised a robust, accurate and reproducible method to measure oil binding/loss capacity of fat structurants.17 In the present study very extensive work was done to prepare and study most of the possible variations of fully saturated TAG molecules (ten in total). The relative oil binding/loss propensity of blends of these TAGs in canola oil (CO) has been measured using the methodology presented in our previous studies.17 TAG molecules were chosen as model systems because of the numerous studies available on them which provide the best baseline knowledge, and because of their widespread use as structuring agents in many lipid-based products.1,4,22,23
In order to develop a better understanding of oil binding/loss mechanisms and their relationship to physical properties, crystal structure, melting behavior, microstructure, and solid fat content (SFC) of the CO–TAG mixtures have been investigated using X-ray diffraction (XRD), differential scanning calorimetry (DSC), polarized light microscopy (PLM), and wide-line pulsed nuclear magnetic resonance (pNMR) techniques. Furthermore, the oil binding capacity (OBC) of these TAGs have been compared to that of previously studied fully hydrogenated canola oil (FHCO) and fully hydrogenated soybean oil (FHSO)17 as well as to a commercially available fat structurant, so-called MF50.
In order to simplify the discussion, we have chosen to represent the results as a function of the relative difference in the total number of carbon atoms (SSS–TAG), here called Excess Molecular Carbon (EMC, in carbon atoms), as a substitute for total number of carbons or molar mass. For example, the EMC of SSS and LPP would be 0 and 10 carbon atoms, respectively. For brevity in the description and discussion of our results, the units for EMC have been dropped in the manuscript.
2. Materials and methods
2.1 Materials
The ten purified symmetrical and asymmetrical TAGs (i.e., tristearin (SSS); 1,3-dipalmitoyl-2-stearoyl-sn-glycerol (PSP); 1,3-dilauroyl-2-stearoyl-sn-glycerol (LSL); 1,3-dimyristoyl-2-stearoyl-sn-glycerol (MSM); and 1,3-dilauroyl-2-palmitoyl-sn-glycerol (LPL), 1,2-stearoyl-3-palmitoyl-sn-glycerol (PSS); and 1,2-dipalmitoyl-3-stearoyl-sn-glycerol (PPS); 1,2-dilauroyl-3-stearoyl-sn-glycerol (LLS), 1,2-dimyristoyl-3-stearoyl-sn-glycerol (MMS); and 1,2-dipalmitoyl-3-lauroyl-sn-glycerol (PPL)) were synthesized according to known procedures.24,25 Their purity exceeded 98%. Purity was determined using a GC equipped with a universal flame ionization detector (FID) having a range of 0–10 V. The sample was run in chloroform, using a Chromo Pack-Triglycerides Analysis Phase column (CP-TAP, Varian, USA), specifically designed for TAG analysis. MF50 was provided by Bunge Oils, Bradley, IL.The TAGs were blended in canola oil (refined, bleached, and deodorized) purchased from Canbra Food LTD (Lethbridge, Alberta, Canada) in an 85/15 wt/wt ratio. The TAG weight fraction in the CO–TAG mixtures were chosen because it compares reasonably with the usual concentrations and is high enough to be well above the critical concentration, Ccr, at which, based on our previous findings,17 saturated TAGs are effective in binding a liquid phase.
2.2 Sample preparation and crystallization protocol
All the samples were prepared under sheared conditions. The CO–TAG mixture was first weighed carefully in a glass tube (25 mm diameter) then placed in a temperature controlled circulating bath (DTRC-620, Jeio Tech Co., Ltd., Korea) where it was heated to 90 °C. The sample was kept at this temperature for 1 hour after which it was mixed vigorously using a Vortex mixer (Fisher Scientific, Ottawa, ON) for about 1 min then immediately transferred into a cooling circulating system (JULABO F25 combined with JULABO F95, Allentown, PA) already set up at 90 °C where it was equilibrated for another 5 min under shear (100 rpm) using a home-made mixer. The sample in the glass tube was then cooled down at a constant rate (3.0 °C min−1) to a final crystallization temperature, TC, of 25 °C. The mixture was finally stored at 25 °C in a temperature controlled incubator for 24 h. All measurements were performed after this incubation period which is reported as the zero measurement time. Because only mass fraction X = 0.15 was used, the samples will be simply referred to as CO–TAG for ease of reading.2.3 Oil binding/loss capacity measurements
The oil binding capacity (OBC) of the TAGs was measured using a method developed by our group and detailed in our previous publication.17 An especially machined aluminum hollow cylinder (10 mm diameter, 5 mm height) was placed at the center of a Whatman paper (number 4, 15 cm diameter) then carefully filled with 0.40 ± 0.02 g of the already processed TAG–oil mixture (as described in Section 2.2). The filter paper with the sample was placed on an o-ring so that the oil does not leak outside the filter paper. The assembly (sample in the cylinder on the paper) was carefully stored in a controlled environment at 25 °C for a period of time (ts) after which OBC was measured. Note that the Whatman filter paper as well as the empty cylinder was subjected to the same temperature profile as the mixture.OBC is calculated using the following equation:
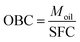 | (1) |
M oil is the mass of entrapped liquid oil in the crystal network after the ts. It is determined from the mass balance eqn (2):
| Moil = Mmix − Mfat − Mmig oil | (2) |
OBC measurements were done after ts of 10, 20, 30, 40, 50, 60 min and 24 h. A different sample was used for each ts. Note that a blank Whatman filter paper subjected to the same storage conditions was used to correct for the influence of the humidity of the environment. The reported values and the uncertainties attached are the calculated means and standard deviations, respectively, of three replicates.
2.4 X-ray diffraction
X-ray diffraction was performed using a Bruker AXS (Milton, Ontario) X-ray diffractometer equipped with a filtered Cu-Kα radiation source (λ = 0.1542 nm) and a 2D detector. The procedure was automated and controlled by the Bruker AXS's “General Area Detector Diffraction System” (GADDs V4.1.08) software. The samples processed as described in Section 2.2 were transferred to the XRD stage where the temperature was already set up and maintained at 25 ± 0.5 °C, by an air jet cooling system (Kinetics-Thermal Systems, USA). The frames were processed using GADDS V.4.1.08 software and the resulting spectra analysed using “Topas V2.1” (Bruker AXS) software. The spectra were decomposed into individual pseudo-Voigt lines using X'Pert HighScore v3.0 Software (PANalytical B.V., Almelo, The Netherlands).The arrangement of two TAGs determines the tilt of the hydrocarbon chains with respect to the plane through the methyl end groups, and this tilt determines the small-angle (long-spacing) reflections in an XRD powder diagram. TAGs generally stack in either a double chain length (DCL) or a triple chain length (TCL) structure, although they can display more complicated layering.28
The β-polymorph is the most stable crystal form with the highest melting temperature of the three polymorphic states and the α-polymorph, is the least stable crystal form with the lowest melting temperature.29–31
2.5 Differential scanning calorimetry
The heating profiles of the crystallized mixtures (processed as described in Section 2.2), before, and after 1 and 24 h of oil binding measurement, were obtained using a Q100 model DSC (TA Instruments, New Castle, DA). Approximately 5.0–8.0 (±0.1) mg of the sample was placed in an aluminium pan which was then hermetically sealed. An empty aluminium pan was used as a reference and the experiments were performed under a nitrogen flow of 50 mL min−1. The sample was quickly transferred to the DSC cell where it was equilibrated at 25 °C then heated to 90 °C at a constant rate of 5 °C min−1. The “TA Universal Analysis” software coupled with a method developed by our group32 was used to analyse the data and extract the main characteristics of the endotherms (onset temperature of melting, Ton, offset temperature of melting, Toff, temperature at maximum heat flow, TM, and enthalpy of melting, ΔHM).2.6 Polarized light microscopy
A polarized light microscope, PLM, (Leica DMRX, Leica Microsystems, Wetzlar, Germany) fitted with a Hamamatsu (C4742-95) digital camera was used for image capture. A Linkam LS 350 temperature-controlled stage (Linkam Scientific Instruments, Tadworth, Surrey, United Kingdom) fitted to the PLM was used to process the samples as described in Section 2.2.2.7 Solid fat content determination
The SFC measurements have been carried out on a Bruker Minispec mq 20 (Milton, Ontario, Canada) wide-line pNMR spectrometer equipped with a temperature-controlled chamber. SFC of the processed sample (protocol described in Section 2.2) was measured after several different storage time periods (ts) at 25 °C. The SFC values are reported as ratios of the intensity of the NMR signal of the solid phase to the total detected NMR signal in percent. Uncertainties attached are the calculated standard deviations of at least three runs.3. Results and discussion
3.1 Polymorphism and microstructure of the CO–TAG mixtures
XRD patterns obtained at 25 ± 0.5 °C for the symmetrical and asymmetrical TAGs are shown in Fig. 1a and b, respectively. As can be seen, despite the presence of a large liquid phase, well defined XRD patterns were obtained for the CO–TAG mixtures. The XRD patterns of CO–SSS, CO–LSL, CO–PPS and CO–LLS mixtures prominently presented the characteristic lines of the β-form whereas those of the others clearly showed additional lines characteristic of the common subcell packing of the β′-form.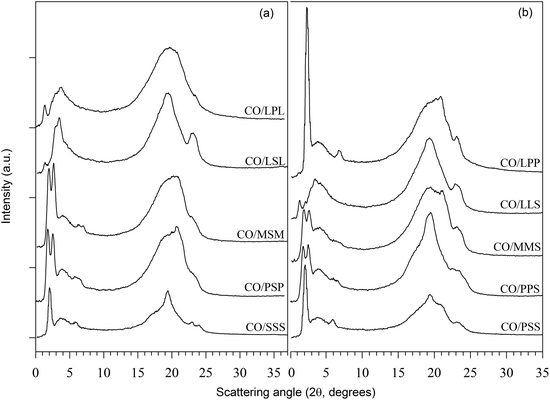 | ||
| Fig. 1 XRD patterns of the CO–TAG mixtures crystallized as described in Section 2.2 and measured at 25 °C. (a) Symmetrical and (b) asymmetrical TAGs. | ||
The width, intensity and overall shape of the wide-angle lines did not reveal wide differences in domain size, density and homogeneity of the solid phase, probably due to the presence of the large amounts of oil. The small-angle diffraction region displayed relatively better resolved peaks (Fig. 1a and b), attributable to large differences in stacking periodicities. The XRD data indicated clearly that the lamellar packing of the TAGs in the β-phase was quite different from those in the β′-phase. Unfortunately, we were not able to accurately estimate the type of lamellar packing of the crystalline structures because of the low number of peaks detected (peaks were probably buried in the liquid and background signals).
PLMs of the mixtures processed as described in Section 2.2 and taken at 25 °C with a magnification of 100× (bar = 100 μm) are shown in Fig. 2a–j, respectively. The inserts are PLMs of the same sample taken at a magnification of 500× (bar = 20 μm). Fig. 2k and l are micrographs of LLS and LSL, respectively, taken at magnification 50× (bar = 20 μm). All the PLMs indicated a radial growth, with CO–PSP showing typical Maltese Crosses and the others either dendrites or needles with branching. The crystalline fibres are, in fact, the primary elements of spherulites, which grow separately, each from a different nucleus. This is not surprising as TAGs often crystallize from the liquid state in the form of spherulites even if non-spherulitic microstructures are not uncommon.33 Note that the formation of needles and dendrites indicates a rapid growth of the crystals through selected facets.
 | ||
| Fig. 2 (a–j) Polarized light micrographs (magnification = 100×; bar = 100 μm) of the mixtures processed as described in Section 2.2 and taken at 25 °C. Insets are PLMs with 500× magnification; same bar = 20 μm. (k and l) Micrographs at magnification 50× (bar = 20 μm) of LLS and LSL, respectively. | ||
As can be seen in Fig. 2a–l, the fat networks presented different crystal shapes and sizes, and a variety of spatial distributions of the solid and liquid phase. Overall, PLM of blends with asymmetrical TAGs showed smaller crystals compared to blends with symmetrical TAGs, due to higher nucleation rates, and a faster initial overall crystal growth. The crystal networks of blends with symmetrical TAGs, with the occurrence of very large crystals, were relatively non- uniform compared to the networks of blends with asymmetrical TAGs. This might be explained by the slower nucleation of the more stable polymorph and its subsequent growth at the expense of the smaller surrounding crystals. A brief description of the microstructure (crystal shape, particle size and distribution, density, and liquid distribution) is reported in Table 1.
| Crystal shape | Crystal size (μm) | Network description | Oil channels width – pockets | % β′ | |
|---|---|---|---|---|---|
| SSS | Dispersed, medium sized highly branched dendrites. Dense core with long branches | Core: 70–120 μm | Relatively uniform | Narrow channels traversing the network. “Oil pockets” were formed between the impinging crystals | 12 ± 3 |
| PSP | Typical Maltese Crosses | 100–120 μm | Relatively heterogeneous; very dense | Interconnected long and narrow (10–20 μm wide). Few pockets of oil. | 53 ± 3 |
| MSM | Star-like dendritic | Large distribution in crystal size, some were large (240 μm) | Very homogeneous. Crystals sparcely distributed | Crystals practically unconnected swimming in a sea of oil. | 46 ± 5 |
| LSL | Long, and highly branched dendrites | Very large, only 1 crystal observed on a slide | Uniform with medium density. Crystal constituted of intricate and wavy, branches | Relatively long and narrow interconnected channels streaming from secondary branches. | 10 ± 3 |
| LPL | Highly branched fibrils intertwined closely at contact | 100–1000 μm | Relatively dense network with irregularly distributed crystals | 40 ± 3 | |
| PSS | Uniformly distributed, nonconnected dendrites | 20–40 μm | Homogeneous and uniform. Densely packed | Narrow channels of ∼10 μm. No oil pockets | 49 ± 4 |
| PPS | Highly branched dendrites | 100–120 μm | Homogeneous and uniform. Densely packed | Very narrow open channels (<10 μm). No oil pockets | 15 ± 7 |
| MMS | Spherical core from which few (an average of ∼5) highly branched long needles grew radially | Core: 100 μm, long needles: ∼800 μm | Heterogeneous with intricate distribution of the branches. Medium density (cores are about 100 μm apart) | Few oil channels. Oil mostly trapped between the branches. Few pockets of oil | 42 ± 3 |
| LLS | Large and highly branched dendrites with multitude of stemming bifurcations | Very large, only 1 crystal observed on a slide | Uniform. Densely branched. | Relatively narrow oil channels | |
| LPP | Radial needle-like fibres originating from the nucleus almost without further forking | ∼200 μm | Uniform. Low density | Isolated crystallites in the sea of oil | 47 ± 3 |
3.2 Melting behavior of the CO–TAG mixtures
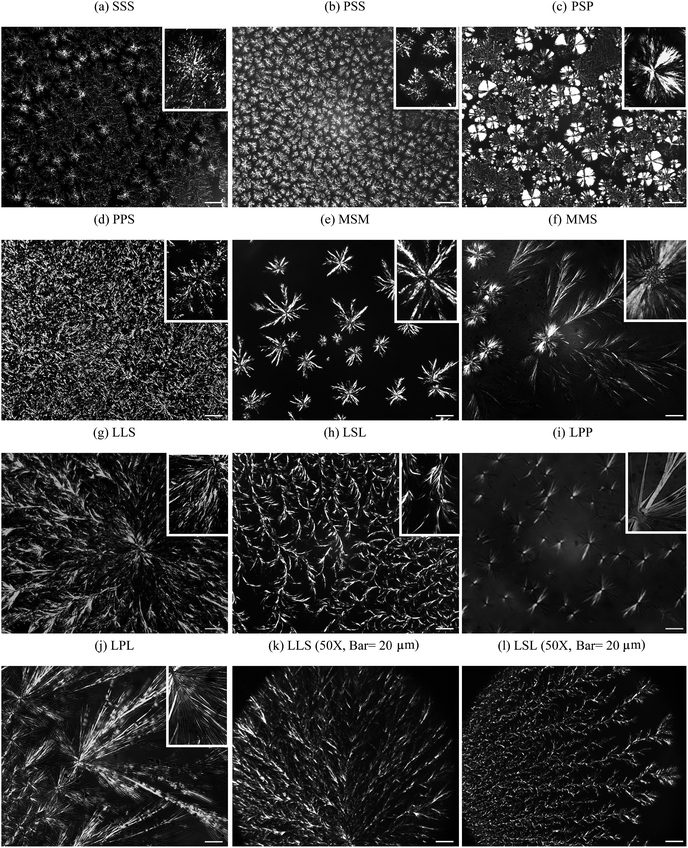
The DSC heating profiles obtained 1 h and 24 h after oil binding experiments are shown in Fig. 3. Except for CO–MSM, which showed two endotherms, the DSC heating thermograms of all the mixtures presented a unique, asymmetrical and relatively broad endotherm. As oil is lost to the Whatman paper during the oil binding experiment, the melting peak sharpened, and its intensity, as well as associated enthalpy, increased noticeably with storage time; this is particularly evident for the CO–SSS sample. The changes observed in the melting profiles of the CO–TAG mixtures are probably linked to rearrangements at different structural levels as a consequence of liquid phase migration and loss. The oil loss is most probably accompanied, at varying degrees, depending on the nature of the TAG, with changes in solid phase distribution and homogeneity, rearrangements and development of the fat crystal network and microstructure, increase in density of the crystallites, and evolution of the crystal packing and order. | ||
| Fig. 3 DSC heating profiles (5 °C min−1) run before oil binding experiments (upper curve), after 1 h and 24 h of oil binding experiment (middle and bottom curve, respectively) of the mixtures: (a) CO–SSS, (b) CO–PSS, (c) CO–PSP, (d) CO–PPS, (e) CO–MSM, (f) CO–MMS, (g) CO–LSL, (h) CO–LLS, (i) CO–LPP and (j) CO–LPL. | ||
Overall, peak temperature (TM), onset of melt (Ton), offset of melt (Toff) of both symmetrical and asymmetrical TAGs showed linear variations with EMC (Fig. 4a–c, respectively) with higher values for the mixtures with symmetrical compared to asymmetrical TAGs whether measured before or after oil binding experiments. The span of melting, as illustrated by the difference ΔT = Toff − Ton, also increased steadily with excess molar carbon (Fig. 4d) but leveled for CO–LPP, CO–LLS and CO–LSL, the mixtures with TAGs with the smallest molar masses. Melting enthalpy ΔHM also increased linearly with molar mass but with no significant difference between the symmetrical and asymmetrical TAGs except for CO–LSL and CO–LLS (Fig. 4e). The linear behavior reflects the high sensitivity of the melting values to molar mass.
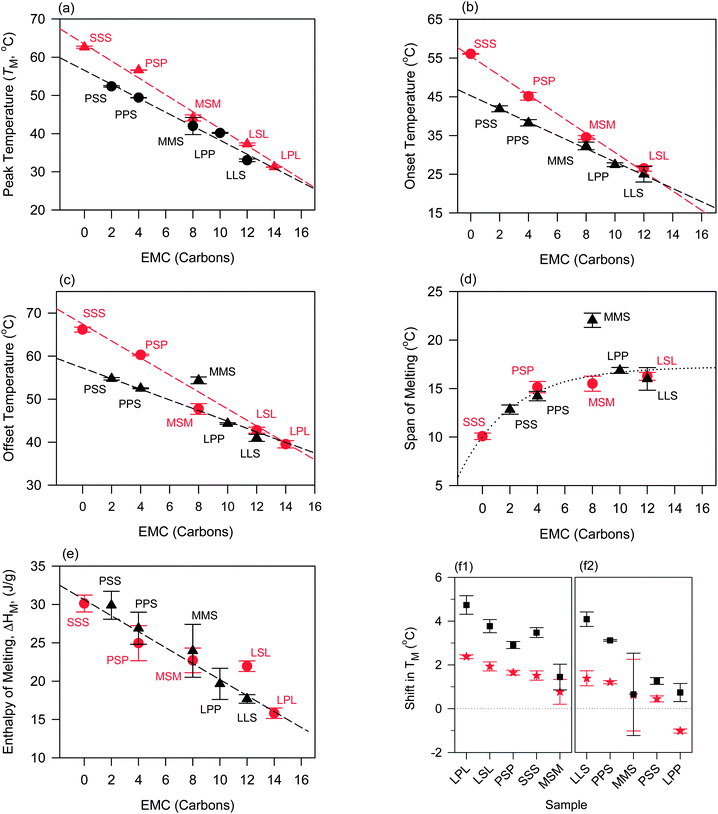 | ||
Fig. 4 Melting values of the mixtures versus excess molar carbon (EMC, carbon atoms) of symmetrical ( ) and asymmetrical (▲) TAGs measured by DSC (heating rate = 5 °C min−1) before oil binding experiments. (a) Peak (TM), (b) onset (Ton), (c) offset (Toff), (d) span (ΔTM) temperature and (e) enthalpy (ΔHM) of melting. (f1 and f2) Shift in TM after 1 h ( ) and asymmetrical (▲) TAGs measured by DSC (heating rate = 5 °C min−1) before oil binding experiments. (a) Peak (TM), (b) onset (Ton), (c) offset (Toff), (d) span (ΔTM) temperature and (e) enthalpy (ΔHM) of melting. (f1 and f2) Shift in TM after 1 h ( ) and 24 h (■) of oil binding experiment in symmetrical and asymmetrical TAG–CO blends, respectively. Dashed lines in (a–c) and (e) are linear fits. ) and 24 h (■) of oil binding experiment in symmetrical and asymmetrical TAG–CO blends, respectively. Dashed lines in (a–c) and (e) are linear fits. | ||
The effect of oil binding experimental time on the melting behavior of the CO–TAG mixtures is most noticeable through the changes observed in the thermal behavior of the mixture after one hour. After 24 hours of experiment, the thermodynamic values shifted comparatively much less, probably due to rapid reorganization and firming of the solid network during the steady state of oil migration and oil loss, and much slower evolution of its characteristics as oil migration and loss proceeds further. The evolution with time of TM, the parameter most affected by oil loss, is shown in Fig. 4f1 and f2 for the symmetrical and asymmetrical TAGs, respectively.
3.3 Oil binding capacity of the CO–TAG mixtures
The OBC as defined in eqn (1) is a measure of the quantity, in grams, of liquid oil bound by one gram of solid network, and is a direct representation of the extent of “Oil Loss”. A quantitative measure of the relative propensity for oil loss can be defined by evaluating the OBC during the initial period of oil loss, when the relative driving and retarding forces for oil loss are at steady state.OBC measured as a function of time (at a constant temperature of 25 °C) depended greatly on the TAG type as shown in Fig. 5a and b for pure fully saturated (hard) symmetrical and asymmetrical TAGs in CO samples, respectively. OBC as a function of time of MF50, FHSO and FHCO in CO samples which have the same fat to oil mass fraction as the TAGs is also represented in the figures. We found that within one hour after measurements have begun (see experimental), the OBC versus time curve is a straight line (R2 > 0.9885) for all the structurants investigated, reflecting steady state conditions. The linear behavior indicates that gravity, and therefore, the actual concentration of liquid oil remaining in the solid matrix, provides the driving force for oil flow. The negative slope indicates simply that as the amount of liquid decreases, the driving force decreases.
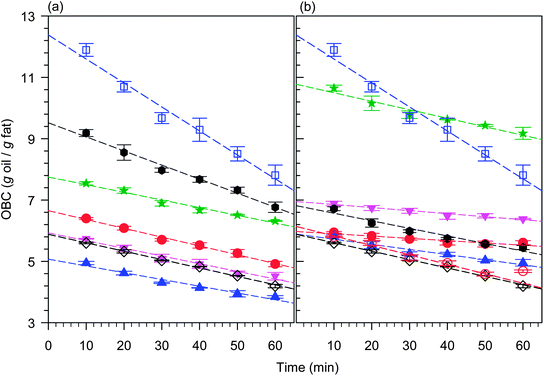 | ||
Fig. 5 OBC measured as a function of time for pure (a) symmetrical and (b) asymmetrical TAGs in CO samples. Dashed lines are linear fits. (a) From top to bottom: MF50 ( ); LPL ( ); LPL (![[hexagon filled, point down]](https://www.rsc.org/images/entities/char_e124.gif) ); LSL ( ); LSL ( ); SSS ( ); SSS ( ); MSM ( ); MSM ( ); FHSO (◇); and PSP ( ); FHSO (◇); and PSP ( ). (b) From top to bottom: LLS ( ). (b) From top to bottom: LLS ( ); MMS ( ); MMS ( ); LPP (); PSS ( ); LPP (); PSS ( ); PPS ( ); PPS ( ); FHCO ( ); FHCO ( ); and FHSO (◇). ); and FHSO (◇). | ||
The linear fit of the OBC versus time plots provided a way to define two key parameters that quantitatively characterize the oil loss phenomenon and therefore describe the relative propensity of the fat network to bind oil. The value of the slope of an OBC versus time curve, when steady state conditions prevail, provides the rate (K) at which oil is lost. It is a quantified representation of the kinetics of oil loss. The value of extrapolated OBC at t → 0 provides the initial amount of oil susceptible to be lost and is a measure of the Propensity for Oil Loss (POL). POL is a function of the network characteristics of the sample before the oil loss begins. It is therefore, dependent on the thermodynamic state and inherent structural hierarchies of the initial network. K and POL can be thought of as the “kinetic and thermodynamic doublet” that characterizes the oil binding capacity of a given structurant. Lower K and higher POL reflect a higher propensity to bind and hold oil, respectively. Note that a high POL reveals a propensity for higher driving forces and may be coupled with a high K.
OBC of hard fats (TAG) should not be considered as the ultimate benchmark for edible oil structuring as the stability of the solid network over time plays a large role in the final property of the product. For example, higher values of OBC do not always mean that the given fat or TAG is an excellent candidate for structuring as it may not be stable over the shelf life of the product.
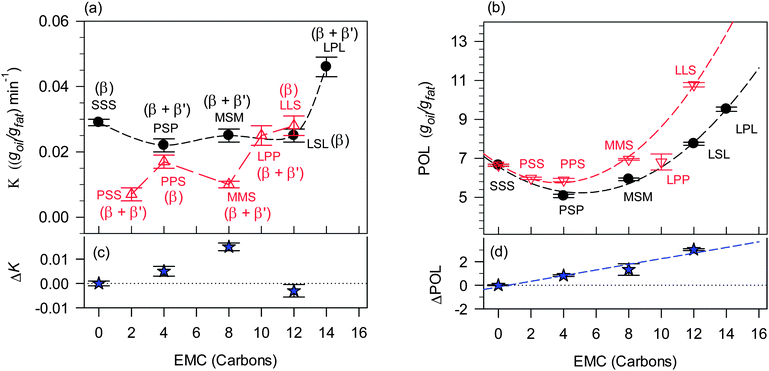
K and POL are listed in Table 2 and plotted as a function of EMC in Fig. 6a and b, respectively. TAG type is shown in Fig. 6a and b above or below the corresponding data point. The nature of the phases detected by XRD in each sample is added above or below the data points to highlight the effect of polymorphism on K and POL. Among all the symmetrical TAGs, the POL of LPL was the highest followed by LSL, SSS, MSM and PSP. LLS had the largest POL among the asymmetrical TAGs, followed by MMS, LPP, PSS and PPS. Note that K did not follow the same trend for either. For example, PSS had the lowest K but not the lowest POL, and PSP had the lowest POL but not the lowest K. This suggests complex relationships between the thermodynamics and kinetics of oil loss and the chemistry of the structurant and the different levels of structure presented by its crystallized network. Indeed, the habit of the crystals of the TAGs plays a key role. In addition to providing insight into the molecular influences of oil binding, Fig. 6a and b also provide an index of structuring propensity. Understandably, for a good structurant, one would expect relatively high levels of POL and relatively low levels of K.
| EMC (carbon atoms) | Molar mass (g mol−1) | K (goil/gfat) min−1 | POL (goil/gfat) | |
|---|---|---|---|---|
| SSS | 0 | 891.4800 | 0.029 ± 0.003 | 6.651 ± 0.050 |
| PSS | 2 | 863.4280 | 7.0 × 10−3 ± 0.002 | 5.972 ± 0.061 |
| PSP | 4 | 835.3680 | 0.022 ± 0.002 | 5.069 ± 0.085 |
| PPS | 4 | 835.3680 | 0.017 ± 0.002 | 5.886 ± 0.088 |
| MSM | 8 | 779.2698 | 0.025 ± 0.002 | 5.925 ± 0.071 |
| MMS | 8 | 779.2698 | 0.010 ± 0.001 | 6.949 ± 0.044 |
| LPP | 10 | 751.2057 | 0.025 ± 0.003 | 6.814 ± 0.410 |
| LLS | 12 | 723.1635 | 0.028 ± 0.003 | 10.775 ± 0.114 |
| LSL | 12 | 723.1635 | 0.025 ± 0.002 | 7.750 ± 0.078 |
| LPL | 14 | 695.1035 | 0.046 ± 0.003 | 9.521 ± 0.111 |
| FHCO | — | — | 0.031 ± 0.002 | 6.136 ± 0.077 |
| FHSO | — | — | 0.027 ± 0.001 | 5.878 ± 0.033 |
| MF50 | — | — | 0.078 ± 0.006 | 12.382 ± 0.233 |
 | ||
| Fig. 6 (a) K and (b) POL plotted as a function of Excess Molar Carbon (EMC). (c and d) Difference in K and POL between symmetrical and asymmetrical TAGs. Dashed lines are guides for the eye in panel (a), polynomial fits in panel (b) and a linear fit in panel (d). The dotted line in panels (c and d) show the zero reference. | ||
Fig. 6a and b show that, overall, the symmetrical TAGs have a weaker ability to bind the oil compared to the asymmetrical TAGs. They demonstrate high POL and K values. Indeed, as discussed before, it could be because they demonstrate high POL values that they also record high K values due to the increased propensity for higher driving forces. Note the different trends in POL and K between symmetrical and asymmetrical TAGs. Overall decreasing POL-values are associated with relatively similar K-values in the case of symmetrical TAG (SSS, MSM, LSL and PSP), and with sharply decreasing K-values in the case of asymmetrical TAGs (from LLS, LPP, PPS, MMS to PSS). This can be explained by the difference in their crystallization behaviour, which leads to differences in polymorphism and subsequent microstructure, which then impacts OBC.
The dramatic effects of symmetry and chain-length mismatch on physical properties are well documented in our studies of the phase behaviour of symmetrical–asymmetrical binary systems of TAGs (LSL–LLS,34 PSP–PPS,35 CSC–CCS,36 and MSM–MMS37). Asymmetrical TAGs have higher nucleation and growth rates at the early stages of the crystallization process compared to symmetrical TAGs. The crystals of asymmetrical TAGs impinge earlier while those of the symmetrical TAGs continue to grow larger due to a smaller number of centers of growth. In the presence of oil, the crystal network of symmetrical TAGs, therefore, matures earlier and ripening occurs relatively rapidly, compared to that of asymmetrical TAGs, which ripens more slowly due to unstable thermodynamic states being initially formed in the rapidly formed network. The differences observed in the microstructure of networks composed of symmetrical and asymmetrical TAGs as a result of such considerations are described and discussed in detail in a later section in this paper (Section 3.3).
As can be seen in Fig. 6a, the K-value of mixtures with symmetrical TAGs started with a somehow higher value for SSS (EMC = 0), plateaued for PSP, MSM and LSL (EMC = 4, 8 and 12, respectively) then rose dramatically for LPL (EMC = 14). The oil loss behavior of the samples with TAGs having EMC of up to 12 (Fig. 6a), suggests that the rate at which oil is lost in mixtures structured by saturated TAGs having at least one stearic acid does not significantly depend on the nature of the two other constituent fatty acids, but rather on the polymorphic phase and ultimately on the details of the crystal network. Note that LSL with an EMC of 12 had almost the same K-value as the other symmetrical TAGs which have a stearic acid.
Mixtures with asymmetrical TAGs all have lower K-values compared to mixtures with symmetrical TAGs and displayed comparatively more irregular behaviour as a function of molar mass (Fig. 6a). PSS (EMC = 2) demonstrated the lowest K, followed by MMS (EMC = 8) then PPS (EMC = 4). The asymmetrical TAGs with large EMC, including LLS which has a stearic acid, displayed much higher K-values, also consistent with their polymorphism. Note that LPP and LLS had a K value which is similar to the K-values of the symmetrical TAGs.
POL versus EMC follows quadratic polynomial functions for both symmetrical and asymmetrical saturated TAGs (Fig. 6b). Overall, POL of symmetrical TAGs was lower than those of asymmetrical TAGs and passes through minima at EMC = 4 for both, i.e. PSP and PPS, respectively. Note that PSS has as low a POL-value as PPS.
The impact of molecular symmetry on K and POL can be appreciated through the differences in these values between symmetrical and asymmetrical molecules. The difference ΔK = KAsym − KSym between asymmetrical and symmetrical TAGs (Fig. 6c) did not have any apparent defined trend, whereas, ΔPOL = POLAsym − POLSym (Fig. 6d) increased almost linearly with increasing EMC. It is obvious that polymorphism and microstructural details had a larger impact than molecular symmetry on K, whereas, molecular symmetry and intersolubility seemed to largely determine the POL-value.
Note that the trends observed in the pair of POL and K values indicate that, overall, symmetrical TAGs demonstrate weaker ability to hold the oil than the asymmetrical TAGs. The relative differences observed in OBC between symmetrical and asymmetrical TAGs suggest that the interactions between TAGs are affected by positional isomerism. The differences measured in OBC between the symmetrical and asymmetrical TAGs seem to indicate that having a long fatty acid chain, such as the stearic acid, in the sn-1 or sn-3 position makes it more available for attraction than in the sn-2 position. Moreover, symmetrical TAGs do not experience the same geometric restrictions to ordering inflicted on the system by the asymmetrical molecular structures.
The CO/MF50 system demonstrated much higher K- and POL-values compared to CO/FHCO, CO/FHSO, or any other pure TAG studied, confirming the link between higher POL and higher driving forces. As reported in our previous studies,17 the differences in propensity to bind oil between FHCO and FHSO can be understood in terms of differences in crystal network details, particularly evident in crystal size and shape. Also, one can expect FHCO to have a relatively higher wettability in canola compared to FHSO because of molecular similarity between CO and FHCO, which would result in a higher oil binding capacity.
One might expect the structuring capability of FHCO and FHSO to be determined mainly by their two main TAG constituents, SSS and PSS (FHCO consists of 80% SSS and 16% PSS and FHSO consists of 68% SSS and 27% PSS). Indeed, there are measurable differences in the K and POL of FHSO and FHCO due to their relatively large difference in SSS and PSS content (Table 2). However, SSS displayed a slightly higher POL than FHCO and FHSO and a similar K-value whereas PSS presented a POL similar to that of the two fully hydrogenated fats and a lower K-value (Table 2). This suggests that there are limits in composition at which these two TAGs are effective in structuring oil, although the differences may possibly be further maximized by enhancing network differences through differences in processing conditions. The differences in OBC values between FHSO and FHCO relate well with the weighted K- and POL-values of their main constituents SSS and PSS, confirming the differentiated contribution of PSS and SSS to the thermodynamics and kinetics of oil loss. The K- and POL-values suggest that while PSS is the determinant factor in setting the thermodynamics of oil binding, SSS is the dominant driver for the kinetics of oil loss.
OBC behaviour of the saturated TAG structurants can be partially explained by simple intermolecular interactions. The behavior of fat crystals in solution is largely determined by the balance between effective attractions and excluded volume repulsions. The volume exclusion is caused by either high concentration of solute, prior to the nucleation stage or when the molecular size of the solute is larger than the solvent. The intermolecular interactions are believed to be responsible for different stabilization38 and solvation39 properties of fatty acids. Comparing the behaviour of saturated and non-saturated mono-carboxylic acids in a non-polar solvent,38 found that interactions of myristic and oleic acid in solutions are repulsive, whereas, interactions between stearic acid molecules are attractive (most likely due to van der Waals forces). Attraction in stearic acid may be strong enough to be the major contribution that provides in total the effective attraction. This could explain in part the relatively higher ability of TAGs containing stearic acid to bind oil more effectively compared to the other fatty acids. After allowance has been made for symmetry and polymorphism effects, the suggestion that the attractive contribution increases as chain length increases from lauric, myristic, palmitic to stearic acids40 may explain the “smooth polynomial’ changes observed in POL versus EMC and the proportional differences observed in POL between saturated TAGs. Note that, when the network is set, this contribution is only effective in the close vicinity of the walls of the solid phase, and that higher attractions do not necessarily mean higher POLs.
The data shown in Fig. 6a suggest that the presence of a β′-phase is much more prone to promote a lower oil loss rate than the β-phase. It may be possible, for example, that SSS had a larger K-value compared to the other symmetrical TAGs because it was mostly crystallized in the β-form rather than the β′-form. The large difference in β′-phase content between MMS (42% β′) and PPS (15% β′) may explain the lower K-value of MMS even if its molar mass is smaller (4 less carbon atoms). Also, the large β′-phase content (∼50% β′) combined with the presence of a second stearic acid in PSS was probably instrumental in it displaying the lowest K-value. Note that the effect of polymorphism on the rate of oil loss was less effective for higher molar mass as prominently highlighted for TAGs which do not have stearic acid, i.e., LPP (EMC = 10) and LPL (EMC = 14) which displayed the highest K-values.
There are obvious strong correlations between the microstructure and the thermodynamics and kinetics of oil loss. The relationships between the details of the solid network observed by PLM (size and distribution of the crystals, solid network homogeneity and density, size of the oil channels), and K and POL are obviously complex. However, prominent network characteristics of the different mixtures, such as the surface area to volume ratio, inherent capillaries and lipid channels are well correlated with K and POL. This highlights that although molecular ensemble dictates the structuring capacity of a solid TAG for a given oil, changes in the propensity to bind oil, or changes in POL and K, can also be procured through processing-induced changes in polymorphism and microstructure.
Smaller crystals lead to higher surface area to volume ratio and therefore to higher binding capacity. Solid networks consisting of densely packed and uniformly distributed small crystals demonstrated the best capacity to bind and hold the oil. The capacity to hold the oil was optimal when narrow channels of oil were also observed, as for example in the case of CO–PSS, which displayed the lowest K-value. The presence of “oil pockets” was correlated with higher POL and comparatively adversely affected the K-value, as observed in SSS compared to PSS in which no oil pockets were observed.
It is worth noting that regardless of the shape of the crystals, the mixtures which displayed a homogeneous microstructure with crystals, having large (SSS and PPS: 120 μm to LSL and LLS: >1200 μm) and narrowly distributed sizes (±10 μm), have been detected mostly in the β-form, whereas, those which displayed less homogeneous microstructures and had networks which were constituted of a combination of small (∼20–40 μm) to medium and large sized crystals (120–1000 μm) were detected in both the β- and β′-form.
The distribution of large and small crystals in the networks may correspond to that of the coexisting polymorphic phases in these mixtures: the small crystals were in the β′-phase and the large crystals in β-phase. A higher nucleation rate followed by a slowing down then halting of growth has probably led to the formation of the small crystals concomitantly with a slower nucleation and an accelerated growth, which has led to the formation of the large crystals. This analysis is only valid because all of our mixtures have been crystallized under the same conditions, because as is well known, the shape and size of the crystallites may vary widely depending on the TAG constituents and processing conditions of the materials.41
The overall thermal behavior of the CO–TAG mixtures, as shown clearly by the thermograms of MMS and LLS, and by the weakly shouldered events in the others, is probably due to the development of more stable polymorphic forms via rearrangement and ordering of existing polymorphs or/and via solid–solid transitions, and changes in the solid–liquid interface energy. The appearance of distinguishable shoulders and their location in the melting profiles is a good indication of the formation of such more stable forms and a support for our analysis.
The case of CO–SSS and CO–PSS highlights the complex relationships between the OBC and thermal behaviour. The melting behaviour of CO–SSS and CO–PSS can explain in part the differences observed between their K and POL, and shed some light into the relative differences between K and POL of FHCO and FHSO. CO–SSS, which had a larger POL than PSS, displayed a sharper peak with a higher peak temperature and enthalpy of melting than CO–PSS at time zero of oil binding experiments, confirming a close relationship between these parameters. The differences observed in K-values closely followed the differences in evolution of the respective thermal behaviour of the mixtures as a function of storage time. The higher K-values of CO–SSS, for example, can be related to the significant change of its melting values during oil loss compared to CO–PSS in which melting values did not change substantially. Again, such “kinetic” indicators are expected to correlate closely with K.
The mixtures made of CO and TAGs that have the lowest molar masses, such as LLS, LSL and LPP, presented the lowest thermodynamic values and the largest span of melting. Colligative effects cannot be excluded in these cases and sizable solubility in CO has probably occurred. The largest changes in thermal values observed for these samples after 1 hour of oil binding experiment, particularly dramatic in the enthalpy of melting, can be linked to their high K-values. Obviously competing effects (molar mass, symmetry, and polymorphism) are at play in determining the OBC of these low molar mass TAGs.
As can be seen in Fig. 7, the SFC versus fat content plots of CO/FHCO, CO/FHSO as well as CO/MF50, are linear with linear fits (R2 > 0.9934) yielding slopes of 0.91, 0.87 and 0.64, respectively. This indicated that CO/FHCO and CO/FHSO have limited solubilities that are close to the ideal case, whereas, CO/MF50 deviated significantly from an ideal behavior. Obviously relatively large amounts of low melting point (LMP) species are present in MF50 and dissolve readily in the liquid and hence contribute to oil loss rather than binding.
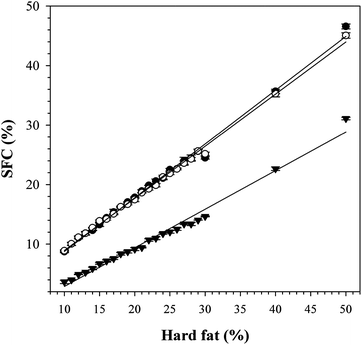 | ||
| Fig. 7 SFC versus fat content of CO/MF50 (▼), CO/FHCO (●) and CO/FHSO (○). The solid lines are linear fits. | ||
The high propensity of MF50 for oil loss is therefore probably due, for a large part, to its high solubility in CO. Because PSS and SSS are the main components in FHCO and FHSO, they are expected to have similar solubility behavior as the hydrogenated oils and therefore, limited contribution of solubility to oil loss. Note that SFC versus solid TAG content of the pure TAGs have not been measured for reasons of cost.
4. Conclusions
Several pure saturated triacylglycerols (TAGs) in canola oil (CO) model systems have been utilized to analyze oil loss in structured oil both from the thermodynamic and kinetic aspects. For the first time, two parameters which represent the kinetics and thermodynamics of oil binding capacity (OBC) of structurants have been defined. The rate of oil loss, K, and the initial amount of oil susceptible to be lost, i.e., the propensity for oil loss (POL), provided an effective and predictive measure of the kinetics and thermodynamics of oil binding capacity (OBC) of structurants, respectively. Furthermore, the thermodynamic and kinetic dependency of the oil loss to the nature of the crystalline TAG has been elucidated. For example, comparison of the OBC versus time plots of the hydrogenated canola and soybean oils with those of their main constituent suggested that while one component (the asymmetrical PSS) is the determinant factor in setting the thermodynamics of oil binding, the other (the symmetrical SSS) is the dominant driver for the kinetics of oil loss.Strong and complex relationships between OBC and the microstructure of the solid network in CO, the liquid phase, have been evidenced. The prominent network characteristic such as the surface area to volume ratio, inherent capillaries and lipid channels were well correlated with K and POL. Solid networks having the highest surface area to volume ratio, i.e., those consisting of densely packed and uniformly distributed small crystals separated by narrow channels of oil, demonstrated the optimal capacity to bind oil. The presence of “oil pockets” was correlated with higher POL and adversely affected the K-value. Small and uniformly distributed solid fat crystals are found to be better structurants of edible oil than large crystals, while parameters such as symmetry of the TAG molecules and their melting profiles are only guiding parameters leading the changes in the TAG habits and network formation. The data suggest that the presence of a β′-phase is much more prone to promote a lower oil loss rate than the β-phase, due to induced habit. The microstructure of mixtures predominantly in the β-form was homogeneous with crystals having large and narrowly distributed sizes, whereas, that of a mixture in the β- and β′-form was less homogeneous and had networks which were constituted of a combination of small to medium and large sized crystals.
The solubility of the hard fat is an important factor to be considered as it greatly affects the OBC. If the fat used as a structurant has a high solubility in edible oil, the amount of oil per SFC will be significantly high and will show high OBC values. Low melting TAGs favor interactions between solvent and solute (oil and fat, respectively), demonstrate a typical dilution effect and result in fewer TAG molecules available to crystallize and consequently, in a smaller solid fraction.
The kinetic and thermodynamic “doublet” (K and POL) is critical to understanding TAG structuring and choosing the adequate fat structurant in optimal concentration that achieves a desirable OBC.
Acknowledgements
The financial support of Elevance Renewable Sciences, NSERC, Grain Farmers of Ontario, GPA-EDC, Industry Canada, and Trent University is gratefully acknowledged.References
- M. A. Rogers, Food Res. Int., 2009, 42, 747–753 CrossRef CAS.
- M. A. Rogers, A. J. Wright and A. G. Marangoni, Soft Matter, 2008, 4, 1483–1490 RSC.
- P. Wassell, G. Bonwick, C. J. Smith, E. Almiron-Roig and N. W. G. Young, Int. J. Food Sci. Technol., 2010, 45, 642–655 CrossRef CAS.
- M. Pernetti, K. F. van Malssen, E. Floter and A. Bot, Curr. Opin. Colloid Interface Sci., 2007, 12, 221–231 CrossRef CAS.
- R. Mezzenga and S. Ulrich, Langmuir, 2010, 26, 16658–16661 Search PubMed.
- A. I. Romoscanu and R. Mezzenga, Langmuir, 2006, 22, 7812–7818 CrossRef CAS.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- D. Rousseau and P. Smith, Soft Matter, 2008, 4, 1706–1712 RSC.
- V. Ghosh, G. R. Ziegler and R. C. Anantheswaran, Crit. Rev. Food Sci. Nutr., 2002, 42, 583–626 CAS.
- P. C. Bomba, in Shelf Life Studies of Foods and Beverages, ed. G. Charalambous, Elsevier, London, UK, 1993 Search PubMed.
- G. Løvoll, Y. Méheust, K. J. Måløy, E. Aker and J. Schmittbuhl, Energy, 2005, 30, 861–872 Search PubMed.
- G. Ziegleder and I. Schwingshandl, Fett/Lipid, 1998, 100, 411–415 CrossRef CAS.
- G. Ziegleder, C. Moser and J. GeierGreguska, Fett/Lipid, 1996, 98, 196–199 CrossRef CAS.
- G. Ziegleder, C. Moser and J. GeierGreguska, Fett/Lipid, 1996, 98, 253–256 CrossRef CAS.
- Y. J. Choi, K. L. McCarthy, M. J. McCarthy and M. H. Kim, Appl. Magn. Reson., 2007, 32, 205–220 Search PubMed.
- J. M. Aguilera, M. Michel and G. Mayor, J. Food Sci., 2004, 69, R167–R174 Search PubMed.
- T. S. Omonov, L. Bouzidi and S. S. Narine, Chem. Phys. Lipids, 2010, 163, 728–740 CrossRef CAS.
- S. Marty, K. Baker, E. Dibildox-Alvarado, J. N. Rodrigues and A. G. Marangoni, Food Res. Int., 2005, 38, 1189–1197 CrossRef CAS.
- J. R. Galdámez, K. Szlachetka, J. L. Duda and G. R. Ziegler, J. Food Eng., 2009, 92, 261–268 CrossRef CAS.
- E. O. Afoakwa, A. Paterson, M. Fowler and J. Vieira, J. Food Eng., 2009, 91, 571–581 Search PubMed.
- S. S. Narine and A. G. Marangoni, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 59, 1908 CrossRef CAS.
- A. Bot, E. Flöter, J. G. Lammers and E. G. Pelan, in Understanding and Controlling the Microstructure of Complex Foods, ed. D. J. McClements, Woodhead Publishing, Cambridge, 2007, ch. 575–599, p. 833 Search PubMed.
- D. W. de Bruijne and A. Bot, in Food Texture: Measurement and Perception, ed. A. J. Rosenthal, Aspen Publishers, Inc., Gaithersburg, MD, USA, 1999, ch. 7, pp. 185–227 Search PubMed.
- D. V. Chandran and R. K. Bhatnagar, J. Am. Oil Chem. Soc., 1968, 45, 581–582 Search PubMed.
- P. H. Bentley and W. McCrae, J. Org. Chem., 1970, 35, 2082–2083 CrossRef CAS.
- K. Larsson, in The Lipid Handbook, ed. F. D. Gunstone, J. L. Harwood and F. B. Padley, Chapman and Hall, London, 1986, pp. 335–377 Search PubMed.
- D. Precht and E. Frede, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 381–388 Search PubMed.
- D. A. Fahey, D. M. Small, D. R. Kodali, D. Atkinson and T. G. Redgrave, Biochemistry, 1985, 24, 3757–3764 Search PubMed.
- R. E. Timms, Prog. Lipid Res., 1984, 23, 1–38 CrossRef CAS.
- M. Kellens, W. Meeussen, C. Riekel and H. Reynaers, Chem. Phys. Lipids, 1990, 52, 79–98 Search PubMed.
- B. S. Ghotra, S. D. Dyal and S. S. Narine, Food Res. Int., 2002, 35, 1015–1048 CrossRef CAS.
- L. Bouzidi, M. Boodhoo, K. L. Humphrey and S. S. Narine, Thermochim. Acta, 2005, 439, 94–102 Search PubMed.
- M. Kellens, W. Meeussen and H. Reynaers, J. Am. Oil Chem. Soc., 1992, 69, 906–911 Search PubMed.
- L. Bouzidi, M. V. Boodhoo, T. Kutek, V. Filip and S. S. Narine, Chem. Phys. Lipids, 2010, 163, 607–629 Search PubMed.
- M. V. Boodhoo, L. Bouzidi and S. S. Narine, Chem. Phys. Lipids, 2009, 160, 11–32 Search PubMed.
- M. V. Boodhoo, L. Bouzidi and S. S. Narine, Chem. Phys. Lipids, 2009, 157, 21–39 Search PubMed.
- M. V. Boodhoo, T. Kutek, V. Filip and S. S. Narine, Chem. Phys. Lipids, 2008, 154, 7–18 Search PubMed.
- V. I. Petrenko, M. V. Avdeev, L. Almásy, L. A. Bulavin, V. L. Aksenov, L. Rosta and V. M. Garamus, Colloids Surf., A, 2009, 337, 91–95 Search PubMed.
- R. Tadmor, R. E. Rosensweig, J. Frey and J. Klein, Langmuir, 2000, 16, 9117–9120 CrossRef CAS.
- M. V. Avdeev, D. Bica, L. Vékás, V. L. Aksenov, A. V. Feoktystov, O. Marinica, L. Rosta, V. M. Garamus and R. Willumeit, J. Colloid Interface Sci., 2009, 334, 37–41 Search PubMed.
- A. G. Marangoni and S. Narine, Physical Properties of Lipids, Marcel Dekker, New York, 2002 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |