Bubble bursting as an aerosol generation mechanism during an oil spill in the deep-sea environment: laboratory experimental demonstration of the transport pathway
Franz S.
Ehrenhauser†
,
Paria
Avij
,
Xin
Shu
,
Victoria
Dugas
,
Isaiah
Woodson
,
Thilanga
Liyana-Arachchi‡
,
Zenghui
Zhang
,
Francisco R.
Hung
and
Kalliat T.
Valsaraj
*
Cain Department of Chemical Engineering, Louisiana State University, Baton Rouge, LA 70803, USA. E-mail: valsaraj@lsu.edu
First published on 5th November 2013
Abstract
Oil spills in the deep-sea environment such as the 2010 Deep Water Horizon oil spill in the Gulf of Mexico release vast quantities of crude oil into the sea-surface environment. Various investigators have discussed the marine transport and fate of the oil into different environmental compartments (air, water, sediment, and biota). The transport of the oil into the atmosphere in these previous investigations has been limited to only evaporation, a volatility dependent pathway. In this work, we studied the aerosolization of oil spill matter via bursting bubbles as they occur during whitecaps in a laboratory aerosolization reactor. By evaluating the alkane content in oil mousse, crude oil, the gas phase, and particulate matter we clearly demonstrate that aerosolization via bursting bubbles is a solubility and volatility independent transport pathway for alkanes. The signature of alkane fractions in the native oil and aerosolized matter matched well especially for the less volatile alkanes (C20–C29). Scanning electron microscope interfaced with energy dispersive X-ray images identified the carbon fractions associated with salt particles of aerosols. Theoretical molecular dynamics simulations in the accompanying paper lend support to the observed propensity for alkanes at air–salt water interfaces of breaking bubbles and the produced droplets. The presence of a dispersant in the aqueous phase increased the oil ejection rate at the surface especially for the C20–C29 alkanes. The information presented here emphasizes the need to further study sea-spray aerosols as a possible transport vector for spilled oil in the sea surface environment.
Environmental impactThe adsorption of alkanes at the air–water interface and the subsequent ejection of the interface via bursting bubbles is a volatility and solubility independent transport vector for organic oil spill matter into the atmosphere. The application of dispersants facilitates the aerosolization and evaporation predominantly by enhancing the dispersion of the oil in the water column and improves therefore the flotation capacity of the bubbles. This aerosolization of oil spill matter via bursting bubbles of whitecaps might be of particular importance for the fate of semi-volatile organic compounds as dissolution, microbial degradation, and evaporation are negligible for these compounds. |
Introduction
Crude oil spills in the deep-sea environment are increasingly frequent since offshore oil and gas exploration has become more technologically feasible. The Deep Water Horizon oil spill in 2010 is one of the largest marine oil spills to occur (4.9 × 106 barrels) and is further unique because of additional facts, viz., the location of the blowout (1500 m below the surface), the distance from the coast (approximately 80 kilometers), and the unprecedented sub- and surface application of dispersants (1.8 × 106 gallons).1,2 Dispersants broke up the oil into smaller droplets. Considering the large amount of oil released and the long distances, the fate and transport vectors are of particular interest as short- and long-term consequences for the ecosystem depend on the location and the type of exposure (dispersed, dissolved, bulk) to the oil.Fig. 1 depicts schematically the transport and fate vectors of the oil during the spill. The release of oil and gas happened 1500 m below sea level, allowing for several sub-surface pathways such as the oil partially dissolving, sinking, levitating, and largely rising to the surface.3 The best mass balance estimate for the spilled oil accounted for 20% of oil skimmed or recovered, 5% burned at the site, 5% of oil evaporated and the rest dispersed in the water column.4–6 Since all possible transport processes were not explicitly characterized, there is still debate regarding the exactness of the oil and dispersant mass balances. Once the oil–gas mixture reaches the sea surface, we can envisage a series of atmospheric transport processes besides the marine transport processes. Firstly, volatile organic compounds (VOC) will quickly evaporate and potentially undergo transformations in the air. Many intermediate volatile compounds (IVOC) and semi-volatile organic compounds (SVOC) will also appear in the air; these can also undergo oxidation and further condense/adsorb on existing aerosols and grow in size.7,8 Since the oil loses its volatile fraction quickly as the plume moves further, the organic carbon (OC) profile in air shows an exponential decline from the spill. Field observations by de Gouw et al.9 showed that substantial amounts of organic aerosol (OA) were found downwind of the surface oil sheen. The observed OA was exclusively ascribed to the transport across the marine/atmospheric boundary layer attributable to evaporation, a transport vector that exponentially declines with time or distance traveled.
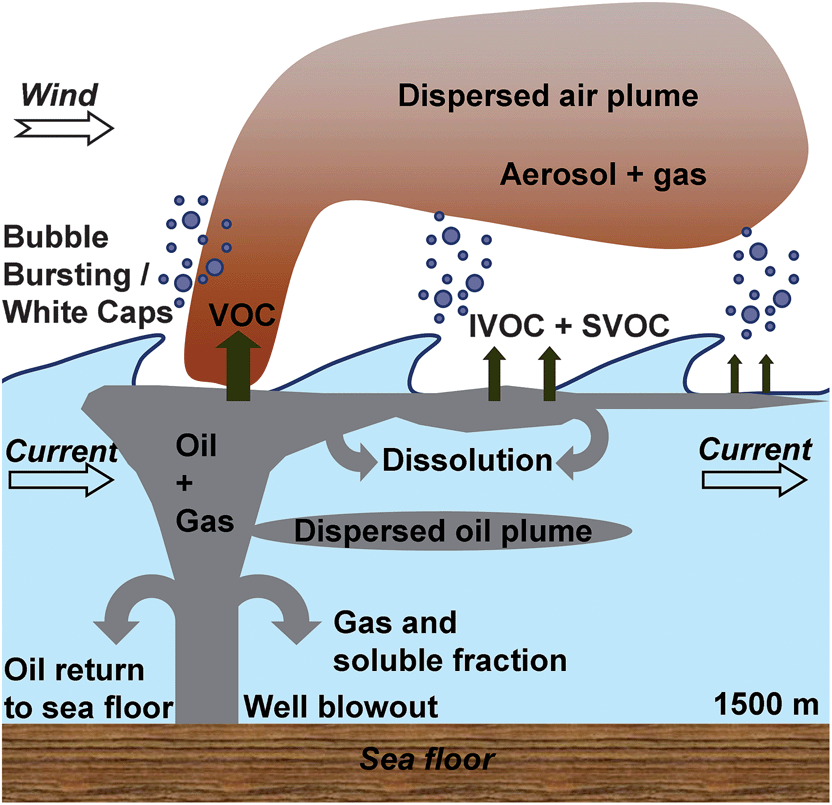 | ||
| Fig. 1 Fate of oil during a deep oil spill (schematic). | ||
The transport via aerosolization, the non-evaporative ejection of material into the atmosphere, has been largely ignored. This is surprising in so far as the generation of ocean spray and mist by whitecaps caused by waves is well-known, and one of the primary production mechanisms for particulate matter in the atmosphere, capable of generating 3.5 × 1012 kg per year.10 This forms the major mechanism of production of the so-called cloud condensation nuclei (CCN) in the atmosphere.11,12
Bubble bursting is capable of ejecting organic matter into the atmosphere and has been shown to cause bacteriological enrichment of aerosols over the ocean, the ejection of surface-active compounds and hydrophobic particles.13–17 The exact mechanism of spray generation at the surface of water by breaking bubbles and the various conditions that affect the phenomena have been a subject of much discussion in the marine chemistry literature.18–22 Although the effect of surfactants on sea spray aerosol generation has been studied, there is contradictory evidence regarding the size and quantity of aerosol generation.23,24 In all such studies typical water-soluble surfactants have been used. However, the use of dispersants in oil spill remediation is quite different in that dispersants are formulations of mainly lipophilic surfactant mixtures and organic solvents, and they are used to not only disperse the oil slick, but also prevent coalescence of the oil. To the best of our knowledge the ejection of solely crude oil by bubble sprays and the effect of added dispersants have not been investigated extensively.11 We adapted a well-known reactor configuration that was used for understanding the aerosol generation via the bubble bursting mechanism.25 Our work addresses the need for evaluating the pathway of aerosolization and in the following we present our results concerning the ejection of alkanes from crude oil, representing 10% (w/w) of the oil.26 In addition we also present observations with regard to the influence of the dispersant Corexit 9500A on the efficacy of aerosol generation. However, we conducted these experiments in a solution of sodium chloride with ionic strength analogous to natural seawater. Finally, classical molecular dynamics (MD) simulations presented in the accompanying paper provide a complementary molecular-level picture of the phenomena taking place between oil alkanes and dispersants at the air–salt water interface.
Experimental
Chemicals and oil samples
Oil mousse was collected on 04/30/2010, ten days after the onset of the Deep Water Horizon spill on a beach close to Port Fourchon, LA, USA. Surrogate oil was provided by the BP Gulf Coast Restoration Organization (Houston, TX, USA). Details of the properties of the surrogate oil used are available from the BP Gulf Coast Restoration Organization. Ethyl acetate (HPLC grade), anhydrous sodium sulfate (pesticide grade) and sodium chloride (per analysis) were procured from Mallinckrodt Chemicals (St Louis, MO, USA). Reference standard solutions of alkanes, methyl arachidate and methyl octacosanoate were procured from Sigma Aldrich (St Louis, MO, USA). All chemicals were used as received. Corexit 9500A was used as received from Nalco Environmental Solutions, LLC (Sugar Land, TX, USA). Details of the composition of Corexit 9500A are available directly from Nalco Environmental Solutions.Aerosolization reactor
In this work, we adapted a bubble column reactor described by Smith et al.27 to simulate the aerosol generation of oceanic whitecaps in the laboratory. We based this on the earlier work of Keene et al.25 Although there have been several evaluations of sea spray aerosol generation in the laboratory,28,29 we chose the method used by Keene et al.25 in this work because of its ability to produce small-sized aerosol, with enhanced transport capacity and environmental relevance. Fig. 2 shows the schematic of the aerosolization reactor. It consists of a 11.5 cm wide and 1.5 m long glass tube which houses an annular shear sparger at the bottom of the column, an external peristaltic circulating pump (Masterflex I/P 77600-62, Thermo Fisher Scientific, Barrington, IL, USA) providing the liquid shear flow, a ring shaped air lift inside the column, and an electrostatic precipitator (Ionic Spore Trap, DS Scientific, Baton Rouge, LA) or a constant mass flow sampling system at the top of the column for collecting particulate matter. The shear sparger incorporates a cylindrical porous frit, which generates small-sized bubbles (0.25 ± 0.04 mm) that are being sheared off by the strong annular liquid flow. Around the frit, the uniform bubbles rise to the surface, burst at the air–water interface (0.3–4 mm bursting diameter), and generate aerosol droplets which are dried up to particulate matter and transported to the top of the column by the air lift (compressed air, dew point below −10 °C).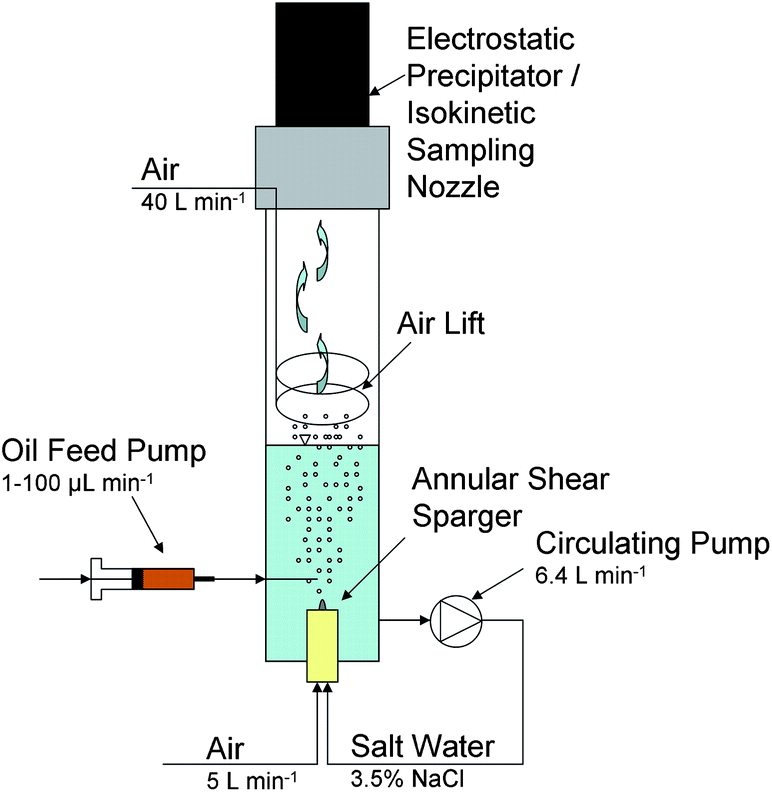 | ||
| Fig. 2 Schematic of bubble column reactor adapted from Smith et al.27 | ||
The gas phase samples were collected at the top of the column by either of two methods. The first method is utilizing the electrostatic precipitator (ESP; Ionic Spore Trap, DS Scientific, Baton Rouge, LA, USA) collecting only particles on aluminum targets. These aluminum targets were covered with carbon pads to allow for direct analysis in a scanning electron microscope (SEM). The second method is designed to procure the ejection rates with a constant mass-flow air-sampling system. The particles were sampled through a conical inlet with the same air velocity as the overall air velocity in the reactor to provide isokinetic sampling condition. The sampling stream was drawn into a bubbler filled with deionized water, allowing for the dissolution of the sodium chloride particles. Measuring the conductivity of the solution in the bubbler over time, and comparing it with the conductivity of sodium chloride solutions of known concentrations determined the mass of the ejected sodium chloride particles.
The experimental protocol consisted of the following steps: 5 L sodium chloride solution filled the bubble column reactor placing the air–water interface of the water column 5 cm below the air lift. A 3.5% (w/w) sodium chloride solution simulated the salinity of seawater. The operating variables of the reactor, namely, the liquid shear flow rate, lift air, and bubble air flow rate were varied to assess their effects on the size of the bursting bubbles and the particle size distribution of the generated particles. Pictures of the particles were obtained by collecting the generated particulate matter at the top of the column for 10 to 20 minutes with the ESP following direct (without sputtering) analysis with a SEM (FEI Quanta, 3D FEG, FIB/SEM dual beam operated at 20 kV). The average particle size and distribution were determined by measuring the crystal sizes (at least 500 particles per experiment) via the data processing software ImageJ from the SEM pictures. The bubble size distribution was assessed at the air–water interface by taking pictures with a digital camera (Casio Exilim ZR1000, Norderstedt, Germany) and manually measuring the bubble diameter via ImageJ of at least 200 bubbles. Elemental compositions of the particulates collected on aluminum targets without carbon pads were determined using an energy dispersive X-ray spectrometer (FEI Quanta 3D FEG, FIB/SEM dual beam interfaced with an EDAX EDS system).
We conducted the experiments on the ejection of alkanes from the weathered oil mousse as follows: a small amount of oil mousse (approx. 1 g) was placed on top of the water column in the bubble column reactor (shear flow 6.4 L min−1, bubble flow 5 L min−1 and, air lift 40 L min−1). Aerosolization was achieved by breaking bubbles at the surface and the particles were collected at four different time intervals with the ESP. The organic fraction of the collected particles on the aluminum targets was dissolved in dichloromethane. The resulting solution was analyzed with a gas chromatograph coupled to a mass spectrometer (GC-MS).
We tested the ejection of alkanes originating from non-weathered crude oil by injecting the crude oil slowly into the stream of bubbles originating from the sparger with a syringe pump (KD scientific Model # 210, Holliston, MA, USA). The injection flow rate varied stepwise from 1 μL min−1 to 100 μL min−1 to approximate the capacity of the bubble column to eject material. Two series of experiments were conducted, one with only the surrogate oil provided by the BP Gulf Coast Restoration Organization and another with 1% Corexit 9500A and the surrogate oil. We measured the ejection rate of alkanes analogously to the sodium chloride ejection by using ethyl acetate as the bubbler solvent. The ethyl acetate was dried over sodium sulfate and analyzed with a gas chromatograph with a flame ionization detector (GC-FID). The alkane content of the particulate matter was assessed by collecting particulate matter (PM) for 15 min at 50 μL min−1 oil flow rate and dissolving the alkanes off the aluminum target with a mixture of ethyl acetate and deionized water. The alkane concentration of the ethyl acetate phase was determined after drying over anhydrous sodium sulfate via GC-MS. Measuring the conductivity of the aqueous phase and comparing to sodium chloride solutions of known concentration quantified the amount of the salt collected.
GC-MS analysis
The samples were analyzed with an Agilent 6890 GC-MS (Agilent Technologies, Wilmington, DE, USA) with a 1 μL splitless injection onto a HP-5msUI (Agilent) 30 m × 250 μm × 0.25 μm column with a trap column (5 m × 250 μm × 0.25 μm) of identical surface chemistry. UHP Helium (Airgas, Baton Rouge, LA, USA) as the mobile phase was provided at a linear velocity of 36 cm s−1. The injection was conducted splitless at 300 °C. The oven program started at 50 °C for 5 min with a 10 °C min−1 to 300 °C and 24 min hold. Methyl arachidate and methyl octacosanoate were used as internal standards for quantification. The mass spectrometer detected alkanes by monitoring the fragment ions at a m/z of 57 and 85 and the internal standard compounds by monitoring m/z of 74 and 87.GC-FID analysis
The samples were analyzed on an Agilent 5890 II GC-FID with a 1 μL splitless injection onto a DB-1HT (JW Scientific, Folsom, CA, USA) 30 m × 250 μm × 0.1 μm column. Helium as the mobile phase was provided at a constant pressure of 120 kPa (linear flow rate at 50 °C approx. 36 cm s−1). We used splitless injection at 300 °C. The oven program started at 50 °C for 1.4 min with a 20 °C min−1 to 350 °C and 3 min hold. Concentrations were determined from external calibration standards.Results and discussion
Bubble and aerosol generation
The size distribution of aerosols generated by bursting bubbles of whitecaps and its environmental relevance depend on their formation. Jet droplets, viz., droplets induced by the bursting bubble, produce rather few, large sized drops (droplet diameter approximately 10 to 15% of the bursting bubble diameter), which settle quickly and are of less importance when considering atmospheric transport.21 The film drops, stemming from the film covering the bubble, are dominant in the aerosol, as they produce many small droplets (<30 μm) with substantial atmospheric lifetime.21,30 To ensure a consistent aerosol production in our aerosolization reactor the reactor parameters bubble-air flow rate, circulating-liquid flow rate, and air lift flow rate were varied to evaluate their effect on the bubble size and particle size. Table 1 yields an overview of the measured physical characteristics of the generated aerosols.| Bubble–air flow rate [L min−1] | Circulating-liquid flow rate [L min−1] | Air-lift flow rate [L min−1] | Bubble diameter at the surface [mm] | Particle diameter [μm] | Ejection rate of NaCl [μg min−1] | ||||
|---|---|---|---|---|---|---|---|---|---|
| 10%ile | Median | 90%ile | 10%ile | Median | 90%ile | ||||
| 5 | 6.4 | 40 | 0.66 | 0.87 | 1.13 | 0.63 | 1.06 | 2.41 | 27.5 ± 1.7 |
| 7 | 6.4 | 40 | 0.48 | 0.63 | 0.92 | 0.81 | 1.54 | 3.26 | 67.7 ± 3.1 |
| 9 | 6.4 | 40 | 0.39 | 0.55 | 0.86 | 0.74 | 1.57 | 3.08 | 74.4 ± 7.7 |
As seen in Table 1 the bubble-air flow rate directly relates to the amount of aerosol produced as more, and smaller bubbles burst at the constant column cross-section within the same time frame. Simultaneously the bubble size is larger with decreased bubble-air flow rate. The particle size does not strongly depend on the bubble-air flow rate, and a bubble-air flow rate of 5 L min−1 was chosen for further experimentation. This flow rate also allowed for improved reproducibility of the ejection rate. The air-lift flow rate, which transports and dries the droplets, had very little influence on the particle size distribution, and the highest tested flow rate of 40 L min−1 was chosen for further experiments to achieve dry particles for the evaluation via SEM. The circulating pump flow rate had no discernible effect on the diameter of the bursting bubbles, a key parameter for the generation of the aerosol and was set to 6.4 L min−1 to ensure sufficient mixing and limit the strain on the peristaltic tubing.
The size of the particles generated in the reactor falls within the range (0.1 to 10 μm) of the ones produced by oceanic whitecap bubbles.31–34 Although the column also produces larger jet droplets (>100 μm) the classification inside the column resulting from wall scavenging effects (visible salt precipitation) due to turbulence and settling allows only smaller particulate matter to be collected at the top. These smaller particles are of more environmental interest as they are subject to potential long-range transport33 (up to 330 km for a 10 μm particle) and have therefore, implications for the off- and onshore environment. Following the sea salt aerosol size distributions of multiple researchers summarized in Lewis and Schwartz33 our aerosol generator generates salt particles, which can be encountered at wind speeds from 0 to 20 m s−1. The flux of 5.65 × 10−9 g cm−2 s−1 of sodium chloride from our reactor is about two to three orders of magnitude higher than the fluxes measured by Cipriano and Blanchard35 most likely because of more intense bubble generation in our system. Extension of laboratory-measured fluxes to the environmental production flux is challenging as the flux depends on whitecap coverage, salinity, wind speed, temperature, and location33,36,37 and is not attempted in the presented work. Nonetheless, we consider our results to be of a proof-of-concept type regarding the effects of bubble bursting on flux.
Ejection of alkanes from oil mousse
Approximately 1 g of oil mousse was liquefied by warming it to 40 °C and added on the top of the water column in the reactor. The oil mousse was subjected to several hours of bursting bubbles, during which only particulate matter was collected with the ESP. The oil mousse as well as the collected particles was analyzed via GC-MS.Fig. 3 shows the GC-trace of the oil mousse and the collected aerosol. The oil mousse contains only alkanes larger than pentadecane (C15), which is expected due to the loss of volatile compounds during the long transport (80 km) to the shoreline. Both the oil mousse (Fig. 3a) and the aerosol (Fig. 3b) exhibit a nearly identical alkane distribution, ranging from pentadecane to triacontane (C30), with the dominant alkanes being centered around icosane (C20). This identical distribution is somewhat surprising, as the transfer to the gas phase via evaporation should cause a change of the alkane distribution towards the lighter and shorter alkanes, i.e., triacontane would not be expected in the aerosol phase. Clearly, the distribution of the hydrocarbons is practically independent of the chain length and vapor pressure, indicating that the evaporation vector for oil mousse is only a minor transport vector. The low vapor pressure (0.62 Pa for C15) and the insolubility of large alkanes in water negate the possibility of these compounds either being evaporated or found dissolved in the aerosol droplets. As such an alternative transport mechanism has to be responsible for the ejection.
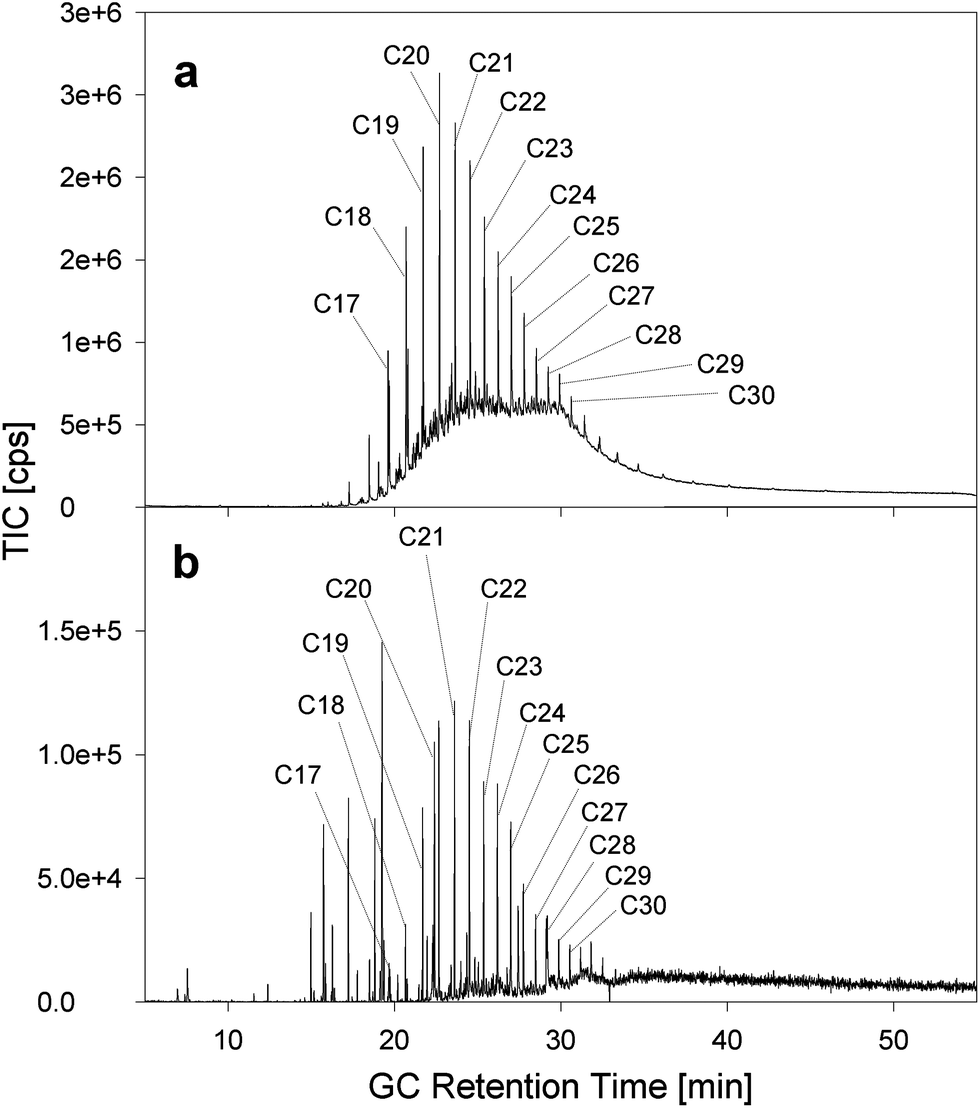 | ||
| Fig. 3 Comparison of the GC-MS chromatogram of (a) oil mousse and (b) ejected aerosol. | ||
Physical ejection by bubbles bursting through an oil film as suggested by Fontana11 is quite unlikely in our situation as the contact area of the floating oil mousse is very limited. The initially flat oil mousse droplets semi-solidify in the cold reactor (20 °C) and take on sausage like forms due to the rolling motion of the water surface, eliminating the possibility of bubbles bursting through the oil mousse. Furthermore, the typically large particles (>100 μm) generated by this mechanism would settle quickly back into the column and not reach the top of the column.
For atmospheric aerosols, the air–water interface is never negligible and the specific surface area can reach dramatic values (1 mL of water in 1 μm droplets yields 6 m2 of surface). An analogous situation arises for bursting bubbles, as an abundant area of air–water interface can be encountered. Therefore, surface driven processes such as adsorption should be considered. Several researchers have shown38–43 that for atmospheric aerosols such as fog and clouds (analogous to ocean spray), non-polar compounds can adsorb at the air–water interface surpassing aqueous solubility levels predicted by Henry's law. This surface adsorption might yield a suitable explanation of the oil ejection in the reactor. For surface active substances and hydrophobic particles, this vector is known and has been shown to be a viable transport pathway.14,17,44 Analogously, the “surface adsorption” of bacteria is suspected to be the cause of the bacterial enrichment on ocean spray.13,16
Ejection of alkanes from crude oil
To assess the air–water interface driven ejection of oil spill matter in further detail we injected crude oil (surrogate oil) at a constant flow rate into the center of the column. By varying the flow rate from 1 to 100 μL min−1 we can assess the capacity of the column of bubbles to eject material. We evaluated the composition of the oil, the gas phase, and the particulate matter via GC-FID and GC-MS.Fig. 4 shows the observed alkane ejection rate (grey symbols) in the gas phase (air and particulate matter) grouped by different chain lengths in response to the injected amount of oil. As the ejection rate of C10–14 (scale on the right hand side) and the C15–C19 portion of the oil is orders of magnitude greater than that of the larger homologues (C20–C29), clearly, evaporation is an important path for these alkanes. For larger alkanes, the ejection is limited to below 1 μg min−1. Nonetheless, even at a very low injection rate of only a few μL crude oil per minute large alkanes are detectable. The shape of the curves also indicates that transport for C10 to C19 is predominantly resulting from evaporation as the ejection rises nearly linearly up to 10 μL min−1 oil injection rate and then flattens out at 20 μL min−1, whereas for larger alkanes the ejection rate flattens below 2 μL min−1 oil injection rate. This flattening of the ejection curve is attributable to the limited capacity of the utilized gas streams/bubble surface to carry out alkanes. As the column simulates two transport mechanisms – evaporation and surface adsorption of alkanes – obviously the evaporative transport has more capacity for compounds with large vapor pressure (C10 to C19). The generated bubble surface area limits the capacity of the air–water interface as the ejection vector, which is a smaller vector for C10 to C19. The higher capacity for more volatile materials is therefore linked to gas flow, whereas the available surface area limits the uptake of the oil on the air–water interface.
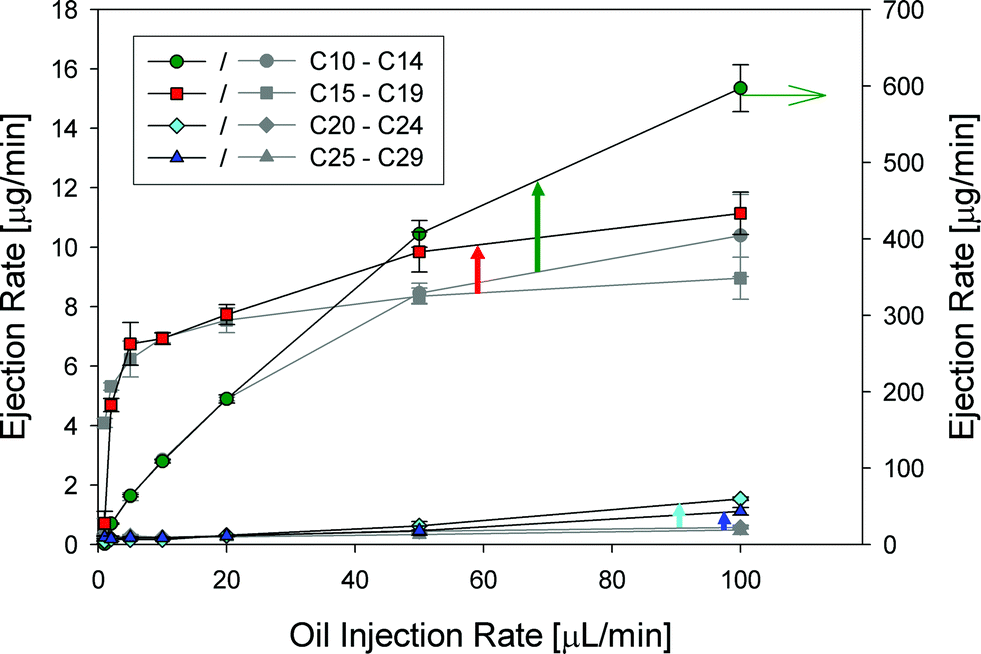 | ||
| Fig. 4 Ejection rate of alkanes (grey symbols) from pure surrogate oil and ejection rate of alkanes in the presence of a dispersant (colored symbols). | ||
Also shown in the figure are data that involve the crude oil ejection rate in the presence of 1% Corexit 9500A. Clearly the presence of dispersants augmented the ejection rate of all the classes of alkanes: C10–C14 (47.7%), C15–C19 (24.4%), C20–C24 (168.2%), and C25–C29 (126.1%) at 100 μL min−1 injection rate. At a lower oil injection rate (e.g. 20 μL min−1), i.e. when the column is not saturated, the addition of a dispersant does not enhance the ejection. As the hydrophobic dispersant is delivered with the oil, it affects the air–water interface only marginally. In the case of a non-saturated reactor the addition of dispersant does not show any effect as practically all injected material is carried out even without dispersant present. Once the limit of the ejection is reached, the dispersant helps to overcome the limited transport capacity by enhancing the transfer across the oil–water–bubble boundary in the water column and also potentially increases the capacity of the air–water interface to adsorb alkanes. As both effects are surface driven processes, the effect on the non-volatile fraction C20–C29 is larger than on the volatile fraction (C10–C19).
The addition of the dispersant clearly increased the dispersion of the oil in water (observed visually in the reactor and also described in the literature45). As described earlier in the Introduction, the present results with the oil phase and dispersants are at variance from results that show contradictory data on aerosol generation from sea sprays in the presence of conventional, water-soluble surfactants.23,29,44,46 Conventional surfactants aggregate at the aqueous surface and reduce surface pressure, making the surface less mobile. However, dispersant formulations (e.g., the Corexit family) are designed with the specific purpose of mixing with only the oil droplets and dispersing it well within the aqueous phase with minimal coalescence. As smaller droplets are formed by dispersant mixing with the oil, the bubbles that rise to the surface have higher flotation capacities and therefore eject more oil-laden jet and film droplets which lead to higher organic aerosol number in the air above the surface. It is well understood in the ore flotation literature that air bubbles in the aqueous phase generally have higher capture efficiencies for finely dispersed, surface active particles and droplets.47,48 Therefore, the bubbles and droplets produced at the surface will be capable of transporting a larger fraction of the oil via aerosolization.
To further illustrate the ability of the surface of bursting bubbles to eject oil spill matter we have plotted the ratio of either the concentration of the alkanes in the particulate matter (collected via an ESP) or their concentration in the gas phase (collected via a funnel) to their respective concentration in the oil spill matter as a measure of volatility versus the vapor pressure of the individual alkane in Fig. 5. If evaporation is the main ejection pathway for a group of compounds, a linear dependency of the volatility and the vapor pressure is expected. Fig. 5 shows that there is indeed a linear dependency of the observed volatility of alkanes up to C18 on their vapor pressure. Therefore, we can conclude that alkanes up to C18 predominantly evaporate. For alkanes with a chain length larger than eighteen carbons the concentration in the gas phase as well as the particulates is independent of vapor pressure. Clearly evaporation becomes negligible for these compounds. Nonetheless, large alkanes have significant thermodynamic free energy minima at the air–salt water interface and can therefore adsorb at the surface of bubbles. In the accompanying paper (Part 2 of this series) we describe molecular dynamics (MD) simulations of the properties of large n-alkanes between air and saltwater interfaces. For example, alkanes such as n-pentadecane, n-icosane and n-triacontane exhibit a deep free energy minimum at these interfaces, where they tend to adopt a flat orientation and form aggregates. Once the bubble breaks and forms atmospheric particulate matter in the form of droplets, these large, non-volatile alkanes are very likely to adsorb at the surface of these bubbles or droplets and be simultaneously aerosolized. In addition, the free energy minimum observed in the MD simulations for n-alkanes at the air–salt water interface becomes even deeper when dispersants are present in the system, which is consistent with the experimental observation that the addition of Corexit enhances the ejection of alkanes. Another interesting aspect shown in Fig. 5 is the fact that the bursting-bubble aerosolization presents a chain-length independent transport pathway for alkane ejection, a feature most likely resulting from the adsorptive nature of the transport.
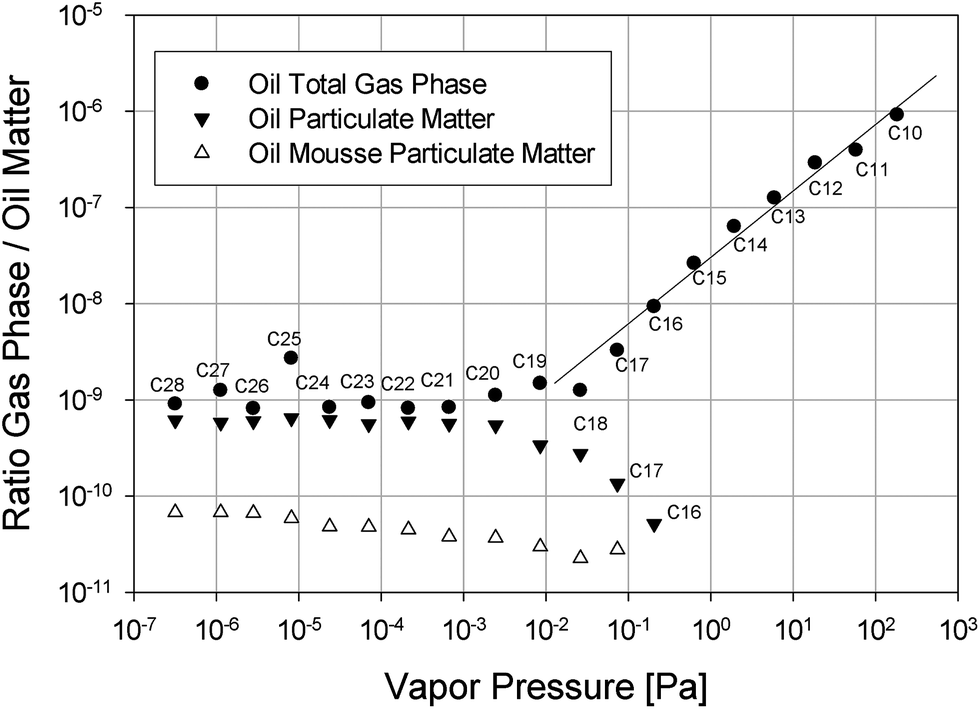 | ||
| Fig. 5 Ratio of the total air concentration for surrogate oil, the concentration of alkanes in particles for surrogate oil and oil mousse to the alkane oil matter concentration. | ||
The collected particulate matter from the crude oil experiments is rich in alkanes with up to 1.2% (w/w) of the aerosol droplet at a low oil injection rate of 50 μL min−1. Fig. 6 shows the SEM of the particles generated and the elemental composition determined by SEM/EDS for the experiment involving only the crude oil. SEM images of the particulate matter showed the crystalline phase of NaCl left after water loss from the wet aerosol droplets. SEM/EDS images of the same dry aerosols showed the co-located C fraction originating from the IVOC and SVOC fractions that were originally associated with the surfaces of the saltwater film in the wet aerosol. Note that this is a tiny fraction of the overall PM mass. As earlier stated, the accompanying paper on molecular dynamics simulations showed the clear preference of large molecular weight alkanes for the air–salt water interface. As the alkanes only represent 10% of the organics of the crude oil it is plausible that the ejection via bursting bubbles can account for particles with a high organic fraction as they were encountered during the Deep Water Horizon oil spill.9,49,50
 | ||
| Fig. 6 Collected salt aerosol: (a) SEM image and (b) elemental composition (EDS mapping). | ||
Two sampling flights were conducted by the National Oceanic and Atmospheric Administration (NOAA) during the spill,51 which measured several meteorological, physical, and chemical parameters. Aerosol was characterized as organic (OA) and total aerosol-mass-spectrometer-amenable aerosol. The OA content found in the atmosphere was further split into contributing portions of oxygenated organic aerosol (OOA) and hydrocarbon-like organic aerosol (HOA). The measurements reported by de Gouw et al.9 found a large plume of organic material in the atmosphere and attributed it to SOA formation from IVOC. De Gouw et al.'s9 study neglected the possibility of entrainment of SVOC or non-volatile materials and ruled out aerosolization based on the appearance of growth of particulate matter from approximately 0.02 μm to 0.1 μm downwind.52 In the upwind side of the spill a background maritime aerosol centered around an aerosol size of 0.1 μm was found. Though the increase in the organic aerosol was predominantly observed at the lower end of the size spectrum, the size fraction of the maritime background doubled downwind, indicating that further aerosolization occurred.
Downwind of the spill site the OA concentration also increased twofold. This increased OA level was attributed by de Gouw et al. to SOA formation.9 The ratio between OOA and HOA can be used to track the oxidative state of atmospheric matter.8,42,53 For SOA and aged atmospheric particulate matter, ratios of OOA to HOA are expected to be greater than one. The ratio of OOA and HOA close to the spill site was approximately one, indicating the strong influx of volatile organic material. Further, atmospheric SOA formation is an oxidative process,7 yielding compounds with higher oxidative state than their parent compounds. Nonetheless, the ratio of HOA to OOA increased downwind of the spill site to higher levels (∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) than at the spill site (1:1), which is in contradiction to a purely oxidative SOA formation. A possible explanation for this discrepancy is either the condensation/adsorption of evaporated native materials onto smaller SOA particles formed or the continuous emission of non-oxidized materials into the atmosphere downwind of the (surface) spill site. As volatile materials up to C18 will quickly evaporate in a matter of a few hours to days,5,54 continuous aerosolization of less volatile materials downwind of the spill site provides a suitable, complementary vector for the transport of organic material from the sea to the atmosphere, potentially explaining the observed reduced degree of oxidation downwind. Clearly, the transport of oil spill matter into the atmosphere is a complex process, where multiple transport vectors will take place and have to be considered.
:1) than at the spill site (1:1), which is in contradiction to a purely oxidative SOA formation. A possible explanation for this discrepancy is either the condensation/adsorption of evaporated native materials onto smaller SOA particles formed or the continuous emission of non-oxidized materials into the atmosphere downwind of the (surface) spill site. As volatile materials up to C18 will quickly evaporate in a matter of a few hours to days,5,54 continuous aerosolization of less volatile materials downwind of the spill site provides a suitable, complementary vector for the transport of organic material from the sea to the atmosphere, potentially explaining the observed reduced degree of oxidation downwind. Clearly, the transport of oil spill matter into the atmosphere is a complex process, where multiple transport vectors will take place and have to be considered.
Conclusions
Although large alkanes have been reported to be found in marine aerosols55 clear experimental evidence for their transport from the marine aquatic environment to the atmospheric environment has been missing. From the experimental data presented here, and the simulation results shown in the accompanying paper, obviously the adsorption of alkanes at the air–water interface and the subsequent ejection of the interface via bursting bubbles is a volatility and solubility independent transport vector for organic oil spill matter into the atmosphere. This vector might be of particular importance for the fate of SVOC such as alkanes with more than eighteen carbons, as dissolution, microbial degradation, and evaporation are negligible for these compounds.During an oil spill both evaporative transport and bubble-bursting induced aerosolization occur. Organic compounds with a comparable or higher volatility like octadecane will predominantly evaporate. However, larger compounds will be ejected via film droplets resulting from bursting bubbles. The application of dispersants facilitates the aerosolization and evaporation predominantly by enhancing the dispersion of the oil in the water column and improves therefore the flotation capacity of the bubbles.
The capability of breaking waves and whitecaps to eject non-volatile material provides an abundance of important consequences; it positively facilitates the dispersion of the oil, but simultaneously transforms a marine environmental problem into an atmospheric issue. Further, research is necessary to determine the effect of the aerosolization transport vector on the complete oil budget and its environmental short- and long-term consequences.
Acknowledgements
This work was supported by the Gulf of Mexico Research Initiative (GoMRI) as part of the Consortium for the Molecular Engineering of Dispersant Systems (CMEDS – LSU GRI Sub award TUL-629-11/12). The authors would like to thank Dr Louis Thibodeaux and Bonnie McLindon for acquiring the oil mousse sample.References
- J. Lubchenco, M. K. McNutt, G. Dreyfus, S. A. Murawski, D. M. Kennedy, P. T. Anastas, S. Chu and T. Hunter, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20222–20228 CrossRef PubMed.
- One Year Later Press Pack, http://www.restorethegulf.gov/release/2011/04/10/one-year-later-press-pack, 2013.
- L. J. Thibodeaux, K. T. Valsaraj, V. T. John, K. D. Papadopoulos, L. R. Pratt and N. S. Pesika, Environ. Eng. Sci., 2011, 28, 87–93 CrossRef CAS.
- M. K. McNutt, R. Camilli, T. J. Crone, G. D. Guthrie, P. A. Hsieh, T. B. Ryerson, O. Savas and F. Shaffer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20260–20267 CrossRef PubMed.
- T. B. Ryerson, R. Camilli, J. D. Kessler, E. B. Kujawinski, C. M. Reddy, D. L. Valentine, E. Atlas, D. R. Blake, J. de Gouw, S. Meinardi, D. D. Parrish, J. Peischl, J. S. Seewald and C. Warneke, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20246–20250 CrossRef CAS PubMed.
- B. Lehr, S. Bristol and A. Possolo, Oil Budget Calculator—Deepwater Horizon. Technical Documentation: A Report by The Federal Interagency Solutions Group, Oil Budget Calculator Science and Engineering Team, National Oceanic and Atmospheric Administration, 2010 Search PubMed.
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- N. M. Donahue, K. M. Henry, T. F. Mentel, A. Kiendler-Scharr, C. Spindler, B. Bohn, T. Brauers, H. P. Dorn, H. Fuchs, R. Tillmann, A. Wahner, H. Saathoff, K.-H. Naumann, O. Möhler, T. Leisner, L. Müller, M.-C. Reinnig, T. Hoffmann, K. Salo, M. Hallquist, M. Frosch, M. Bilde, T. Tritscher, P. Barmet, A. P. Praplan, P. F. DeCarlo, J. Dommen, A. S. H. Prévôt and U. Baltensperger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13503–13508 CrossRef CAS PubMed.
- J. A. de Gouw, A. M. Middlebrook, C. Warneke, R. Ahmadov, E. L. Atlas, R. Bahreini, D. R. Blake, C. A. Brock, J. Brioude, D. W. Fahey, F. C. Fehsenfeld, J. S. Holloway, M. Le Henaff, R. A. Lueb, S. A. McKeen, J. F. Meagher, D. M. Murphy, C. Paris, D. D. Parrish, A. E. Perring, I. B. Pollack, A. R. Ravishankara, A. L. Robinson, T. B. Ryerson, J. P. Schwarz, J. R. Spackman, A. Srinivasan and L. A. Watts, Science, 2011, 331, 1295–1299 CrossRef CAS PubMed.
- M. C. Spillane, E. C. Monahan, P. A. Bowyer, D. M. Doyle and P. J. Stabano, in Oceanic Whitecaps and Their Role in Air-Sea Exchange Processes, ed. E. C. Monahan and G. M. Niocaill, D. Reidel Publishing Company, Doordrecht, Netherlands, 1986, p. 294 Search PubMed.
- M. Fontana, Water, Air, Soil Pollut., 1976, 5, 269–280 CAS.
- S. R. Massel, Ocean Waves Breaking and Marine Aerosol Fluxes, Springer Verlag, New York, NY, USA, 2007 Search PubMed.
- D. C. Blanchard, in Air-Sea Exchange of Gases and Particles, ed. P. S. Liss and W. George.Slinn, Kluwer Academic Publishers, Hingham, MA, USA, 1982, vol. 108, pp. 407–454 Search PubMed.
- M. A. McInnes, D. A. Ellis, E. M. Webster and A. Peters, Atmos. Environ., 2013, 69, 304–312 CrossRef CAS PubMed.
- P. Schmitt-Kopplin, G. Liger-Belair, B. P. Koch, R. Flerus, G. Kattner, M. Harir, B. Kanawati, M. Lucio, D. Tziotis, N. Hertkorn and I. Gebefügi, Biogeosciences, 2012, 9, 1571–1582 CrossRef CAS.
- K. A. Prather, T. H. Bertram, V. H. Grassian, G. B. Deane, M. D. Stokes, P. J. DeMott, L. I. Aluwihare, B. P. Palenik, F. Azam, J. H. Seinfeld, R. C. Moffet, M. J. Molina, C. D. Cappa, F. M. Geiger, G. C. Roberts, L. M. Russell, A. P. Ault, J. Baltrusaitis, D. B. Collins, C. E. Corrigan, L. A. Cuadra-Rodriguez, C. J. Ebben, S. D. Forestieri, T. L. Guasco, S. P. Hersey, M. J. Kim, W. F. Lambert, R. L. Modini, W. Mui, B. E. Pedler, M. J. Ruppel, O. S. Ryder, N. G. Schoepp, R. C. Sullivan and D. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7550–7555 CrossRef CAS PubMed.
- F. MacIntyre, J. Geophys. Res., 1972, 77, 5211–5228 CrossRef CAS.
- D. C. Blanchard, Science, 1964, 146, 396–397 CAS.
- D. C. Blanchard, in Applied Chemistry at Protein Interfaces, American Chemical Society, 1975, vol. 145, pp. 360–387 Search PubMed.
- E. C. Monahan, D. E. Spiel and K. L. Davidson, in Oceanic Whitecaps, ed. E. Monahan and G. Niocaill, Springer, Netherlands, 1986, vol. 2, pp. 167–174 Search PubMed.
- E. Andreas, J. Edson, E. Monahan, M. Rouault and S. Smith, Boundary-Layer Meteorol., 1995, 72, 3–52 CrossRef.
- C. D. O'Dowd and G. de Leeuw, Philos. Trans. R. Soc., A, 2007, 365, 1753–1774 CrossRef CAS PubMed.
- R. L. Modini, L. M. Russell, G. B. Deane and M. D. Stokes, J. Geophys. Res.: Atmos., 2013, 118, 1388–1400 Search PubMed.
- E. Fuentes, H. Coe, D. Green, G. de Leeuw and G. McFiggans, Atmos. Chem. Phys., 2010, 10, 9295–9317 CrossRef CAS.
- W. C. Keene, H. Maring, J. R. Maben, D. J. Kieber, A. A. P. Pszenny, E. E. Dahl, M. A. Izaguirre, A. J. Davis, M. S. Long, X. Zhou, L. Smoydzin and R. Sander, J. Geophys. Res., 2007, 112, D21202 CrossRef.
- Virtual Oil Dataset Macondo Oil Spill, BP Gulf Coast Restoration Organization, 2012 Search PubMed.
- J. S. Smith, L. F. Burns, K. T. Valsaraj and L. J. Thibodeaux, Ind. Eng. Chem. Res., 1996, 35, 1700–1710 CrossRef CAS.
- A. P. Ault, R. C. Moffet, J. Baltrusaitis, D. B. Collins, M. J. Ruppel, L. A. Cuadra-Rodriguez, D. Zhao, T. L. Guasco, C. J. Ebben, F. M. Geiger, T. H. Bertram, K. A. Prather and V. H. Grassian, Environ. Sci. Technol., 2013, 47, 5603–5612 CrossRef CAS PubMed.
- E. Fuentes, H. Coe, D. Green, G. de Leeuw and G. McFiggans, Atmos. Meas. Tech., 2010, 3, 141–162 CrossRef CAS.
- F. Resch, in Oceanic Whitecaps and Their Role in Air-Sea Exchange Processes, ed. E. C. Monahan and G. M. Niocaill, D. Reidel Publishing Company, Doordrecht, Netherlands, 1986, pp. 101–112 Search PubMed.
- G. B. Deane and M. D. Stokes, Nature, 2002, 418, 839–844 CrossRef CAS PubMed.
- C. W. Fairall, K. L. Davidson and G. E. Schacher, Tellus, Ser. B, 1983, 35, 31–39 CrossRef.
- R. Lewis and E. Schwartz, Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models—A Critical Review, AGU, Washington DC, 2004 Search PubMed.
- E. L. Andreas, in Atmosphere–Ocean Interactions, ed. W. Perrie, WIT Press, Southampton, UK, 2002, vol. 1, pp. 1–47 Search PubMed.
- R. J. Cipriano and D. C. Blanchard, J. Geophys. Res., C: Oceans Atmos., 1981, 86, 8085–8092 CrossRef.
- M. D. Anguelova, PhD dissertation, University of Delaware, 2002.
- E. M. Mårtensson, E. D. Nilsson, G. de Leeuw, L. H. Cohen and H. C. Hansson, J. Geophys. Res.: Atmos., 2003, 108, 4297 CrossRef.
- K. T. Valsaraj, Water Res., 1994, 28, 819–830 CrossRef CAS.
- S. Raja, R. Raghunathan, X.-Y. Yu, T. Lee, J. Chen, R. R. Kommalapati, K. Murugesan, X. S. Y. Qingzhong, K. T. Valsaraj and J. L. Collett Jr, Atmos. Environ., 2008, 42, 2048–2061 CrossRef CAS PubMed.
- D. J. Donaldson and D. Anderson, J. Phys. Chem. A, 1999, 103, 871–876 CrossRef CAS.
- B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2009, 11, 7760–7779 RSC.
- F. S. Ehrenhauser, K. Khadapkar, Y. Wang, J. W. Hutchings, O. Delhomme, R. R. Kommalapati, P. Herckes, M. J. Wornat and K. T. Valsaraj, J. Environ. Monit., 2012, 14, 2566–2579 RSC.
- K. T. Valsaraj, G. J. Thoma, D. D. Reible and L. J. Thibodeaux, Atmos. Environ., Part A, 1993, 27, 203–210 CrossRef.
- C. J. Ebben, A. P. Ault, M. J. Ruppel, O. S. Ryder, T. H. Bertram, V. H. Grassian, K. A. Prather and F. M. Geiger, J. Phys. Chem. A, 2013, 117, 6589–6601 CrossRef CAS PubMed.
- C. Spier, W. T. Stringfellow, T. C. Hazen and M. Conrad, Environ. Pollut., 2013, 173, 224–230 CrossRef CAS PubMed.
- K. Sellegri, C. D. O'Dowd, Y. J. Yoon, S. G. Jennings and G. de Leeuw, J. Geophys. Res.: Atmos., 2006, 111, D22215 CrossRef.
- A. N. Clarke and D. J. Wilson, Foam Flotation: Theory and Applications, Marcel Dekker, Inc., New York, 1983 Search PubMed.
- R. Clift, J. E. Grace and M. E. Weber, Bubbles, Droplets and Particles, Academic Press, New York, 1978 Search PubMed.
- A. R. Ravishankara and J. Goldman, Air Chemistry in the Gulf of Mexico Oil Spill Area NOAA WP-3D Airborne Chemical Laboratory Flights of 8 and 10 June 2010, NOAA, Boulder, CO, USA, 2010 Search PubMed.
- A. M. Middlebrook, D. M. Murphy, R. Ahmadov, E. L. Atlas, R. Bahreini, D. R. Blake, J. Brioude, J. A. de Gouw, F. C. Fehsenfeld, G. J. Frost, J. S. Holloway, D. A. Lack, J. M. Langridge, R. A. Lueb, S. A. McKeen, J. F. Meagher, S. Meinardi, J. A. Neuman, J. B. Nowak, D. D. Parrish, J. Peischl, A. E. Perring, I. B. Pollack, J. M. Roberts, T. B. Ryerson, J. P. Schwarz, J. R. Spackman, C. Warneke and A. R. Ravishankara, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20280–20285 CrossRef CAS PubMed.
- NOAA, Gulf 2010 flights, http://esrl.noaa.gov/csd/groups/csd7/measurements/2010gulf/ Search PubMed.
- R. Bahreini, A. M. Middlebrook, C. A. Brock, J. A. de Gouw, S. A. McKeen, L. R. Williams, K. E. Daumit, A. T. Lambe, P. Massoli, M. R. Canagaratna, R. Ahmadov, A. J. Carrasquillo, E. S. Cross, B. Ervens, J. S. Holloway, J. F. Hunter, T. B. Onasch, I. B. Pollack, J. M. Roberts, T. B. Ryerson, C. Warneke, P. Davidovits, D. R. Worsnop and J. H. Kroll, Environ. Sci. Technol., 2012, 46, 8025–8034 CrossRef CAS PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- T. B. Ryerson, K. C. Aikin, W. M. Angevine, E. L. Atlas, D. R. Blake, C. A. Brock, F. C. Fehsenfeld, R. S. Gao, J. A. de Gouw, D. W. Fahey, J. S. Holloway, D. A. Lack, R. A. Lueb, S. Meinardi, A. M. Middlebrook, D. M. Murphy, J. A. Neuman, J. B. Nowak, D. D. Parrish, J. Peischl, A. E. Perring, I. B. Pollack, A. R. Ravishankara, J. M. Roberts, J. P. Schwarz, J. R. Spackman, H. Stark, C. Warneke and L. A. Watts, Geophys. Res. Lett., 2011, 38, L07803 CrossRef.
- R. B. Gagosian, in The Role of Air-Sea Exchange in Geochemical Cycling, ed. P. Buat-Ménard, D. Reidel Publishing Company, Dordrecht, Netherlands, 1986, vol. 185, pp. 409–442 Search PubMed.
Footnotes |
| † Current address: Audubon Sugar Institute, LSU AgCenter, St Gabriel, LA 70776, USA. |
| ‡ Current address: Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA. |
| This journal is © The Royal Society of Chemistry 2014 |