Iron nanoparticles for environmental clean-up: recent developments and future outlook
Weile Yan*a, Hsing-Lung Lienb, Bruce E. Koelc and Wei-xian Zhangd
aDepartment of Civil and Environmental Engineering, Texas Tech University, Lubbock, Texas, USA. E-mail: weile.yan@ttu.edu
bDepartment of Civil and Environmental Engineering, National University of Kaoshiung, Kaoshiung, Taiwan
cDepartment of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey, USA
dTongji University, State Key Laboratory for Pollution Control & Resources Reuse, Shanghai, China
First published on 6th December 2012
Abstract
Nanoscale zero-valent iron (nZVI) is one of the most extensively applied nanomaterials for groundwater and hazardous waste treatment. In the past fifteen years, progress made in several key areas has deepened our understanding of the merits and uncertainties of nZVI-based remediation applications. These areas include the materials chemistry of nZVI in its simple and modified forms, the nZVI reactivity with a wide spectrum of contaminants in addition to the well-documented chlorinated solvents, methods to enhance the colloidal stability and transport properties of nZVI in porous media, and the effects of nZVI amendment on the biogeochemical environment. This review aims to provide an up-to-date account of advancement in these areas as well as insights gained through field experience.
 Weile Yan | Weile Yan is an assistant professor in the Department of Civil and Environmental Engineering at Texas Tech University. She received her Bachelor and Ph.D. degrees, both in Environmental Engineering, from the National University of Singapore (2002) and Lehigh University (2011), respectively, and spent a brief post-doctoral term at Princeton University (2011). Her past and current research efforts have focused on groundwater remediation using zero-valent iron materials, surface chemistry of metal or metal oxide nanoparticles in aqueous systems, and sustainable materials for environmental separation and catalysis applications. |
 Hsing-Lung Lien | Hsing-Lung Lien received his Ph.D. in Environmental Engineering from Lehigh University, USA in 2000 and worked at the National Laboratory of USEPA Subsurface Protection and Remediation Division in Ada, OK in 2000–2002. He served as an assistant professor at National University of Kaohsiung (NUK) since 2002 and was promoted to full professor in 2010. Prof. Lien's research interest is in the use of nanotechnologies for water treatments. He conducted the first field test of using nanoscale zerovalent iron for groundwater remediation in Taiwan. |
 Bruce E. Koel | Bruce Koel is Professor at Princeton University in Chemical and Biological Engineering, Associated Faculty in Chemistry, MAE, and PRISM, and NSTX Collaborator at the Princeton Plasma Physics Laboratory. He received his B.S. and M.S. degrees from Emporia State University and a Ph.D. degree in Chemistry from UT-Austin. He was a Miller Postdoctoral Fellow at UC-Berkeley with G. A. Somorjai. His research experience is in surface chemistry and interfacial phenomena, surface analysis, catalysis and electrocatalysis, and nanoscience. He received the George A. Olah Award in Hydrocarbon or Petroleum Chemistry from the ACS, and is a Fellow of the AAAS, APS, and AVS. |
 Wei-xian Zhang | Wei-xian Zhang is currently the Director of the State Key Laboratory of Pollution Control and Resource Reuse at Tongji University, China. He received his Ph.D. in Environmental Engineering from John Hopkins University in 1996. He was formerly an associate professor at the Department of Civil and Environmental Engineering at Lehigh University prior to 2010. His group initiated the use of nanoscale zerovalent iron (nZVI) for water treatment in 1997 and conducted the first field assessment of nZVI for in situ groundwater remediation in 2001. His current interest includes assessing and realizing the use of emerging technologies for decontaminating surface and underground environments in developing countries. |
Environmental impactNanoscale zero-valent iron (nZVI) is one of the most prevalently used engineered nanoparticles for groundwater and soil remediation. Over the past 15 years, nZVI-based remediation has grown into a prominent sub-field of environmental nanotechnology, with more than 50 pilot and full-scale implementations conducted worldwide and a vast volume of research literature published on this subject in the past few years. This review aims to provide a critical and updated account of recent research developments in several key technological areas of nZVI as well as notable findings from field experiences. The authors expect this review will facilitate a current and objective evaluation of the potential of nZVI materials in existing and new applications and inform future research efforts to address pertinent engineering challenges. |
Overview: nZVI technology development
“There is plenty of room at the bottom”. This prophetic statement by Richard Feynman in 1959 beckoned the advent of nanotechnology,1 a field that has since then profoundly influenced science and engineering disciplines in which the unusual properties of materials are harnessed in applications impacting virtually all aspects of modern society, e.g., energy production, chemical synthesis, electronics, lighting, biotechnology, and health care. Among these, nanotechnology for environmental remediation is a highly anticipated frontier, as the growing demand for a limited supply of clean water places an urgent need on technologies that can deliver faster, cleaner, and more affordable water treatment measures.Applications of engineered nanomaterials for environmental clean-up came about when a small amount of nanosized iron particles was found to rapidly transform a group of recalcitrant groundwater contaminants including tricholoroethylene (TCE) and polychlorinated biphenyls (PCBs).2 In the ensuing years, nZVI technology has evolved into the most active sub-field of environmental nanotechnology with a series of remarkable advances seen in the areas of fundamental understanding of nZVI reactivity with environmental contaminants, surface engineering for enhanced transport, and pilot and full-scale field implementation (Fig. 1). The technological advancement has been accompanied by a steep increase in publications on the subject, particularly in the last few years. To convey a sense of the breadth of topics covered by this literature, the themes of the publications can be broadly categorized based on the types of contaminants treated (Fig. 2a) and the physicochemical properties and engineering aspects being investigated (Fig. 2b). The size of each segment reflects the number of articles in the respective area. In addition to the well-studied chlorinated solvents, the list of contaminants amenable to nZVI treatment encompasses a variety of deleterious organic pollutants, such as pesticides, azo-dyes, flame retardants, and antibiotics. Also on the list are important inorganic contaminants such as nitrate, arsenic, hexavalent chromium, heavy metals, and radionuclides. With an increasing number of field applications being conducted with nZVI for in situ groundwater and soil treatment, engineering aspects related to the stability, mobility, and long-term eco-toxicological impacts of nZVI have received increasing attention (Fig. 2b).
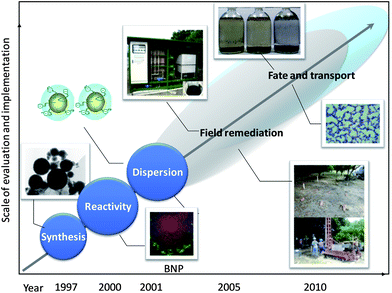 | ||
| Fig. 1 Developmental milestones of nZVI technology over the past 15 years. | ||
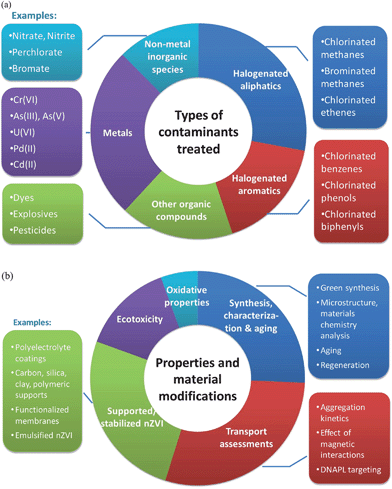 | ||
| Fig. 2 Overview of the nZVI-related literature (based on a total of 445 publications surveyed during the preparation of this article). (a) Breakdown by the type of contaminants treated. (b) Breakdown by the properties and engineering aspects of nZVI investigated. | ||
The development of nZVI technology can be traced back to studies by Gillham and O'Hannesin, who discovered that bulk zero-valent iron (ZVI) was able to reduce a group of halogenated aliphatic compounds in groundwater.3 This property of iron led to the development of iron permeable-reactive barriers (Fe-PRBs), which are vertical trenches filled with granular ZVI materials placed in the flow path of an underground contaminant plume.4 Over one hundred PRB structures have been constructed in the United States in the past two decades with various degrees of success.5–7 A critical concern for the long-term effectiveness of these PRB installations is decreased permeability due to the deposition of iron oxidation products and biofilm growth.5,6 Moreover, high installation costs prohibit the use of PRBs for treating deep aquifers or sites with distributed contaminant sources. Iron nanoparticles, or nZVI, are considered by many as a complementary treatment option to Fe-PRBs.8,9 Owing to their diminutive size, iron nanoparticles can be directly injected into groundwater by gravity or a pressurized feed, rendering a highly flexible treatment technology for sites with complex hydro-geological characteristics.10 The last ten years have seen over 50 pilot or full-scale applications of nZVI for in situ groundwater and soil remediation worldwide, according to recent reviews by Karn et al. and Mueller et al.8,11 Most of the pioneering field work was conducted in the U.S., while interest by countries in Europe is picking up with 15 field demonstrations and full-scale implementations completed or on-going in the Czech Republic, Germany, Italy, and Slovakia.11 In Asia, a pilot test has been completed in Taiwan,12,13 and several are being planned in China and other countries.
In parallel with these field implementations, the progress made in several fundamental areas has deepened our understanding of the merits and uncertainties of nZVI-based remediation applications. These areas include the materials chemistry of nZVI in its simple and modified forms, the reactivity of nZVI with a wide spectrum of contaminants in addition to the well-documented chlorinated solvents, methods to enhance the colloidal stability and transport properties of nZVI in porous media, and the effects of nZVI amendment on the biogeochemical environment. This review aims to provide an up-to-date account of advances in these areas as well as insights gained through field experience. Our intention is to present a current and objective view of the potential of nZVI materials in existing and new applications and inform future research efforts to address key engineering challenges.
Materials chemistry of iron nanoparticles
Synthesis routes
As with other nanoscale materials, nZVI can be made via top-down and bottom-up approaches. Bottom-up approaches involve bringing together iron atoms to form Fe(0) clusters at the nanometer scale. This is achieved by chemical reduction of ferrous or ferric salts,14 or by vapor condensation in a vacuum or inert gas.15 Sodium borohydride reduction of ferric or ferrous salts remains the most common method to produce small quantities of nZVI at the lab scale (eqn (1)). This reaction scheme is easy to adjust for making nZVI with different physical properties. For instance, the addition of surfactants or polyelectrolytes into the precursor solutions allows researchers to make emulsified or surface-stabilized nZVI with precise control of the size, size distribution, and core–shell morphology.16–19 It should be noted that the wet chemistry synthesis is quite expensive (over $200 per kg nZVI) due to the high cost of sodium borohydride and the labor required. Additionally, it is difficult to scale this process up to an industrial scale due to the several separation steps involved and a large amount of wastewater produced.20| 4Fe3+ + 3BH4− + 9H2O → 4Fe0(s) + 3H2BO3− + 12H+ + 6H2(g) | (1) |
Other bottom-up approaches have been reported, such as the decomposition of iron pentacarbonyl (Fe(CO)5) in organic solvents in the presence of high-intensity ultrasonic waves.21,22 While this approach is able to produce ultrafine iron (10–20 nm) with a narrow size distribution, iron pentacarbonyl is a highly toxic reagent. Currently, bottom-up commercial-scale production of nZVI is achieved either by reduction of goethite (α-FeOOH) or hematite (α-Fe2O3) by H2 at elevated temperatures or by electrolytic reduction of ferrous iron using an applied current.23–25
Top-down approaches break down bulk iron materials into nanometer-scale fragments through mechanical means. A precision ball-milling technique, which uses stainless steel balls as the grinding media to fragment the starting iron materials into pieces with dimensions less than 100 nm, has been described by Li et al.20 By using several well-studied chlorinated contaminants as probe molecules, it was shown that such milled nZVI had comparable chemical reactivity to the borohydride-reduced nZVI.20 The main advantages of this milling process over the hydrogen reduction method are a shorter processing time and presumably lower energy consumption. However, milling has issues with mechanical wear-and-tear of the fabrication equipment and a limited control over particle size distribution and morphology.
More recently, the synthesis of iron nanoparticles using naturally derived polyphenolic compounds from tea leaves and sorghum bran extracts has garnered much interest.26–28 This method offers a possible route to synthesize iron nanoparticles in situ by injecting the reductants underground to react with dissolved iron that is either naturally occurring or augmented in the groundwater. Polyphenols are greener reagents than the widely used sodium borohydride. Additionally, the abundant hydroxyl and phenol groups present in these compounds afford them properties of a good capping agent to stabilize the reactive surfaces of the nanoparticles and decrease their bio-toxicity.29 Structural characterization of the polyphenol-reduced nanoparticles suggests that they contain largely α-Fe2O3 or iron oxyhydroxide, with only a minor component of amorphous Fe(0),27,29 which may explain their low reactivity in reduction applications30 and their more favorable use as catalysts for oxidative reactions with H2O2.26–28
The reactivity of nZVI can be catalytically enhanced by amending with a small amount of added metal (Pd or Ni). Pd–Fe nanoparticles are commonly prepared by mixing the as-synthesized nZVI in a palladium salt solution.2,31–33 Because the standard reduction potential of palladium is more positive than that of Fe(0), it is reduced by Fe(0) via classical metal replacement reactions and deposits as metallic palladium onto the nZVI surface.34 Ni–Fe nanoparticles are often prepared via simultaneous reduction of iron and nickel salts in solutions by sodium borohydride (‘co-reduction’ method).35,36 Co-reduced Ni–Fe bimetallic particles have shown a higher reactivity compared to those formed via doping of nickel on the pre-formed nZVI.35 Ethanol or acetone is often used as a co-solvent in the synthesis media to enhance the solubility of palladium or nickel salts and to mitigate extensive surface passivation induced by the galvanic effect during synthesis.31,32
General characterization
A comprehensive review of the characteristics of various commercial and lab-synthesized nZVI materials has been made by Nurmi et al.24 Methods for characterizing the pertinent properties of nZVI are summarized in Table 1. nZVI particles made from borohydride reduction generally adopt a smooth, spherical shape, whereas those from reduction of goethite or hematite by H2 are more angular-shaped with strong surface faceting.24,37 In the absence of surface stabilizing chemicals, these nanoparticles tend to associate into linear or fractal aggregates due to chemical and magnetic interactions.19,38| Property | Assessment method | Reported characteristicsa | References |
|---|---|---|---|
| a For pristine nZVI unless otherwise stated. | |||
| I. General characterization | |||
| Morphology | SEM, TEM | Varying from smooth, spherical shaped for nZVI made in solution phase to angular shaped with surface facets for particles reduced from iron oxides under H2. | 19, 24 and 38 |
| Generally particles associate to form clusters in the absence of surface stabilizers. | |||
| Size and size distribution | Size of dry particles: electron microscopic observation | Primary particles 10–100 nm | 19, 24, 39 and 40 |
| Hydrodynamic radius: acoustic spectrometry, dynamic light scattering (DLS) | Hydrodynamic radii of aggregates on the order of 100 nm depending on the synthesis route, polyelectrolytes used, and background solution | ||
| Specific surface area | Brunauer–Emmett–Teller (BET) isotherm gas adsorption | 10–50 m2 g−1 | 19, 24 and 50 |
| Surface charge | Electrophoretic (zeta-potential) measurement | pHISP 6.5–8.3 | 19, 39 and 95 |
| Crystallographic characterization | Electron diffraction, X-ray diffraction (for grain sizes larger than a few nm) | bcc Fe(0) and Fe3O4 were identified | 24, 43, 45 and 50 |
| Degree of crystallinity varies with synthesis routes | |||
| Fe(0) content | Temperature programmed reduction, redox titration, hydrogen evolution test | Varying from 20% to over 90%, with borohydride-derived nZVI generally having a higher Fe(0) content than nZVI from thermal H2 reduction | 43–45 |
| Oxidation–reduction potential (ORP) | Conventional ORP probe or rotating disk electrode | Although ORP is a lumped parameter and has no direct correlation with nZVI concentration, it is an expedient tool to assess nZVI reactivity in batch reactors | 12, 138, 139 and 143 |
| II. Advanced spectroscopic characterization | |||
| Surface chemical analysis | X-ray photoelectron spectroscopy (XPS) | The surface of solution-made nZVI has a stoichiometry close to FeOOH. Thickness of the (oxyhydr)oxide shell can be calculated from the ratio of the oxidized and metallic Fe signals | 45 and 53 |
| Fine structure analysis | Mössbauer spectroscopy | bcc Fe(0) and disordered ferric oxide were identified | 52 and 144 |
| Fine structure analysis | XANES, EELS | The fine structure of the oxide component does not resemble any known bulk iron oxides, implying a high degree of disorder | 47 |
| Ultra-high resolution chemical mapping | STEM-XEDS | This technique allows visualization of the spatial distribution of catalytic dopants (e.g. Pd) or contaminants in the internal structure of nZVI | 46 and 93 |
Coarse analysis of the size distribution of nZVI is often obtained by direct TEM observations of a statistically significant number of particles.19,24 When particle transport is concerned, it is more relevant to measure the hydrodynamic radii of aggregates using acoustic spectrometry19 or dynamic light scattering (DLS) techniques.39,40 There is no direct comparison of the results from the acoustic and light scattering methods, however, DLS data may be skewed by the presence of a small fraction of large aggregates. Both methods require a stable suspension of the nanoparticles, which entails the addition of polyelectrolyte stabilizers. One can also make an indirect estimation of the particle size based on the specific surface area of the particles by assuming a spherical shape for the particles.20
The particle synthesis history has a profound impact on their crystallographic properties. The electron diffraction pattern of fresh borohydride-reduced nZVI suggests that the metallic iron cores are polycrystalline bcc Fe(0). This corroborates XRD data, in which the broadening of the primary diffraction peak (2θ = 44.6°) is indicative of a grain size smaller than 1.5 nm.24,37 However, analysis of X-ray absorption data suggests that the nature of the Fe(0) core of solution-derived nZVI may be more disordered containing a glassy metal iron phase that is transparent to the diffraction analyses.41,42 This amorphous nature would have direct implications on the nZVI ability to produce and activate hydrogen, which may account for the higher reactivity of solution-derived nZVI in hydrodechlorination reactions compared to the highly crystalline nZVI produced via H2 reduction of iron oxides.37,43
Experiences suggest that the relative amount of metallic and oxidized iron in nZVI is an appropriate quality control parameter since it correlates with the reductive capacity and longevity of nZVI. In practice, the Fe(0) content is determined experimentally from hydrogen evolution in acidic media,43 temperature-programmed reduction analysis,44 or aqueous redox titration.45 Reported values of the mass fraction of Fe(0) in fresh nZVI range from 20% for H2-reduced nZVI to over 90% for nZVI from borohydride reduction (Table 1). In all types of nZVI, the oxide phase is a significant component, which is a unique feature of the nano-sized iron particles.
Core–shell structure revisited
The surface of nZVI mediates virtually all aspects of nZVI chemistry, including iron corrosion, particle stabilization, and contaminant adsorption, and redox transformations. This surface, as we now know, is a thin layer of iron oxide that forms spontaneously during synthesis and undergoes continuous, albeit sometimes slow, evolution during the entire service life of the nanoparticles. Under a high-resolution transmission electron microscope (HR-TEM), the borohydride-made nZVI appears to be clad in a smooth amorphous oxide skin,46 while the H2-reduced nZVI contains distinct domains of crystalline oxide structures.24 The average thickness of the oxide shell on freshly made borohydride-synthesized nZVI is approximately 3 nm.45 This value agrees with the classic iron oxidation model, which predicts that a film of about 2–3 nm is formed instantaneously when metallic iron is first exposed to the atmosphere at room temperature.47,48 A further increase in thickness is exponentially slower due to an increase in diffusion length for the outbound movement of iron.The fine structure of the oxide phase depends on the synthesis process, particle size, and storage conditions.49,50 Characterizations with various spectroscopic and electroscopic techniques (Table 1) seem to agree with the presence of a mixed Fe(II)/Fe(III) phase in proximity to the metallic core and a predominantly Fe(III) oxide phase at the surface of the nanoparticles.47,51,52 For nZVI of an aqueous origin, the surface is covered with a thin layer of hydrated oxide (FeOOH).53,54 This chemical heterogeneity, which is confined in a few nanometers thickness, creates a highly disordered structure with potent chemical reactivity. Surface hydroxyl (–OH) groups may function as coordinative sites to bind with contaminants similar to that on many iron oxide-based adsorbents. Meanwhile, charge transport through the oxide is a facile process owing to its semiconducting nature and defective structure.47,55 Abundant defects such as oxygen and iron vacancies provide conduits for atom or ion diffusion.41 This implies that nZVI can exhibit a multifaceted reactivity reflecting the properties of both iron oxide and metallic iron. Indeed, reduction, adsorption, co-precipitation, and oxidation of contaminants have all been observed during nZVI treatment (Section 3).
Particle aging and structural evolution
The reactive nature of nZVI implies that the structure and chemical properties of nZVI are a strong function of time. In anoxic solutions, Reardon et al. showed that corrosion of nZVI is a bi-phasic process consisting of a rapid initial oxidation stage (∼2 days) followed by a period of several months of further corrosion at a slow and nearly constant rate.56,57 Among common groundwater anions, nitrate is known to greatly impact the aging behavior of nZVI,58,59 and natural organic matter (NOM) or silica may slow down Fe(0) oxidation by forming a protective layer on the anodic sites.56 Limited air exposure during and after synthesis favors the formation of a Fe3O4-like film, which is electron-conducting and permits further oxidation of the iron.60 In comparison, exposure to fully aerated water or a large volume of air triggers the formation of oxides with a more passivating character (e.g. maghemite or hematite).59,60Microscopically, exhaustion of the Fe(0) cores was observed when nZVI was exposed to an anoxic solution for up to 5 months.61 Contrary to a common perception, the native oxide shell of nZVI did not experience appreciable growth over prolonged aqueous immersion. Instead, corrosion products deposit as surface debris or large-scale platelets that are extraneous to the core–shell particles.61 Meantime, aging-induced crystal reformation of the Fe(0) cores gives rise to a highly crystalline bcc iron phase.50 Therefore, the decrease in nZVI reductive activity with time is attributable to the increase in crystallinity (thus lower reactivity) of the metal phase. Over a sufficiently long time, complete loss of Fe(0) cores and emergence of hollowed-out oxide shells were observed. This dynamic structural change is illustrated in Fig. 3.
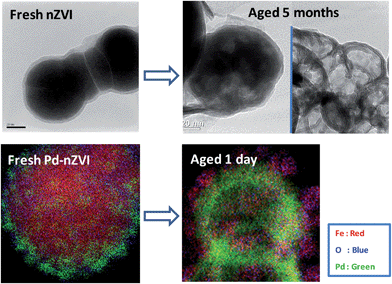 | ||
| Fig. 3 Structural evolution of nZVI during aging in an aqueous environment. (Top) Transmission electron microscopy (TEM) images of fresh nZVI particles and those after aging for 5 months. (Bottom) Scanning TEM-X-ray dispersive spectroscopy (STEM-XEDS) overlay images of fresh palladium-doped nZVI and those after aging for 1 day. Images from ref. 61 and 93. | ||
Aging-induced structural and morphological changes of nZVI are more pronounced for bimetallic nanoparticles because of the galvanic couple formed by the second metal and Fe(0), which greatly accelerates iron oxidation.62 A recent study employing aberration-corrected scanning transmission electron microscopy coupled with energy-dispersive X-ray spectroscopy (STEM-XEDS), which is capable of elemental mapping at sub-nanometer resolution, has provided direct evidence of drastic structural changes of palladium-doped nZVI within a brief aging period. It was observed that palladium nano-islands on the outer surface of nZVI migrated through the oxide layer and segregated at the Fe(0)/Fe-oxide interface during a 24 h aqueous exposure (Fig. 3). This radical change explains the significant loss of dechlorination activity over the course of repeated use of Pd-nZVI nanoparticles32 and highlights the need for further research focusing on prolonging the useful life span of such bimetallic nanoparticles. Strategies such as the use of co-solvents during synthesis or immobilizing particles on solid supports may alleviate the rapid structural breakdown and afford some control over their aging behavior.63,64
Immobilized or surface stabilized nZVI
High surface energy and strong magnetic interactions aggravate self-aggregation issues for unmodified nZVI.65,66 Difficulty in preparing stable, well-dispersed aqueous nZVI suspensions has remained an obstacle that hinders practical applications of nZVI in various environmental systems. Several strategies have been explored to enhance the stability of nZVI, including coating the nanoparticle surface with polyelectrolytes or non-ionic surfactants, conjugating with a solid support, and dispersing the particles in oil–water emulsions.Surface coating of nZVI improves the particle stability via charge and steric stabilization. In this regard, several anionic polymers, such as PAA (polyacrylic acid), PSS (polystyrene sulfonate), and carboxymethyl cellulose (CMC), are attractive candidates because of their large molecular weights and high densities of charged functional groups.40,67–71 Negative surface charges are preferred in these applications because they reduce the affinity of nZVI for the natural soil media.72 Affixing the polymer stabilizers can be achieved by either mixing the as-synthesized nZVI with polymer solutions (post-synthesis method) or adding them into an iron precursor solution prior to the particle synthesis (pre-synthesis method). Table 2 provides a list of the surface modifiers evaluated for nZVI. Phenrat et al. assessed several of these anionic polymers using sedimentation tests, and their results suggest that polymer coatings are less effective for the larger particles, and a greater hydrodynamic thickness of the coating layer conveys more stability.40
| Stabilizing agent | Modification method | Field test | References |
|---|---|---|---|
| I. Surface coated nZVI | |||
| Polyacrylic acid (PAA) | Pre- or post-synthesis coating | Pilot test at a VC manufacturing site | 12, 68 and 70 |
| Carboxymethyl-cellulose (CMC) | Pre-synthesis coating | Pilot test at a secondary source zone contaminated with PCE and TCE | 18, 139, 141 and 145 |
| Polyvinyl alcohol-co-vinyl acetate-co-itaconic acid (PV3A) | Pre- or post-synthesis coating | 65 | |
| PMAA–PMMA–PSS triblock copolymers | Post-synthesis coating | 146 | |
| Polystyrene sulfonate (PSS) | Post-synthesis coating | 40 and 67 | |
| Polyaspartate | Commercial nZVI | 40 | |
| Maleic acid-based polymer | Commercial nZVI | 147 | |
| Natural biopolymers: | Post-synthesis coating | 39, 71 and 148–150 | |
| • Guar gum | |||
| • Xanthan gum | |||
| • Calcium alginate | |||
| • Starch | |||
| II. Emulsified nZVI | |||
| Water-in-oil-in-water (W/O/W) emulsion | Post-synthesis emulsification. nZVI dispersed in aqueous droplets of 1–20 μm surrounded by an oil membrane | Pilot test at a site with TCE DNAPL | 73 |
| Oil-in-water (O/W) emulsion | Post-synthesis emulsification. nZVI dispersed in oil-in-water droplets of 1–2 μm | 74 | |
| III. Supported nZVI | |||
| Functionalized membranes | Soaking membranes in iron salt solutions followed by borohydride reduction | 151 and 152 | |
| Carbon supports | Commercially available granular activated carbon or tailor-made porous carbon supports. Iron ions anchored prior to reduction | 33, 68 and 76 | |
| Silica supports | Bulk or porous silica fabricated from sol–gel or aerosol-based processes. | 77 and 153 | |
| Clay supports | Naturally available or synthetic zeolites and pillared interlayered clays (e.g. bentonite). | 78–80 | |
Dispensing nZVI in emulsified oil–water suspensions was proposed to target nZVI delivery to dense non-aqueous phase liquid (DNAPL) source zones.73–75 Commercially available food-grade vegetable oil and surfactants (e.g. SPAN™, TWEEN™, oleic acid) have been used to formulate the emulsion. Encapsulating nZVI in oil vesicles is expected to improve the particle delivery to the DNAPL phase and to protect nZVI from reacting with passivating groundwater constituents.73 Pilot and full-scale treatments with emulsified nZVI have been conducted at several TCE DNAPL sites,8,73 in which enhanced microbial activity for TCE reduction was observed due to the injected oil and surfactants serving as excellent carbon sources and electron donors.
Immobilizing nZVI on a solid support enhances particle dispersion and enables applications of nZVI in continuous flow systems. By using carboxylic, hydroxylic, or amine groups as chelating sites for the nanoparticles (or their precursor ions), nZVI has been successfully attached to a variety of substrates including carbon, silica, clays, and ultrafiltration or reverse osmosis membranes (Table 2). Mallouk et al. modified the surface of carbon supports by grafting with sulfonic acid groups to impart negative charges for optimal dispersion in the groundwater.68 In other applications, an unfunctionalized carbon may enhance the adsorption of organic contaminants such as PCB through hydrophobic interactions.33 More sophisticated forms of carbon, such as a highly porous carbon fabricated from a sugar-based aerosol process, have been devised by Zhan et al.,76 and similar set-ups were used to prepare porous silica supports.77 nZVI has also been anchored on naturally abundant clay minerals including zeolites, bentonite, and montmorillonite.78–80 The inherent ion-exchange capability of these minerals allows facile incorporation of iron. Intercalation of iron into the pillared layers of bentonite creates interlayer meso- or micro-pores, which provide pollutant transport channels and a large reactive surface area. None of these composite materials have been evaluated in field trials. Presumably, efforts are needed to produce these composite materials in large quantities at a competitive price. A list of these modified nZVI materials is given in Table 2.
Admittedly, the enhanced stability and transport capability of nZVI in its encapsulated or supported forms are at the expense of a reduced chemical reactivity due to the obstruction of reactive sites81,82 and the additional diffusive barriers caused by the encapsulation materials.63 Assessing the overall impact of surface-engineered nZVI under field-relevant conditions would be necessary for evaluation.
Reactivity of nZVI with aqueous contaminants
Reductive dehalogenation
The environmental applications of iron for reductive degradation of halogenated organics are a recent extension of the century-long use of iron as a catalyst for organic synthesis in the chemical industry.83 Zerovalent iron is a moderately strong reductant (eqn (2)). The half-reaction of aliphatic chlorinated compounds shown in eqn (3) has a standard reduction potential ranging from +0.5 to +1.5 V at neutral pH.84 Thus there is a strong thermodynamic driving force for their reduction by zerovalent iron materials.| Fe0 → Fe2+ + 2e−, E0 = −0.44 V | (2) |
| RCl + 2e− + H+ → RH + Cl−, E0 = 0.5–1.5 V at pH = 7 | (3) |
The mechanisms of iron-mediated hydrodehalogenation reactions have been extensively investigated. It is generally accepted that the reaction is heterogeneous in nature, involving adsorption of the contaminants at the iron surface prior to breaking of carbon–halogen bonds.55,85 The core–shell nature of nZVI implies facile electron transfer across the thin oxide shell. Doping a small amount of a second metal on the surface of nZVI causes a significant increase in reactivity. Among many transitional metals studied for catalytic dehalogenation reactions, Pd is the most commonly used catalyst owing to its optimal structural and chemical properties to generate activated hydrogen species and to cleave the carbon–halogen bond.86,87 Compared to monometallic iron, bimetallic iron systems deliver greatly enhanced reaction rates, more saturated end products, and lead to little or no accumulation of chlorinated intermediates.88,89 The reduction process consists of a series of steps, namely, H2 evolution, dissociative adsorption of H2 and the formation of active hydrogen species, and the reduction of surface adsorbed halogenated contaminants.36,86,90Fig. 4 illustrates reaction mechanisms for TCE reduction by nZVI and Pd-nZVI.36,91,92 In this proposed model, there is a synergistic effect between the second metal (e.g. Pd) and Fe, whereby Pd serves as the hydrodehalogenation and hydrogenation catalyst and Fe provides the hydrogen source through water reduction. Unfortunately, the reactivity of the bimetallic particles declines rapidly in aqueous environments due to the accelerated iron oxidation and entrapment of active Pd sites underneath an iron oxide shell.93 Continued research to improve the materials chemistry of bimetallic particles for sustained reactivity in such applications is highly worthwhile.
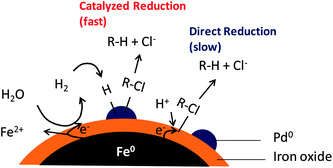 | ||
| Fig. 4 Mechanisms of reductive dechlorination by monometallic nZVI and palladium-doped nZVI. | ||
Sequestration of inorganic contaminants
The versatility of nZVI as an environmental remediation agent is reflected in its ability to capture a variety of inorganic contaminants, including nitrate,58 Cr(VI),68,94 As(III) and As(V),52,95,96 heavy metals,53,97,98 and radionuclides99,100 (Fig. 2a). In all cases, the core–shell nature of nZVI has a profound influence on the sequestration pathways (Fig. 5).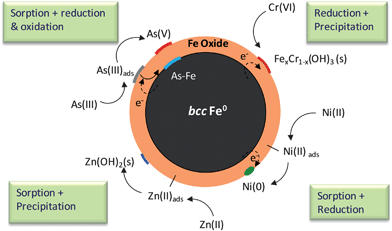 | ||
| Fig. 5 Role of the core–shell structure in contaminant sequestration. | ||
Generation of reactive oxidants
nZVI is used primarily as a reductant in anoxic media, however, when the nanoparticles are exposed to an aerobic environment, the core–shell nature of nZVI imparts an interesting oxidative chemistry producing reactive oxygen species (ROS) including hydrogen peroxide and hydroxy and peroxy radicals.112,113 The oxidative pathway starts with a two-electron oxidation of dissolved oxygen by Fe(0) to form hydrogen peroxide (eqn (4)). H2O2 may accept another two-electron transfer from Fe(0) to form water (eqn (5)), or react with Fe(II) to produce a potent oxidant, OH˙ radical, under acidic conditions (eqn (6)), or Fe(IV) species at neutral to alkaline pH (eqn (7)). Under neutral pH conditions, Fe(II) may react with O2 directly to produce O2˙− radicals (eqn (8)).114| O2 + Fe0 + 2H+ → Fe2+ + H2O2 | (4) |
| H2O2 + Fe0 + 2H+ → Fe2+ + H2O | (5) |
| H2O2 + Fe2+ → Fe3+ + OH˙ + OH− (acidic pH) | (6) |
H2O2 + Fe2+ → Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2+ + H2O (pH > 5) O2+ + H2O (pH > 5) | (7) |
| Fe2+ + O2 → Fe3+ + O2˙− (pH ∼ 7) | (8) |
Compared to bulk iron, nZVI produces more ROS due to the higher rates of iron corrosion that is driven by its large surface area and a more disordered and reactive oxide layer. For this reason, studies have attempted to use nZVI in an oxidative function similar to heterogeneous Fenton reagents.115–117 However, this oxidative reactivity of nZVI tends to diminish with time and with increasing dose of nZVI due to surface passivation by corrosion products.113 The type and quantity of oxidants formed are sensitive to pH and the ligand environment.118–120 OH˙ radical is the dominant oxidant under acidic conditions. At neutral pH, a different but weaker oxidant exists. The identity of this weaker oxidant is subject to speculation, with several species, e.g. Fe(IV) and O2˙−, as possible candidates.114,118,121,122 On the other hand, the oxidant yield of nZVI is enhanced by introducing chelating agents (e.g. EDTA),120 or electron shuttles such as natural organic matter (NOM)123 and polyoxometalates (POMs).119,124 Since corrosion is a surface-mediated process, the nature of the oxide shell of nZVI may exert a significant influence on its oxidizing capacity.113 It is possible to tailor the reducing and oxidizing properties of nZVI such that concurrent or sequential redox processes can be enabled in a single nZVI treatment system. Such combined redox processes have been reported to result in higher degradation of azo-dyes and other contaminants.117,125
Biogeochemical interactions in the environment
Toxicity of nZVI
Concerns over the potential negative impact of nZVI applications have spurred studies examining the toxicological characteristics of nZVI. Very recently, acute bactericidal and virucidal activities of fresh uncoated nZVI were observed in pure culture studies.126–128 Compared to iron oxide nanoparticles (Fe3O4 and γ-Fe2O3), nZVI exhibited a short-term elevated ability of microbial inactivation.129 This toxic effect was significantly lower when nZVI was oxidized126 or in the presence of an environmental matrix.130,131 The bactericidal action of nZVI was partly attributed to an oxidative stress caused by the generation of reactive oxidative species (ROS) from the reduced iron and oxygen.127,132 In addition, direct interactions of nZVI with cell membranes and the interruption of electron and ion transport chains are believed to play important roles.127,129 While these results may suggest potential applications of nZVI as an antimicrobial agent, the implications for the toxicity of nZVI in natural systems are limited due to the use of a relatively high dose of nZVI in these experiments and the absence of an environmental background matrix, which is known to suppress the reactivity of nZVI and reduce its attachment to microbial surfaces.The toxicity of nZVI was greatly mitigated when experiments were conducted in the presence of natural organic matter (NOM), polyelectrolytes, or an aquifer matrix.130,131,133 Li et al. reported that coating nZVI with polyelectrolytes or NOM led to a nearly complete loss of bactericidal activity towards E. coli.130 The minimum nZVI concentration to see an inactivation effect increased from 5 mg L−1 for pristine nZVI to as high as 500 mg L−1 for coated nZVI. For the mixed culture of a TCE-degrading bacterial consortium, the addition of nZVI coated with biodegradable surfactants created a noticeable biostimulatory effect, while pristine nZVI severely inhibited the microbial reductive dehalogenation activity.134 As natural organic matter is ubiquitous in the subsurface environment, these results suggest a probably mild impact associated with nZVI use on microbial communities in groundwater or soil media. While the overall microbial population may not be affected to a great extent by nZVI amendment, changes in population structure and composition have been noted in microcosm studies.131,133 These studies report a clear shift to species able to utilize cathodic hydrogen produced during nZVI corrosion (e.g. sulfate-reducers and methanogens). This composition change may bring about increased degradation of contaminants via biological pathways.
Interactions with soil biogeochemistry
Comprehensive assessment of the impacts of nZVI on soil geochemistry and microbiology under field conditions is difficult to obtain due to resource constraints that have limited the number of monitoring points and the duration and type of parameters recorded at the injection sites. In general, oxidation–reduction potential (ORP), dissolved oxygen, pH, electric conductivity, and iron concentrations (dissolved and total) are the primary parameters measured at field sites. The introduction of nZVI was associated with a marked reduction in ORP,12,133 indicating accumulation of nZVI reaction products (e.g. H2) after injection. pH was reported to be stable over nZVI injection owing to the large natural buffering capacity in groundwater. These conditions have been reported to favor the establishment of a variety of anaerobic bacteria including sulfate-reducers, iron-reducers, and methanogens.133 The relative abundance of these organisms would depend on the site characteristics and the dose of iron injected. For surface-stabilized nZVI, the injection of biodegradable polymeric substances further enhances the anaerobic biological activity. A review of recent field studies does seem to suggest that nZVI treatment can trigger a combination of chemical reduction and anaerobic biodegradation processes.135The impact of nZVI on soil mineral formation and transformation has not been systematically assessed. The release of dissolved iron and the precipitation of ferric/ferrous hydroxide products during nZVI corrosion are expected to influence the concentrations and chemical states of trace metals in the soil media. Such changes are closely tied to microbiological activities (e.g. iron-reducers) and may have profound long-term impacts on the immobilization of metal contaminants (e.g. Cr(VI), U(VI)).
Current status of field deployment
Based on a recent review by Karn et al., nZVI-based materials were used in over 90% of pilot or full-scale groundwater and soil remediation projects.8 As of today, there are 58 completed or on-going nZVI remediation sites worldwide.136 Among these, 36 cases were conducted in the United States, including eight full-scale implementations. Europe is another actively growing region. Seventeen sites, mainly located in Czech Republic and Germany, have been commissioned including three full-scale deployments.11 The publicly available data on these remediation projects are fairly limited and scattered in various sources.8,11,136,137Table 3 shows a list of sites that have disclosed relatively more implementation details (namely, dose of nZVI, total injection mass, treatment volume, and operation duration). Throughout these sites, the contaminants treated were chlorinated aliphatics, implying the establishment of a niche market for nZVI in treating this particular group of contaminants.136| Site location | Contaminants treated | Type of iron | Dose (g L−1) | Total iron used (kg) | Degradation efficiency | Operation duration (mon) | Treated volume (m3) | References |
|---|---|---|---|---|---|---|---|---|
| a — Denotes no data available. | ||||||||
| I. United States | ||||||||
| Cape Canaveral, FL, USA | TCE | Emulsified nZVI | — | — | 57–>99% (TCE) | — | 34 (Pilot) 210 (Full-scale) | 8, 136 and 137 |
| Edison, NJ, USA | TCA, DCA, TCE, DCE, VC | nZVI | 24 | 135 | 40–99% (TCA, top layer), 89–>99% (TCA, bottom layer) | 13 | — | 8, 136 and 137 |
| Hamilton, NJ, USA | TCE, DCE, TCA, DCA | nZVI | <30 | 2000 | <90% (total VOC) | 1 | — | 8, 136 and 137 |
| Lakehurst, NJ, USA | PCE, TCE, TCA, DCE, VC | nZVI + palladium | 2 | 135 | 74% (total VOC) | — | — | 8, 136 and 137 |
| Jacksonville, FL, USA | PCE, TCE, TCA | nZVI + palladium | 4.5–10 | 135 | 65–99% | 9 | 740 | 8, 136 and 137 |
| Passaic, NJ, USA | TCE | nZVI | — | 49 | 90–>99% (total VOC) | 6 | — | 8, 136 and 137 |
| Patrick AFB, FL, USA | TCE | Emulsified nZVI | — | — | >99% (TCE) | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 | 8, 136 and 137 | |
| Research Triangle Park, NC, USA | PCE, TCE, DCE, VC | nZVI + palladium | 1.9 | 11.2 | >90% (total VOC) | 3 | — | 8, 136 and 137 |
| Rochester, NY, USA | DCA, MC, DCP | nZVI | 10–20 | 100 | 90% | — | — | 8, 136 and 137 |
| Rockaway, NJ, USA | CT | nZVI | — | 55 | Rebound to ∼90% of original level after 4 months | 12 | — | 8, 136 and 137 |
| II. Other countries | ||||||||
| Horice v Podkrkonosi, Czech Republic | PCE, TCE, DCE | nZVI | 2.5 | 500 | 60–75% | — | 21600–72000 | 136 |
| Pisecna, Czech Republic | PCE, TCE, DCE | nZVI | — | 300 | 40–80% | — | 25000 | 136 |
| Quebec, Canada | TCE, DCE, VC | nZVI + palladium + stabilizer | — | 4550 | 98% (TCE) | 12 | 4500 | 8 and 136 |
| Kaohsiung, Taiwan | TCA, DCA, TCE, DCE, VC | nZVI + palladium + stabilizer | 18 | 40 | 50–99% (VC) | 3 | 2400 | 8 and 136 |
A close look at the data in Table 3 reveals that large spatial and temporal variations in degradation efficiency exist at individual sites. Many sites reported nearly complete removal of contaminants at the injection wells but much less removal in the periphery.136 The duration of post-injection monitoring also spans from weeks to years. Except for one case that reported a concentration rebound to the original level after weeks of injection, most cases reported a considerable extent of reduction in contaminant concentrations from 40% to nearly complete removal.8 For most field trials, changes in the contaminant concentrations are the only information available to gauge the effectiveness of nZVI treatment (Table 3). However, to allow comparison of field tests on a consistent basis, it is necessary to take into account other factors, including at least the size of treatment zones and the operation duration. In other words, a more general indicator of the treatment effectiveness should reflect the total amount of contaminants transformed, integrated over the entire effective zone (eqn (9)). To arrive at comparable figures, common guidelines on field data acquisition (e.g., the spacing of monitoring wells and the sampling frequency) may need to be followed. Finally, the degradation efficiency normalized to the total amount of iron injected could provide more meaningful statistics on the overall performance (eqn (10)).
| Total contaminants degraded = ∫ΔCcontaminants × volume of effective zone | (9) |
 | (10) |
Before a sizable collection of the above data becomes available, we are restricted to making rather preliminary remarks here. In general, the final outcome is dependent on the nature of treatment (e.g., source removal or pathway management), site hydrogeological characteristics, the type and dose of nZVI used, and the injection scheme. Although nZVI has proven effective in laboratory assessments, its reactivity is less predictable under field conditions and the reactive life-span of nZVI is much shorter than the conventional micro-iron materials. The general consensus among practitioners is that nZVI treatment works best in anaerobic environments with good hydraulic conductivity and a pH range that is neutral to weakly acidic.136 Although nZVI has been deployed for contaminant source control via direct injection, this application is limited by the difficulty in accurately locating the contaminant source zones (i.e., DNAPL phases) in the subsurface.
Currently, our understanding of the interaction between nZVI and the aquifer environment is still limited. The geochemical properties of the aquifer may be changed by the nZVI amendment.11,12,138,139 The field study by He et al. using CMC-stabilized Pd-nZVI for PCE and TCE remediation showed that nZVI amendment triggered rapid abiotic degradation within a few weeks after injection and stimulated slower but persistent biodegradation processes spanning several years.139 In addition, Quinn et al., who used emulsified nZVI targeting underground TCE DNAPL pools, concluded that a significant fraction of TCE removal was due to anaerobic microbial degradation, an unintended but beneficial side effect of modifying nZVI with organic polymer coatings.73 Analysis of the carbon stable isotope signature in the reaction products may allow quantification of the relative contribution of abiotic and biotic pathways.140 Based on the limited number of field studies reported in the literature, the mobility of stabilized nZVI in soil media was rather low139,141 despite good breakthrough results in laboratory column tests. This reflects that bench-scale testing may not fully capture the effects of many important processes controlling the transport and delivery of nZVI in real systems.
From an engineering point of view, there is a need to develop a design protocol for field engineers to estimate the dose of nZVI in the injection stream, the number of injection points, and the total amount of nZVI applied. Oftentimes, the process entails estimating remediation capacities based on literature data. However, it is much more reliable to obtain such information from preliminary lab assessments using groundwater from the site to account for background effects. Similarly, model-assisted prediction of the transport distances of nZVI142 and the impact of dilution effect on the actual concentrations of nZVI and stabilizers reaching the aquifer at a given hydrogeological condition would be immensely valuable.
Another challenge that arises at the post-implementation stage is to delineate the boundary of the effective treatment zone. Although ORP is an expedient measure of the changes in redox conditions, it may not represent the successful delivery of the particles. Other parameters, such as total suspended solids or total iron concentration, may provide more direct information about the extent of nZVI travel.12 Additionally, changes in the contaminant concentrations may be caused by fluid displacement and not necessarily reflect actual degradation efficiency. Correlation analysis of multiple parameters and isotope studies will provide stronger lines of evidence for chemical (and biological) transformations. The discussion here illustrates the importance of close integration between theoretical, laboratory and pilot-scale investigations for optimal design of in situ remediation strategies (Fig. 6). Such integrated efforts tend to be resource intensive, but priority could be given to a few well-characterized sites as prototypical studies. It is anticipated that detailed documentation of these studies with in-depth data analysis will greatly improve our ability to assess the potential of nZVI for large-scale implementation and drive research to address critical technological needs.
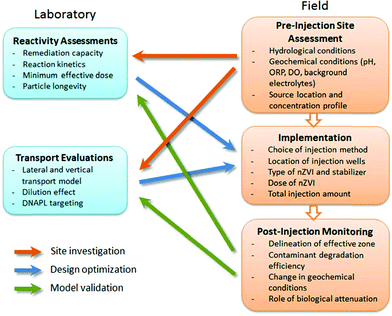 | ||
| Fig. 6 Integrated laboratory and field investigations for optimal design of nZVI remediation technology. | ||
Future perspective and research needs
The growing number of scientific investigations and commercial implementations testify that among the myriad nanomaterials evaluated for contaminant abatement, nZVI stands as one of the most technically mature and economically viable candidates for applications at environmentally relevant scales. This prominent status of nZVI does not lessen the need to address various issues that have emerged by seeking increased knowledge about its chemical and colloidal properties and performing systematic, well-documented field evaluations. From a chemical viewpoint, how to prolong nanoparticle reactivity and improve the selectivity over the target pollutants are salient questions to be addressed. On the engineering front, there is a pressing need to develop models capable of predicting the mobility of particles in porous media and methods for mitigating aggregation and other transport-impeding characteristics of nZVI.Admittedly, some of the uncertainties associated with subsurface remediation result from the heterogeneous nature of groundwater systems and the difficulty in characterizing them accurately. This has been an obstacle to designing an effective clean-up scheme and assessing true remediation performance. Another barrier lies in the difficulty of connecting findings from laboratory studies to field implementation. The complex hydrogeochemical and biological interactions between nZVI and the natural environment are difficult to reproduce in a laboratory setting. To tackle these challenges, it is highly desirable to facilitate close collaborations between the research community and the remediation industry. Specifically, well-planned mesocosmic or pilot studies may bridge the gap between isolated laboratory reactors and real aquifer systems.
While nZVI is applied predominantly to breakdown chlorinated solvents in groundwater and DNAPL phases, the list of contaminants amenable to nZVI treatment can possibly encompass a host of other harmful substances identified at underground sites. Many consider nZVI to be an alternative technology to bioremediation and Fe-PRBs. However, it is our opinion that the heterogeneous composition of groundwater media may call for a combination of multiple treatment arrangements administered concurrently or in different phases. In this context, the flexibility and ease of operation associated with nZVI injection imply that it will play an important part in an integrated remediation scheme. As stimulation of biotic degradation processes by nZVI amendment was noted in several field studies, it is possible to harness these biological interactions to enhance the performance and extend the operative time window of nanoparticle-based remediation. The use of biodegradable surface modifiers with nZVI may offer an effective means to manipulate the chemical and biological activity to suit specific remediation needs. Despite the many leaps forward and steady advancements, the field of underground remediation with nanomaterials is still in its infancy. There is plenty of room available at the bottom awaiting more effective and sustainable engineering inventions.
Acknowledgements
BEK gratefully acknowledges support from the Andlinger Innovation Fund of the Andlinger Center for Energy and the Environment (ACEE) at Princeton University.References
- R. Feynman, There is plenty of room at the bottom, Caltech Institute Archives, 1959, http://www.zyvex.com/nanotech/feynman.html.
- C. B. Wang and W. X. Zhang, Environ. Sci. Technol., 1997, 31, 2154–2156 CrossRef CAS.
- R. W. Gillham and S. F. Ohannesin, Ground Water, 1994, 32, 958–967 CrossRef CAS.
- M. M. Scherer, S. Richter, R. L. Valentine and P. J. J. Alvarez, Crit. Rev. Environ. Sci. Technol., 2000, 30, 363–411 Search PubMed.
- A. R. Gavaskar, N. N. M. Sass, R. J. Janoy and D. O'Sullivan, Battelle Memorial Institute, Columbus, OH, 1998 Search PubMed.
- L. Y. Liang, N. Korte, B. H. Gu, R. Puls and C. Reeter, Adv. Environ. Res., 2000, 4, 273–286 Search PubMed.
- R. T. Wilkin, C. M. Su, R. G. Ford and C. J. Paul, Environ. Sci. Technol., 2005, 39, 4599–4605 Search PubMed.
- B. Karn, T. Kuiken and M. Otto, Environ. Health Perspect., 2009, 117, 1823–1831.
- J. Theron, J. A. Walker and T. E. Cloete, Crit. Rev. Microbiol., 2008, 34, 43–69 CrossRef CAS.
- X. Q. Li, D. W. Elliott and W. X. Zhang, Crit. Rev. Solid State Mater. Sci., 2006, 31, 111–122 CrossRef CAS.
- N. C. Mueller, J. Braun, J. Bruns, M. Cernik, P. Rissing, D. Rickerby and B. Nowack, Environ. Sci. Pollut. Res., 2012, 19, 550–558 Search PubMed.
- Y. T. Wei, S. C. Wu, C. M. Chou, C. H. Che, S. M. Tsai and H. L. Lien, Water Res., 2010, 44, 131–140 CrossRef CAS.
- Y.-T. Wei, S.-c. Wu, S.-W. Yang, C.-H. Che, H.-L. Lien and D.-H. Huang, J. Hazard. Mater., 2012, 211, 373–380 Search PubMed.
- G. N. Glavee, K. J. Klabunde, C. M. Sorensen and G. C. Hadjipanayis, Inorg. Chem., 1995, 34, 28–35 CrossRef CAS.
- H. Hahn, Nanostruct. Mater., 1997, 9, 3–12 CrossRef CAS.
- E. E. Carpenter, S. Calvin, R. M. Stroud and V. G. Harris, Chem. Mater., 2003, 15, 3245–3246 CrossRef CAS.
- F. Li, C. Vipulanandan and K. K. Mohanty, Colloids Surf., A, 2003, 223, 103–112 CrossRef CAS.
- F. He, D. Y. Zhao, J. C. Liu and C. B. Roberts, Ind. Eng. Chem. Res., 2007, 46, 29–34 CrossRef CAS.
- Y. P. Sun, X. Q. Li, J. S. Cao, W. X. Zhang and H. P. Wang, Adv. Colloid Interface Sci., 2006, 120, 47–56 CrossRef CAS.
- S. L. Li, W. L. Yan and W. X. Zhang, Green Chem., 2009, 11, 1618–1626 RSC.
- K. S. Suslick, S. B. Choe, A. A. Cichowlas and M. W. Grinstaff, Nature, 1991, 353, 414–416 CrossRef CAS.
- D. Mahajan, E. T. Papish and K. Pandya, Ultrason. Sonochem., 2004, 11, 385–392 CAS.
- I. Capek, Adv. Colloid Interface Sci., 2004, 110, 49–74 CrossRef CAS.
- J. T. Nurmi, P. G. Tratnyek, V. Sarathy, D. R. Baer, J. E. Amonette, K. Pecher, C. M. Wang, J. C. Linehan, D. W. Matson, R. L. Penn and M. D. Driessen, Environ. Sci. Technol., 2005, 39, 1221–1230 CrossRef CAS.
- H. B. Liu, T. H. Chen, D. Y. Chang, D. Chen, Y. Liu, H. P. He, P. Yuan and R. Frost, Mater. Chem. Phys., 2012, 133, 205–211 Search PubMed.
- G. E. Hoag, J. B. Collins, J. L. Holcomb, J. R. Hoag, M. N. Nadagouda and R. S. Varma, J. Mater. Chem., 2009, 19, 8671–8677 RSC.
- E. C. Njagi, H. Huang, L. Stafford, H. Genuino, H. M. Galindo, J. B. Collins, G. E. Hoag and S. L. Suib, Langmuir, 2011, 27, 264–271 Search PubMed.
- T. Shahwan, S. Abu Sirriah, M. Nairat, E. Boyaci, A. E. Eroglu, T. B. Scott and K. R. Hallam, Chem. Eng. J., 2011, 172, 258–266 Search PubMed.
- M. N. Nadagouda, A. B. Castle, R. C. Murdock, S. M. Hussain and R. S. Varma, Green Chem., 2010, 12, 114–122 RSC.
- V. Smuleac, R. Varma, S. Sikdar and D. Bhattacharyya, J. Membr. Sci., 2011, 379, 131–137 Search PubMed.
- H. L. Lien and W. X. Zhang, J. Environ. Eng., 2005, 131, 4–10 CrossRef.
- B. W. Zhu and T. T. Lim, Environ. Sci. Technol., 2007, 41, 7523–7529 CrossRef CAS.
- H. Choi, S. R. Al-Abed, S. Agarwal and D. D. Dionysiou, Chem. Mater., 2008, 20, 3649–3655 CrossRef CAS.
- R. Muftikian, K. Nebesny, Q. Fernando and N. Korte, Environ. Sci. Technol., 1996, 30, 3593–3596 CrossRef CAS.
- C. L. Chun, D. R. Baer, D. W. Matson, J. E. Amonette and R. L. Penn, Environ. Sci. Technol., 2010, 44, 5079–5085 Search PubMed.
- B. Schrick, J. L. Blough, A. D. Jones and T. E. Mallouk, Chem. Mater., 2002, 14, 5140–5147 CrossRef CAS.
- Y. Q. Liu, H. Choi, D. Dionysiou and G. V. Lowry, Chem. Mater., 2005, 17, 5315–5322 CrossRef CAS.
- R. A. Crane and T. B. Scott, J. Hazard. Mater., 2012, 211, 2931–2942 Search PubMed.
- A. Tiraferri, K. L. Chen, R. Sethi and M. Elimelech, J. Colloid Interface Sci., 2008, 324, 71–79 Search PubMed.
- T. Phenrat, N. Saleh, K. Sirk, H. J. Kim, R. D. Tilton and G. V. Lowry, J. Nanopart. Res., 2008, 10, 795–814 CrossRef CAS.
- W. Yan, R. Vasic, A. I. Frenkel and B. E. Koel, Environ. Sci. Technol., 2012, 46, 7018–7026 Search PubMed.
- S. Calvin, E. E. Carpenter and V. G. Harris, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 4 Search PubMed.
- Y. Q. Liu, S. A. Majetich, R. D. Tilton, D. S. Sholl and G. V. Lowry, Environ. Sci. Technol., 2005, 39, 1338–1345 CrossRef CAS.
- J. S. Cao, X. Q. Li, J. Tavakoli and W. X. Zhang, Environ. Sci. Technol., 2008, 42, 3780–3785 Search PubMed.
- J. E. Martin, A. A. Herzing, W. L. Yan, X. Q. Li, B. E. Koel, C. J. Kiely and W. X. Zhang, Langmuir, 2008, 24, 4329–4334 CrossRef CAS.
- W. Yan, A. A. Herzing, C. J. Kiely and W.-x. Zhang, J. Contam. Hydrol., 2010, 118, 96–104 Search PubMed.
- C. M. Wang, D. R. Baer, J. E. Amonette, M. H. Engelhard, J. Antony and Y. Qiang, J. Am. Chem. Soc., 2009, 131, 8824–8832 CrossRef CAS.
- C. M. Wang, D. R. Baer, L. E. Thomas, J. E. Amonette, J. Antony, Y. Qiang and G. Duscher, J. Appl. Phys., 2005, 98, 094308 CrossRef.
- Q. Wang, S. R. Kanel, H. Park, A. Ryu and H. Choi, J. Nanopart. Res., 2009, 11, 749–755 Search PubMed.
- Q. Wang, S. Lee and H. Choi, J. Phys. Chem. C, 2010, 114, 2027–2033 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe and N. S. McIntyre, Surf. Sci., 2004, 572, 217–227 CrossRef CAS.
- S. R. Kanel, J. M. Greneche and H. Choi, Environ. Sci. Technol., 2006, 40, 2045–2050 CrossRef CAS.
- X. Q. Li and W. X. Zhang, J. Phys. Chem. C, 2007, 111, 6939–6946 CrossRef CAS.
- D. R. Baer, D. J. Gaspar, P. Nachimuthu, S. D. Techane and D. G. Castner, Anal. Bioanal. Chem., 2010, 396, 983–1002 CrossRef CAS.
- M. M. Scherer, B. A. Balko and P. G. Tratnyek, Mineral–Water Interfacial Reactions: Kinetics and Mechanisms. ACS Symposium Series no. 715, ACS, 1998, pp. 301–322 Search PubMed.
- E. J. Reardon, R. Fagan, J. L. Vogan and A. Przepiora, Environ. Sci. Technol., 2008, 42, 2420–2425 Search PubMed.
- V. Sarathy, P. G. Tratnyek, J. T. Nurmi, D. R. Baer, J. E. Amonette, C. L. Chun, R. L. Penn and E. J. Reardon, J. Phys. Chem. C, 2008, 112, 2286–2293 CrossRef CAS.
- K. Sohn, S. W. Kang, S. Ahn, M. Woo and S. K. Yang, Environ. Sci. Technol., 2006, 40, 5514–5519 CrossRef CAS.
- B. C. Reinsch, B. Forsberg, R. L. Penn, C. S. Kim and G. V. Lowry, Environ. Sci. Technol., 2010, 44, 3455–3461 Search PubMed.
- H. S. Kim, J. Y. Ahn, K. Y. Hwang, I. K. Kim and I. Hwang, Environ. Sci. Technol., 2010, 44, 1760–1766 Search PubMed.
- W. Yan, M. A. V. Ramos, B. E. Koel and W.-x. Zhang, J. Phys. Chem. C, 2012, 116, 5303–5311 Search PubMed.
- H. H. Uhlig, Corrosion and Corrosion Control, Wiley, 1971 Search PubMed.
- D. E. Meyer and D. Bhattacharyya, J. Phys. Chem. B, 2007, 111, 7142–7154 CrossRef CAS.
- H. Choi, S. R. Al-Abed and S. Agarwal, Environ. Sci. Technol., 2009, 43, 4137–4142 Search PubMed.
- Y. P. Sun, X. Q. Li, W. X. Zhang and H. P. Wang, Colloids Surf., A, 2007, 308, 60–66.
- T. Phenrat, N. Saleh, K. Sirk, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2007, 41, 284–290 CrossRef CAS.
- T. Phenrat, H. J. Kim, F. Fagerlund, T. Illangasekare, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2009, 43, 5079–5085 CrossRef CAS.
- B. Schrick, B. W. Hydutsky, J. L. Blough and T. E. Mallouk, Chem. Mater., 2004, 16, 2187–2193 CrossRef.
- E. J. Bishop, D. E. Fowler, J. M. Skluzacek, E. Seibel and T. E. Mallouk, Environ. Sci. Technol., 2010, 44, 9069–9074 Search PubMed.
- P. Jiemvarangkul, W.-x. Zhang and H.-L. Lien, Chem. Eng. J., 2011, 170, 482–491 CrossRef CAS.
- F. He and D. Y. Zhao, Environ. Sci. Technol., 2005, 39, 3314–3320 CrossRef CAS.
- A. R. Petosa, D. P. Jaisi, I. R. Quevedo, M. Elimelech and N. Tufenkji, Environ. Sci. Technol., 2010, 44, 6532–6549 CrossRef CAS.
- J. Quinn, C. Geiger, C. Clausen, K. Brooks, C. Coon, S. O'Hara, T. Krug, D. Major, W. S. Yoon, A. Gavaskar and T. Holdsworth, Environ. Sci. Technol., 2005, 39, 1309–1318 CrossRef CAS.
- N. D. Berge and C. A. Ramsburg, Environ. Sci. Technol., 2009, 43, 5060–5066 Search PubMed.
- N. D. Berge and C. A. Ramsburg, J. Contam. Hydrol., 2010, 118, 105–116 Search PubMed.
- J. J. Zhan, I. Kolesnichenko, B. Sunkara, J. B. He, G. L. McPherson, G. Piringer and V. T. John, Environ. Sci. Technol., 2011, 45, 1949–1954 Search PubMed.
- J. J. Zhan, T. H. Zheng, G. Piringer, C. Day, G. L. McPherson, Y. F. Lu, K. Papadopoulos and V. T. John, Environ. Sci. Technol., 2008, 42, 8871–8876 CrossRef CAS.
- W. Wang, M. Zhou, Q. Mao, J. Yue and X. Wang, Catal. Commun., 2010, 11, 937–941 CrossRef CAS.
- Z.-x. Chen, X.-y. Jin, Z. Chen, M. Megharaj and R. Naidu, J. Colloid Interface Sci., 2011, 363, 601–607 Search PubMed.
- L.-n. Shi, X. Zhang and Z.-l. Chen, Water Res., 2011, 45, 886–892 Search PubMed.
- J. Chen, Z. Xiu, G. V. Lowry and P. J. J. Alvarez, Water Res., 2011, 45, 1995–2001 Search PubMed.
- T. Phenrat, Y. Q. Liu, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2009, 43, 1507–1514 Search PubMed.
- F. J. Urbano and J. M. Marinas, J. Mol. Catal. A: Chem., 2001, 173, 329–345 CrossRef CAS.
- L. J. Matheson and P. G. Tratnyek, Environ. Sci. Technol., 1994, 28, 2045–2053 CAS.
- E. J. Weber, Environ. Sci. Technol., 1996, 30, 716–719 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4091 CrossRef CAS.
- M. S. Wong, P. J. J. Alvarez, Y. I. Fang, N. Akcin, M. O. Nutt, J. T. Miller and K. N. Heck, J. Chem. Technol. Biotechnol., 2009, 84, 158–166 CrossRef CAS.
- R. Muftikian, Q. Fernando and N. Korte, Water Res., 1995, 29, 2434–2439 CrossRef CAS.
- G. V. Lowry and M. Reinhard, Environ. Sci. Technol., 1999, 33, 1905–1910 CrossRef CAS.
- I. F. Cheng, Q. Fernando and N. Korte, Environ. Sci. Technol., 1997, 31, 1074–1078 CrossRef CAS.
- H. Song and E. R. Carraway, Appl. Catal., B, 2008, 78, 53–60 Search PubMed.
- H. L. Lien and W. X. Zhang, Appl. Catal., B, 2007, 77, 110–116 CrossRef CAS.
- W. L. Yan, A. A. Herzing, X. Q. Li, C. J. Kiely and W. X. Zhang, Environ. Sci. Technol., 2010, 44, 4288–4294 Search PubMed.
- H. S. Cao and W. X. Zhang, J. Hazard. Mater., 2006, 132, 213–219 CrossRef.
- S. R. Kanel, B. Manning, L. Charlet and H. Choi, Environ. Sci. Technol., 2005, 39, 1291–1298 CrossRef CAS.
- M. A. V. Ramos, W. Yan, X. Q. Li, B. E. Koel and W. X. Zhang, J. Phys. Chem. C, 2009, 113, 14591–14594 CrossRef CAS.
- T. E. Shokes and G. Moller, Environ. Sci. Technol., 1999, 33, 282–287 CrossRef CAS.
- X. Q. Li and W. X. Zhang, Langmuir, 2006, 22, 4638–4642 CrossRef CAS.
- S. Yan, B. Hua, Z. Y. Bao, J. Yang, C. X. Liu and B. L. Deng, Environ. Sci. Technol., 2010, 44, 7783–7789 Search PubMed.
- R. A. Crane, M. Dickinson, I. C. Popescu and T. B. Scott, Water Res., 2011, 45, 2931–2942 CrossRef CAS.
- E. J. O'Loughlin, S. D. Kelly, R. E. Cook, R. Csencsits and K. M. Kemner, Environ. Sci. Technol., 2003, 37, 721–727 CrossRef CAS.
- A. Abdelouas, Elements, 2006, 2, 335–341 CrossRef CAS.
- J. N. Fiedor, W. D. Bostick, R. J. Jarabek and J. Farrell, Environ. Sci. Technol., 1998, 32, 1466–1473 CrossRef CAS.
- D. K. Nordstrom, Science, 2002, 296, 2143–2145 CrossRef CAS.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- J. A. Lackovic, N. P. Nikolaidis and G. M. Dobbs, Environ. Eng. Sci., 2000, 17, 29–39 CrossRef CAS.
- J. Farrell, J. P. Wang, P. O'Day and M. Conklin, Environ. Sci. Technol., 2001, 35, 2026–2032 CrossRef.
- B. A. Manning, S. E. Fendorf and S. Goldberg, Environ. Sci. Technol., 1998, 32, 2383–2388 CrossRef CAS.
- S. Goldberg and C. T. Johnston, J. Colloid Interface Sci., 2001, 234, 204–216 CrossRef CAS.
- A. Manceau, Geochim. Cosmochim. Acta, 1995, 59, 3647–3653 CrossRef CAS.
- W. L. Yan, M. A. V. Ramos, B. E. Koel and W. X. Zhang, Chem. Commun., 2010, 46, 6995–6997 RSC.
- S. H. Joo, A. J. Feitz and T. D. Waite, Environ. Sci. Technol., 2004, 38, 2242–2247 CrossRef.
- S. H. Joo, A. J. Feitz, D. L. Sedlak and T. D. Waite, Environ. Sci. Technol., 2005, 39, 1263–1268 CrossRef CAS.
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 1262–1267 CrossRef CAS.
- A. J. Feitz, S. H. Joo, J. Guan, Q. Sun, D. L. Sedlak and T. D. Waite, Colloids Surf., A, 2005, 265, 88–94 Search PubMed.
- S. E. Mylon, Q. Sun and T. D. Waite, Chemosphere, 2010, 81, 127–131 CrossRef CAS.
- L. Xu and J. Wang, J. Hazard. Mater., 2011, 186, 256–264 Search PubMed.
- I. A. Katsoyiannis, T. Ruettimann and S. J. Hug, Environ. Sci. Technol., 2008, 42, 7424–7430 CrossRef CAS.
- C. Lee, C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 4927–4933 CrossRef CAS.
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 6936–6941 CrossRef CAS.
- S. J. Hug and O. Leupin, Environ. Sci. Technol., 2003, 37, 2734–2742 CrossRef CAS.
- S. Y. Pang, J. Jiang and J. Ma, Environ. Sci. Technol., 2011, 45, 307–312 CrossRef CAS.
- S.-H. Kang and W. Choi, Environ. Sci. Technol., 2009, 43, 878–883 Search PubMed.
- J. Lee, J. Kim and W. Choi, Environ. Sci. Technol., 2007, 41, 3335–3340 CrossRef CAS.
- B.-H. Moon, Y.-B. Park and K.-H. Park, Desalination, 2011, 268, 249–252 Search PubMed.
- C. Lee, J. Y. Kim, W. I. Lee, K. L. Nelson, J. Yoon and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 4927–4933 CrossRef CAS.
- J. Y. Kim, H.-J. Park, C. Lee, K. L. Nelson, D. L. Sedlak and J. Yoon, Appl. Environ. Microbiol., 2010, 76, 7668–7670 CrossRef CAS.
- J. Y. Kim, C. Lee, D. C. Love, D. L. Sedlak, J. Yoon and K. L. Nelson, Environ. Sci. Technol., 2011, 45, 6978–6984 CAS.
- M. Auffan, W. Achouak, J. Rose, M. A. Roncato, C. Chaneac, D. T. Waite, A. Masion, J. C. Woicik, M. R. Wiesner and J. Y. Bottero, Environ. Sci. Technol., 2008, 42, 6730–6735 CrossRef CAS.
- Z. Li, K. Greden, P. J. J. Alvarez, K. B. Gregory and G. V. Lowry, Environ. Sci. Technol., 2010, 44, 3462–3467 CrossRef CAS.
- Z.-m. Xiu, Z.-h. Jin, T.-l. Li, S. Mahendra, G. V. Lowry and P. J. J. Alvarez, Bioresour. Technol., 2010, 101, 1141–1146 CrossRef CAS.
- A. Sevcu, Y. S. El-Temsah, E. J. Joner and M. Cernik, Microb. Environ., 2011, 26, 271–281 Search PubMed.
- T. L. Kirschling, K. B. Gregory, E. G. Minkley, G. V. Lowry and R. D. Tilton, Environ. Sci. Technol., 2010, 44, 3474–3480 Search PubMed.
- Z.-m. Xiu, K. B. Gregory, G. V. Lowry and P. J. J. Alvarez, Environ. Sci. Technol., 2010, 44, 7647–7651 Search PubMed.
- S. Comba, A. Di Molfetta and R. Sethi, Water, Air, Soil Pollut., 2011, 215, 595–607 Search PubMed.
- P. Bardos, B. Bone, D. Elliott, N. Hartog, H. John and N. Paul, Defra Research Project Final Report, 2011 Search PubMed.
- U. S. EPA, Selected Sites Using or Testing Nanoparticles for Remediation, 2008, http://www.clu-in.org/download/remed/nano-site-list.pdf.
- D. W. Elliott and W. X. Zhang, Environ. Sci. Technol., 2001, 35, 4922–4926 CrossRef CAS.
- F. He, D. Y. Zhao and C. Paul, Water Res., 2010, 44, 2360–2370 CrossRef CAS.
- M. Elsner, G. L. Couloume, S. Mancini, L. Burns and B. S. Lollar, Ground Water Monit. Rem., 2010, 30, 79–95 CrossRef CAS.
- P. Bennett, F. He, D. Y. Zhao, B. Aiken and L. Feldman, J. Contam. Hydrol., 2010, 116, 35–46 Search PubMed.
- Y.-T. Wei and S.-C. Wu, Environ. Sci. Technol., 2010, 44, 8996–9002 Search PubMed.
- Z. Shi, J. T. Nurmi and P. G. Tratnyek, Environ. Sci. Technol., 2011, 45, 1586–1592 CrossRef CAS.
- K. Siskova, J. Tucek, L. Machala, E. Otyepkova, J. Filip, K. Safarova, J. Pechousek and R. Zboril, J. Nanopart. Res., 2012, 14, 805 Search PubMed.
- S. R. Kanel, R. R. Goswami, T. P. Clement, M. O. Barnett and D. Zhao, Environ. Sci. Technol., 2008, 42, 896–900 CrossRef CAS.
- N. Saleh, K. Sirk, Y. Q. Liu, T. Phenrat, B. Dufour, K. Matyjaszewski, R. D. Tilton and G. V. Lowry, Environ. Eng. Sci., 2007, 24, 45–57 CrossRef CAS.
- T. Phenrat, F. Fagerlund, T. Illangasekare, G. V. Lowry and R. D. Tilton, Environ. Sci. Technol., 2011, 45, 6102–6109 Search PubMed.
- A. N. Bezbaruah, S. Krajangpan, B. J. Chisholm, E. Khan and J. J. Elorza Bermudez, J. Hazard. Mater., 2009, 166, 1339–1343 CrossRef.
- S. Comba and R. Sethi, Water Res., 2009, 43, 3717–3726 Search PubMed.
- E. Dalla Vecchia, M. Luna and R. Sethi, Environ. Sci. Technol., 2009, 43, 8942–8947 Search PubMed.
- D. E. Meyer, K. Wood, L. G. Bachas and D. Bhattacharyya, Environ. Prog., 2004, 23, 232–242 CrossRef CAS.
- J. Xu, A. Dozier and D. Bhattacharyya, J. Nanopart. Res., 2005, 7, 449–467 CrossRef CAS.
- B.-W. Zhu, T.-T. Lim and J. Feng, Chemosphere, 2006, 65, 1137–1145 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |