Silver nanoparticles in the environment
Su-juan
Yu
,
Yong-guang
Yin
and
Jing-fu
Liu
*
State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, P.O. Box 2871, Beijing 100085, China. E-mail: jfliu@rcees.ac.cn; Fax: +86-10-62849192; Tel: +86-10-62849192
First published on 6th December 2012
Abstract
Silver nanoparticles (AgNPs) are well known for their excellent antibacterial ability and superior physical properties, and are widely used in a growing number of applications ranging from home disinfectants and medical devices to water purificants. However, with the accelerating production and introduction of AgNPs into commercial products, there is likelihood of release into the environment, which raises health and environmental concerns. This article provides a critical review of the state-of-knowledge about AgNPs, involving the history, analysis, source, fate and transport, and potential risks of AgNPs. Although great efforts have been made in each of these aspects, there are still many questions to be answered to reach a comprehensive understanding of the positive and negative effects of AgNPs. In order to fully investigate the fate and transport of AgNPs in the environment, appropriate methods for the preconcentration, separation and speciation of AgNPs should be developed, and analytical tools for the characterization and detection of AgNPs in complicated environmental samples are also urgently needed. To elucidate the environmental transformation of AgNPs, the behavior of AgNPs should be thoroughly monitored in complex environmental relevant conditions. Furthermore, additional in vivo toxicity studies should be carried out to understand the exact toxicity mechanism of AgNPs, and to predict the health effects to humans.
 Su-juan Yu | Su-juan Yu is a PhD student in Research Center for Eco-Environmental Sciences (RCEES), Chinese Academy of Sciences. Her research focuses on the analysis, and environmental behavior and effects of silver nanoparticles. |
Environmental impactThere is a growing production and application of silver nanoparticles (AgNPs) in various areas including catalysis, consumer products, food technology, textiles/fabrics, as well as medical products and devices. It was reported that about 25% of the >1300 nanomaterial-containing consumer products contain AgNPs. The rapid growth in the commercial use of AgNPs will inevitably increase silver exposure in the environment and the general population. To correctly forecast their environmental and human health risks, a comprehensive understanding of the source, distribution, transformation and toxicity of AgNPs is needed. This article reviews the available information on the environmental and toxicological chemistry of AgNPs. There are still many gaps our knowledge that have to be filled to fully understand the benefits and risks of AgNPs. |
1. Introduction
1.1 History
Metallic silver (Ag) is a durable transition element and because of is rarity (67th in abundance among the elements) and its attractive white metallic luster, silver has long been used as jewellery, currency coins and silverware. Among its wide applications its antimicrobial activity is of great interest. The use of silver vessels to keep water and wine clean probably dates back to ancient times. Silver's medicinal use is also of great antiquity. Silver nitrite was applied for the treatment of ulcers in the 17th and 18th centuries,1 and around 1884, 1% silver nitrite was introduced by German obstetrician C. S. F. Crede as an eye solution to prevent gonococcal conjunctivitis for new born babies.2 In 1967, Fox introduced silver sulfadiazine in the treatment of burn patients, and even today silver sulfadiazine cream remains the most widely used medicine for serous burn wounds.3However, prolonged exposure to silver may cause silver deposition in the body, resulting in irreversible discoloration of skin or eyes, i.e. argyria or argyrosis.4 Because of this and with the advent of more available antibiotics such as penicillin and cephalosporin, medicinal interest in silver faded around the Second World War. But it did not take many years for interest in silver to revive, under the large increase in the number of multiple-resistant bacterial strains due to the abuse of antibiotics and the discovery that silver nanoparticles (AgNPs) showed excellent performance in antibacterial application. It was reported that AgNPs show biocidal action by the slow release of Ag+, and by multiple mechanisms (such as interaction with thiol groups in proteins and enzymes, inhibition of DNA replication, induction of oxidative stress) making it more difficult for bacteria to produce resistant strains.5 Also, the large surface area, which promotes the reactivity and sorption with pathogens, makes AgNPs an ideal candidate for antibacterial application.
Actually, nanosilver is not new. As early as 1889, Lea had reported the first synthesis of a silver colloid stabled by citrate.6 Though not formally registered or under the name of “nano”, literature shows that silver colloids have been used in the medical area for more than 100 years since 1897 by the name of “Collargol”. The first biocidal silver product “Algaedyn” was registered in 1954 in the U.S., which is still used in disinfectants today.7 During the past two decades, the advancement of nanotechnology has opened new avenues for AgNPs. Being in the nano-scale dimension, AgNPs exhibit many novel properties relative to the bulk metal, which has aroused intense interest in the development of new applications.
1.2 Properties and applications
Pure silver has high thermal and electrical conductivity and relatively low contact resistance, which makes it a popular option in electronics. Silver nanoparticles or nanowires have been used to fabricate thin-film transistor electrodes,8 as pastes and inks for printed circuit boards,9 optoelectronics, data storage devices and battery-based intercalation materials.10By virtue of their extremely small size, AgNPs possess large surface area, which offers them high surface energy and more possible reactive sites. These characteristics qualify AgNPs as one of the most promising materials in catalysis. AgNPs and nanocomposites are capable of catalyzing a number of reactions, such as CO and benzene oxidation,11 reduction of 4-nitrophenol in the presence of NaBH4,12 reduction of Rhodamine B (RhB),13 and reduction of 4-nitrophenol to 4-aminophenol.14
Different from the bulk metal, AgNPs also show surface plasmon resonance (SPR) under irradiation of light, which induces SPR peaks in the UV-vis wavelength range. Typically, the width and position of the SPR peaks are influenced by the size, shape and dispersion of the nanoparticles.10 AgNPs are also used for surface-enhanced Raman scattering (SERS). It is reported that they can enhance the efficiencies of SERS by as much as 1014 to 1015 fold, which allows detection and identification of single molecules.15 As a result of these unique properties, AgNPs are used in sensing and imaging applications, including the detection of DNA,16 selective colorimetric sensing of cysteine,17 sensing purine nucleoside phosphorylase activity,18 and selective colorimetric sensing of mercury(II).19
For years, knowledge about nanosilver's ability to kill harmful bacteria has drawn extensive attention, making it popular for incorporation into various products. Nanosilver exhibits a broad spectrum of antimicrobial activity, and can inhibit the growth of both Gram-positive and Gram-negative bacteria (including Escherichia coli, Pseudomonas aeruginosa and Staphylococcus aureus).20,21 The antibacterial activity on different drug-resistant pathogens of clinical importance, such as multidrug-resistant Pseudomonas aeruginosa, ampicillin-resistant E. coli O157:H7 and erythromycin-resistant Streptococcus pyogenes, was also reported.22 A study also revealed that the antimicrobial activity of several antibiotics was increased in the presence of AgNPs.23
Nanosilver is also an effective fungicide. AgNPs can kill a number of ordinary fungal strains, including Aspergillus fumigatus, Aspergillus fumigatus, Mucor, Saccharomyces cerevisiae, and Candida tropicalis.24
Nanosilver also has antiviral properties; it was reported that AgNPs synthesized in Hepes buffer could inhabit HIV-1 replication, and the anti-HIV activity (98%) was much higher than that of gold nanoparticles (6–20%).25 The inhibition of hepatitis B virus26 and herpes simplex virus27 was also assessed.
Due to the excellent antimicrobial activity, nanosilver is becoming a blossoming field of research and has been highly commercialized. It is found in a wide category of products available in the consumer market. It is reported that of the 1317 products containing nanomaterials in the market (March 10, 2011), 313 were claimed to contain AgNPs.28 The products include food packaging materials, food storage containers, water purificants, odor-resistant socks and underwear, room sprays, laundry detergents, washing machines, lotions and soaps. Also, AgNPs are widely used in medical applications including wound dressings, female-hygiene products, surgical instruments, bone cements and implantable devices.
1.3 Environmental concerns
The widespread application of AgNPs in our daily life will inevitably increase human and ecosystem exposure. Also, during the production, transport, erosion, washing or disposal of AgNP containing products, AgNPs may be released to the environment. Though the long historical use of silver has not shown obvious adverse effects, there is concern about the potential risks of AgNPs in the environment.A number of literature reports have appeared on the leaching and fate of AgNPs. Benn et al.29 revealed that AgNPs and Ag+ could be easily leached into water by simply immersing commercial AgNP containing socks into water with shaking. Some brands of socks could even lose nearly 100% of the total silver contents after four consecutive washings. Consumer silver nanotextiles were subjected to surfactants, oxidizing agents, different pH, washing machines and simulated perspiration fluids to test the normal laundering process and human skin sweat on the release of AgNPs.30–33 It was shown that low pH, mechanical stress and the presence of bleaching largely enhanced the dissolution of Ag, and the nature of incorporation determined the amount and form of Ag release. The considerable release of Ag in simulated perspiration fluids suggested the potential human risk in the use of textiles containing AgNPs. Release of AgNPs from outdoor facades under ambient weather conditions and washing machines was also reported.34 Commercial laundry silver nanowashing machines, which claimed to produce AgNPs to kill bacteria were found to release silver in their effluent at an average concentration of 11 μg L−1.35 Recently, Cleveland et al.36 studied the long-term release and fate of AgNPs from consumer products in a modular estuarine mesocosm system. They found that nanosilver consumer products showed an extended leaching of a large amount of Ag during the experimental period of 60 days and the silver was either taken up by the estuarine biota or adsorption by sediment, sand and biofilm. The increased exposure and the accumulation of Ag in organisms such as hard clams, grass shrimps, mud snails and cordgrass stalks and leaves has alerted the community to pay more attention to the potential hazards of AgNPs. Given the vast use in the market and the potential risk of nanosilver, a systematic study of the environmental and toxicological chemistry of AgNPs is needed before a headlong rush to use nanosilver products. The purpose of this article is to provide a critical review of the state-of-knowledge about AgNPs, including the analysis, source, fate and transport, and potential risks of AgNPs, and some open questions are also discussed.
2. Analysis of AgNPs
In sharp contrast to the increasing attention to the application of AgNPs, information on their occurrence, fate and transport is limited. To achieve insights into their behavior in complex environmental media, appropriate methods for separation and determination of AgNPs are highly demanded. As techniques to detect and characterize engineered nanoparticles in the environment have been reviewed recently,37–39 we emphasize results on the analysis of AgNPs in real samples in this section.As detecting real environmental samples is always challenged by complex matrices and low concentrations, a preconcentration procedure is always needed before analysis. Liu's group40 reported for the first time the extraction of trace AgNPs in environmental waters by cloud point extraction (CPE). Based on the interaction of AgNPs and a non-ionic surfactant Triton X-114 (TX-114), AgNPs could be trapped in the micelles of the surfactant. Then by changing the temperature to help attain the cloud point of TX-114, the solution separates into two phases. AgNPs, which are retained in the surfactant-rich phase, can be concentrated and separated after centrifugation. Results suggested that AgNPs could be enriched by 100 times by adding 0.2% (w/v) TX-114, and that the presence of humic acids (as high as 30 mg L−1) and Ag+ did not disturb the extraction. For environmental waters spiked with 0.1–146 μg L−1 of AgNPs, 57–116% of the total AgNPs could be recovered after the extraction. Additionally, transmission electron microscopy (TEM)/scanning electron microscope coupled with energy-dispersive X-ray spectroscopy (SEM-EDS)/ultraviolet-visible spectroscopy (UV-vis) results all showed the presence of AgNPs in the surfactant-rich phase, and their size and shape did not change during the extraction, which offers a promising method to trace AgNPs in the environment.
Hyphenated techniques, which are capable of providing multidimensional information of test samples, emerge as one of the most promising tools for the characterization of nanomaterials. Hydrodynamic chromatography (HDC) coupled with ICP-MS was successfully applied to investigate AgNPs in sewage sludge.41,42 By spiking AgNPs into sewage sludge and shaking for a few hours, AgNPs could be concentrated in the supernatant. HDC-ICP-MS chromatography revealed that AgNPs could then be directly separated from the supernatant even without a preparative step, and the analysis process was complete within 10 min per sample.
Field-flow fractionation (FFF) has proved to be another popular tool to isolate NPs due to its excellent separation efficiency and the capability to couple with various detectors. The FFF-ICP-MS technique was successfully applied to separate and characterize AgNPs from biological tissues.43 After AgNPs exposure for 28 days, the tissues of freshwater oligochaete Lumbriculus variegates were extracted by sonication and analyzed by FFF-ICP-MS. Results revealed that the average size of AgNPs increased from 31 to 46 nm, suggesting AgNPs may change notably during biological exposure. Another study also used FFF-ICP-MS to separate AgNPs from surface waters and untreated wastewater,44 showing its prospective use in analyzing environmentally relevant samples.
As AgNPs are widely used in consumer spray products for their antibacterial ability, there is a risk that nanoparticles may be directly inhaled and deposited in the respiratory tract during product use. Marr et al.45 explored the emission behavior of three consumer spray products containing AgNPs. In the study, a polyethylene chamber was used to simulate an airtight room and after a constant spraying scheme, several techniques (e.g., ultrafine condensation particle counter, dynamic light scattering (DLS), TEM, SEM-EDS and ICP-MS) were conducted to measure the concentration and size distributions of the aerosols. It was shown that emitted aerosols ranged from nanoscale up to 10 μm, and 0.24–56 ng of silver could be released per spray action. It is also estimated that up to 70 ng of silver may deposit in the respiratory tract according to the usual use of the spray products.
Speciation analysis of AgNPs and Ag+ in commercially available products was also reported recently by cloud point extraction based on TX-114 (Fig. 1).46 By adding Na2S2O3 as a complexing agent with Ag+, AgNPs and Ag+ could be separated from each other, with AgNPs extracted into the surfactant-rich phase, and Ag+ preserved in the aqueous phase. The spiked recoveries in different consumer products were in the range of 71.7–103% for AgNPs and 1.2–10% for Ag+, showing AgNPs and Ag+ were efficiently separated. TEM/SEM-EDS/UV-vis techniques were applied to characterize the presence of AgNPs, and the concentration of AgNPs and Ag+ was determined by ICP-MS after microwave digestion.
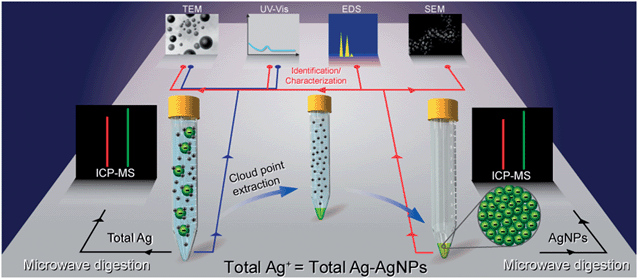 | ||
| Fig. 1 Cloud-point extraction (CPE) protocol and the different techniques for characterization (reproduced from ref. 46 with permission, © 2007 American Chemical Society). | ||
Although a great number of techniques have been developed to characterize and quantify AgNPs, distinguishing Ag+ and AgNPs is still one of the greatest challenges due to their common co-occurrence. To achieve the speciation analysis of Ag+ and AgNPs, multiple steps of pretreatment such as filtration, centrifugation, extraction, is needed, which is not only tedious and energy intensive, but can also lead to undesirable artifacts. Single particle inductively coupled plasma-mass spectrometry (spICPMS) is an emerging method that has received considerable attention. As each AgNP was detected as a single pulse and dissolved Ag+ produced pulses of average stable intensity under spICPMS detection mode, the forms of silver could be identified based on the pulse type.47 The size of AgNPs can also be assumed by calculating the ion count relative to the mass of standards of Ag+ introduced into the plasma.48 Furthermore, the high sensitivity of spICPMS offers the opportunity to detect environmentally relevant samples, such as wastewater effluent samples. An example is 9568 Ag particles per mL found in wastewater samples collected from a wastewater treatment plant (WWTP) in Sweden, showing the capability of this technique in monitoring AgNPs in the environment.
3. Source of AgNPs
3.1 Natural sources
The increasing use of nanosilver has generated substantial enthusiasm in developing methods to fabricate different AgNPs, which may eventually elevate the amount of nanosilver in the environment. However, it seems that not all AgNPs are produced by humans. It is reported that silver as nanoparticles was found in an old silver mining area of Mexico,49 and before the manufacture of engineered AgNPs, colloidal and particulate silver was also discovered in river and estuarine waters of Texas.50In fact, there are many natural reducing agents in the environment, such as humic acids (HAs). It is well known that HAs occur ubiquitously and contain many functional groups, including quinines, ketones, aldehydes, phenolic and hydroxyls, which facilitate them to reduce metal ions. Earlier literature has reported the preparation of AgNPs using peat fulvic acids, and SPR peaks in the UV-vis spectroscopy confirmed the presence of AgNPs.51 Sharma et al.52 further investigated the reduction of Ag+ in the presence of different sources of HAs under environmentally relevant conditions. They found that Ag+ (concentration as low as 1 mg L−1) incubated with sedimentary HAs and river HAs readily formed AgNPs at room temperature (22 °C). When raising the temperature to 90 °C, SPR peaks appeared within 90 min, indicating the quick production of these nanoparticles. DLS, TEM and atomic force microscopy (AFM) images clearly showed the formation and morphology of AgNPs (Fig. 2).
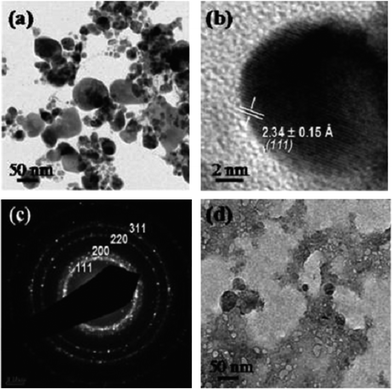 | ||
| Fig. 2 Low-resolution (a) and high-resolution (b) TEM images of AgNPs produced in marine sediment humic acid solution with the corresponding SAED pattern (c) of the AgNPs. A low-resolution TEM image of the as-prepared marine sediment humic acid solution is shown for comparison (d) (reproduced from ref. 52 with permission, © 2007 American Chemical Society). | ||
Our study53 also showed that ionic Ag could be photochemically reduced to AgNPs by dissolved organic matter (DOM) in natural water under sunlight within several hours. The formation of AgNPs could be identified by TEM, selected-area electron diffraction (SAED), EDS and X-ray diffraction (XRD) analysis. However, the newly formed AgNPs were not stable and easily coalesced due to the presence of inorganic cations such as Ca2+ and Mg2+ in environmental waters. Further experiments demonstrated that the photo-reduction was pH-dependent and was mediated by superoxide generated from photoirradiation of the phenolic group of HAs, and the dissolved oxygen dramatically enhanced the reduction of Ag+. As this process occurred under environmentally relevant conditions, it once again demonstrates that not all AgNPs are of anthropogenic origin, and they can form spontaneously in nature.
Another study revealed that AgNPs could be generated from silver objects through oxidative dissolution and subsequent reduction.54 Researchers discovered that when different capping-agent stabilized AgNPs immobilized on positively charged SiO2 grids were exposed to ambient laboratory conditions, many new smaller particles appeared around the original nanoparticles. TEM, EDS, X-ray photoelectron spectroscopy (XPS) and SAED results all confirmed that the newly formed particles were AgNPs. Further investigation showed that new nanoparticles could also be generated from bulk objects such as silver wire, jewellery and eating utensils, proving this phenomenon was general, and also implying that macroscale elemental silver objects are a potential source of AgNPs in the environment.
It is also reported that plants have the capability to take up metal ions and form nanoparticles.55 Jose-Yacaman and co-workers found that alfalfa roots could absorb silver atoms and transfer them through specific channels to different areas. STEM/TEM-EDS and extended X-ray absorption near-edge structure (XANES) analysis proved that the Ag atoms arranged to coalesce and nucleate to form AgNPs inside plants. Recently, the green synthesis of AgNPs involving environmentally benign reducing agents and nontoxic stabilizing agents has attracted much attention and have been thoroughly reviewed by Sharma et al.56 Glucose,57 coffee and tea extract,58 unicellular green alga extract,59 and bacteria60 were successfully explored for the synthesis of AgNPs, indicating that many natural substances or organisms could produce AgNPs.
3.2 Anthropogenic sources
Though it has been proved that AgNPs could be formed naturally, there is no doubt that anthropogenic activities play a key role in potential silver pollution. The widespread use of AgNPs has stimulated a flourishing development of the silver industry. It is estimated that about 500 t/a nanosilver is produced worldwide, and this amount is still steadily increasing.61 AgNPs are used as electronic devices, incorporated into textiles, dressing and medical devices, or directly added into disinfectants. However, during the production and manufacturing of nanosilver products, they could be directly released into the environment.62 The synthesis process often involves mixing, centrifugation and filtration steps to remove impurities and the wastewater may be directly discharged into the environment. Also, the powder nanoparticles occur as aerosols in workshops and escape through open windows to air. Additionally, other activities, such as sampling for quality control, leaking from broken packaging and other accidents could lead to unintentional release of AgNPs.The uncontrollable release of silver during the use, recycling and disposal process gives rise to public concerns. Ideally the released AgNPs and Ag+ would undergo WWTP processing. Though previous study29 has shown that WWTP biomass is able to partition high levels of heavy metals and with treatment largely reduce the silver concentration in the effluent stream, unfortunately, in some instances untreated sewage sludge is often used as an agricultural additive or fertilizer, which results in the recycling of AgNPs. This would be an important source of AgNPs to the environment.
4. Fate and transport of AgNPs
4.1 Influencing factors
Once released into the environment, AgNPs would undergo different pathways during transport. They may remain as individual particles in suspension and be delivered long distances, or tend to aggregate at high ionic strength. After contact with oxygen and other oxidants, partial oxidation and Ag+ dissolution is also expected. Most probably, AgNPs would react with sulfide, chloride or other natural substances, altering the original properties of the nanoparticles.5 The behaviors of the AgNPs largely depends on the surface properties of the nanoparticle themselves and the surrounding environment, involving capping agents, electrolyte composition, solution ionic strength, pH and natural organic matter (NOM).Due to the large surface area and high surface energy of nanoparticles, they are prone to coalesce to form larger clusters. As a consequence, capping agents are always added to control the final size of the product. Different capping agents exploit their advantages by steric repulsion, electrostatic repulsion or both, resulting in a number of AgNPs modified with various functional groups. The capping agents showed to significantly affect the stability of AgNPs. Charge-stabilized AgNPs (e.g. citrate) are more susceptible to external conditions than sterically stabilized AgNPs (e.g. PVP or PEG).63 Suspended in standard OECD media for 21 days, citrate coated NPs were almost completely reacted, while PEG and PVP coated AgNPs only suffered from little change in particle size.
Solution composition and ionic strength are also important factors in determining the stability of AgNPs. The presence of divalent cations such as Ca2+ and Mg2+ greatly enhance the coagulation of AgNPs. High ionic strength tends to weaken the electrostatic repulsion between particles and reduce the electric double layer on the surface of AgNPs, resulting in colloidal aggregation.64
The mobility of AgNPs is inseparable from the water chemistry such as the pH of the suspension. The pH influences the surface potential of particles and therefore dominates the coagulation size. Nanoparticles exhibit different aggregation states over a wide range of pH, and aggregate sizes increase when the pH comes near pHzpc (the pH of the point of zero charge).65 Also, AgNPs are able to adsorb charged species in the environment by electrostatic interaction, affecting their fate in the environment.
NOM, ubiquitously existing in aquatic systems, is also a key factor influencing the fate of AgNPs. As mentioned above, the presence of various functional groups of NOM facilitates surface binding of AgNPs, resulting in more stable AgNPs suspensions. AgNPs coated by NOM are able to stay dispersed for months52 and the existence of humic acid greatly affected the aggregation kinetics of citrate coated AgNPs in NaCl solution, increasing the critical coagulation concentration from 47.6 to 72.1 mM NaCl.66
4.2 Transport and distribution in the environment
As mentioned above, AgNPs can escape from the manufacturing factory during the production process, including drying the solution, mechanical grinding, mixing and packaging, resulting in the release of AgNPs into the atmosphere.67 Also, the widespread use of AgNPs in disinfection sprays promotes the emission of AgNPs to the air. Surface disinfectants, which can be used on walls, tables, chairs and floors, often cause AgNPs to deposit on these surfaces, and AgNPs would probably transfer to a duster cloth after cleaning and then go down the drain during laundering. Added in anti-odor sprays and used in rooms, AgNPs could also find their way in the open air by transport. According to Fick's first law that the diffusion coefficient is inversely related to the particle diameters, AgNPs would diffuse rapidly because of their small size. If they are stable enough, long distance mobility in the air would be expected. Additionally, their large surface area provides abundant reactive sites for dusts, microbes and pollution, making the AgNPs much more toxic than the original particles. The airborne particles may also coalesce to large agglomerates during transport, and deposit on surfaces by gravitation, or be washed down to terrestrial or aquatic systems by rain.Processes such as direct disposal of AgNP product, waste incineration or landfill, AgNPs suspended in air depositing on the land, and sewage sludge recycling as a fertilizer to agricultural soils could cause AgNPs to enter soils.68 The fate and transport of AgNPs in soils is governed by a number of variables, such as the particle size, surface charge and the soil environment. NPs may also adsorb organic contaminants and act as carriers for transport of contaminants.69 As AgNPs are always modified by the stabilizing agents and possess a surface charge, the electrostatic interaction with different soil types alters the mobility of AgNPs.10 For example, citrate capped AgNPs mainly bear negative charges in ambient conditions. When passing through a positive charged soil, the attraction forces may prevent the long distance mobility of AgNPs. On the contrary, a negative charged soil may cause AgNPs to be more mobile in such soils. It is also documented that AgNPs could strongly adsorb onto soils. The sorption experiment of three different sized AgNPs (10, 20, 50 nm) was conducted in Toccoa entisols (ionic strength = 0.05 mol L−1, soil pH = 5.2) from the southeastern United States. Results showed that all the three AgNPs have high affinity for the soil surface, with 97–100% AgNPs adsorbed on the soils in the concentration range of 10–500 mg L−1.68
AgNPs may enter the aquatic system in several ways: (1) silver leached out from nanosilver consumer products, and eventually end up in streams and rivers;29 (2) suspended AgNPs in air finally depositing on water; (3) runoff scouring AgNP polluted soils or landfill sewage sludge could result in AgNPs migrating to surface water. The water environment could largely affect the mobility of AgNPs. It has been mentioned that NOM could adsorb on nanoparticles, and act as stable agents to make AgNPs more mobile in aquatic systems. Besides, the NOM type and source, molecular weight, concentration and functional groups influence the stability of AgNPs.70 On the other hand, divalent cations (e.g. Ca2+ and Mg2+), commonly present in natural waters, could easily induce the aggregation of AgNPs. Colloidal clusters would probably deposit in the sediment, reducing the bioavailability for aquatic organisms and plants.
As AgNPs are not highly stable and can easily be oxidized, a slow dissolution of Ag+ would be expected in aquatic environments.71 Positively charged free Ag+ occurs only in extremely low concentrations in natural water. Ag+ binds with negatively charged ligands, such as S2−, SO42− and CO32−.5 In marine waters, as sodium chloride is the dominant salt, Ag+ associates with Cl− to a great extent, and the main species observed are AgCl2−, AgCl32− and AgCl43−, making silver more mobile in seawater.72 In a recent paper reviewing the transformation of AgNPs on the stability and toxicity,73 the Eh (oxidation-reduction potential)–pH diagrams for the system Ag–S–Cl–CO2–H2O and Ag–S–Cl–Cyst–H2O in freshwater and seawater were provided to describe the speciation of Ag in different conditions. The main forms of Ag+ varied significantly at varied pH, Eh, the type and concentration of ligands, and the strength of silver binding with these ligands. When dealing with real environment samples, the speciation would be much more complicated due to complex ion mixtures. As speciation greatly influences the fate and transport of AgNPs, they would behave quite differently in various water conditions.
4.3 Transformation in environment
Liu et al.71 investigated the ion release kinetics of AgNPs in solution, and examined the parameters that affect Ag+ dissolution including dissolved oxygen, pH, temperature and salinity. They found that by removing dissolved oxygen in AgNPs suspension, the release of Ag+ was completely inhibited, and the dissolution of Ag+ was greatly enhanced with decreasing pH in air-saturated water, showing that both protons and dissolved oxygen played key roles in controlling Ag+ release. Another study also showed that the Ag+ release rates were closely related to the primary particle size and concentration when other environmental factors were kept the same.75 Liu et al.76 also revealed that the release rate constant k (from −(dm/dt) = km) of two different sized AgNPs (4.8 and 60 nm) and a silver foil varied greatly: 4.1 per day for 4.8 nm AgNPs, 0.74 per day for 60 nm AgNPs, and 1.1 × 10−5 per day for silver foil.
Recently, sunlight induced nanoparticle aggregation was shown by Cheng and co-workers.80 PVP and gum arabic (GA) coated AgNPs were exposed to natural sunlight, and obvious aggregation was observed after several days compared with control particles in the lab. Evidence showed that UV in sunlight induced this destabilization, possibly due to the oscillating of electrons at resonant conditions. Furthermore, they also observed that the toxicity of AgNPs was reduced significantly after sunlight irradiation, indicating the aggregation state of AgNPs is an important parameter influencing their bioavailability and cytotoxicity.
In another study, time-resolved dynamic light scattering was applied to monitor the aggregation kinetics of AgNPs in Hoagland medium under anoxic and anaerobic conditions.81 It was shown that the aggregation rates were 3–8 times faster in the presence of dissolved oxygen (DO) than those in the absence of DO, revealing that DO also greatly influenced the stability of AgNPs in aqueous environments.
Levard et al.86 monitored the sulfidation process of PVP-coated AgNPs in Na2S solutions, and found significant changes of their surface properties and morphology. TEM images showed that the original dispersed particles turned into chain-like structures, implying AgNPs were partially oxidized, and then dissolved and forming Ag2S nanobridges between adjacent particles (Fig. 3). Another study also focused on the oxysulfidation of AgNPs, in which the mechanism of this process was systematically examined.87 Two different pathways were proposed: (1) at high sulfide concentration, AgNPs would directly convert into Ag2S nanoparticles by particle–fluid reaction; (2) whereas at low sulfide concentration, AgNPs were likely to be oxidized and release Ag+ first, and then react with sulfide to form Ag2S.
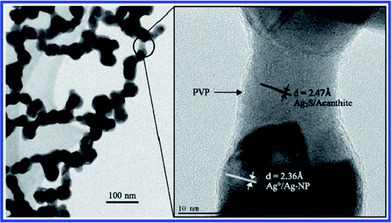 | ||
| Fig. 3 TEM images of partly sulfidized AgNPs. The right image is at higher magnification and is centered on one of the nanobridges observed at low magnification (left image) (reproduced from ref. 86 with permission, © 2007 American Chemical Society). | ||
Given the wide occurrence of sulfidation and the low solubility of Ag2S (Ksp = 5.92 × 10−51), the reaction with sulfur would greatly influence the fate of AgNPs in the environment, especially in their bioavailability and toxicity. According to Levard et al.,88 the sulfidation of AgNPs could largely reduce the release of Ag+, and decrease the growth inhibition of Escherichia coli. Moreover, the growth inhibition closely correlates with the degree of sulfidation (by changing the HS−/Ag ratio).
Impellitteri et al.91 assessed the chemical transformation of AgNPs by immersing an antimicrobial sock containing AgNPs into a hypochlorite/detergent solution, and the speciation of AgNPs was studied by X-ray absorption near edge spectroscopy (XANES) spectra. It was revealed that more than 50% AgNPs in socks were converted to AgCl in hypochlorite/detergent solution. However, when exposed to 1 mol L−1 NaCl solution, almost no AgCl was detected after a long time, suggesting the oxidation might be the limiting step in the formation of AgCl.
5. Effects of AgNPs
5.1 Mechanism of toxicity
Although a number of studies have tried to fully elucidate the mechanism behind the biocidal action of AgNPs, no universal conclusion has been drawn so far. There is no doubt that the antibacterial activity of AgNPs is a complex process, and several possible modes of action are proposed, involving (1) generation of reactive oxygen species (ROS),92–94 (2) direct attachment to cell membrane and disruption of membrane integrity,95 (3) changes in membrane permeability,96 (4) interaction with proteins and disruption of their regular function,97,98 and (5) interference with DNA replication and causing DNA damage.99Generally, ROS are natural byproducts of normal cellular metabolism of oxygen, and can be cleared by cell's radical-scavenging activities. However, increased production of ROS is beyond the ability of antioxidant defences, and may result in oxidative stress due to the accumulation of excess ROS. These free radicals may attack cell membranes, react with lipids, proteins and nucleic acids, and disrupt the normal cellular transport system.5,100
In an investigation of the AgNP toxicity to human HepG2 cells, Kim et al. found that the toxicity was directly related to the oxidative stress.94 In the study, dose-dependent production of cellular oxidants and DNA double-strand breaks were detected in HepG2 cells when exposed to AgNPs or Ag+. However, when cells were pretreated with an antioxidant N-acetylcysteine, both of the oxidative stress and AgNPs induced DNA damage were absent, indicating that the toxicity of AgNPs was dependent on the production of ROS. Similarly, Choi and Hu also observed that the inhibition extent of nitrifying bacteria was correlated well with the production of ROS in AgNP exposure, though no direct evidence was obtained.92
A panel of recombinant bioluminescent bacteria was also studied to analyze the toxic modes of AgNPs. As the bacterial strains could specifically respond to protein/membrane, oxidative stress, and DNA damage, promoter activities of the bacteria could directly indicate different pathways of toxicity. Results showed that AgNPs could cause the production of superoxide radicals and damage the membrane protein, but no DNA damage was observed.93
However, in another report that assessed the effect of AgNPs on rainbow trout hepatocytes, no excess ROS was detected after exposure. The cytotoxicity was mainly associated with the reduced mitochondrial activity and membrane integrity.97 Membrane disruption related toxicity of AgNPs was also reported in many other studies. The formation of pits and pores in the cell membrane of fungus C. albicans was observed, and the authors suggested that AgNPs may attack cell membrane lipid bilayers and destroy the membrane permeability barrier, resulting in the leakage of ions, formation of pores and cell death.96E. coli membrane damage caused by AgNPs was also confirmed by Sondi and co-workers.95 TEM/SEM/EDS results all showed that AgNPs accumulated on cell membranes and some were successfully incorporated into the lipid bilayer structure, forming irregular shaped pits in the outer membrane. It was speculated that AgNPs may lead to the progressive release of lipopolysaccharide molecules and proteins, causing changes in membrane integrity and permeability, and finally inducing cell malfunction and death.
In another proteomic analysis, several envelope protein precursors were observed to accumulate in E. coli after exposure to AgNPs, suggesting AgNPs may destabilize the bacterial membrane, induce collapse of proton motive force, and decrease the cellular ATP levels.98 AshaRani et al. also proposed that the AgNPs toxicity may possibly be associated with the disruption of the mitochondrial respiratory chain, which leads to the reduction of ATP content and in turn causing DNA damage.99
5.2 Toxicity to mammals
To date, very few data are available on the effects of AgNPs in mammals in vivo, but existing results have showed that AgNPs can cause potential toxicity to test animal models. A summary of adverse effects of AgNPs to mammals is given in Table 1.| Ag NP size/nm | Organism | Dose concentration | Exposure method | Effect measured | Ref. |
|---|---|---|---|---|---|
| 14.6 ± 1.0 | Female Fischer 344 rats | 3 × 106 particles cm−3 (133 μg m−3) | Inhalation exposure: 6 h per day, 0,1, 4, 7 days | AgNPs detected in lung, blood, liver, kidney, spleen, brain, and heart; rapid clearance of AgNPs from lung | 101 |
| 13–15 | Sprague–Dawley rats | Low-dose (1.73 × 104 particles cm−3, 0.5 μg m−3); medium-dose (1.27 × 105 particles cm−3, 3.5 μg m−3); high-dose (1.32 × 106 particles cm−3, 61 μg m−3) | Inhalation exposure: 6 h per day, 5 times per week, 4 weeks | No remarkable changes in nasal cavity and lungs; size and number of goblet cells containing neutral mucins increased | 103 |
| 22.18 ± 1.72 | C57BL/6 mice | 1.91 × 107 particles cm−3 | Inhalation exposure: 6 h per day, 5 times per week, 2 weeks | Expression of several genes associated with motor neuron disorders, neurodegenerative disease, and immune cell function | 107 |
| 18 | Sprague–Dawley rats | Low-dose (0.7 × 106 particles cm−3); medium-dose (1.4 × 106 particles cm−3); high-dose (2.9 × 106 particles cm−3) | Inhalation exposure: 6 h per day, 5 times per week, 13 weeks | Decreased tidal volume and minute volume, lung inflammation | 104 |
| 12–16 | Sprague–Dawley rats | Low-dose (1.73 × 104 particles cm−3); medium-dose (1.27 × 105 particles cm−3); high-dose (1.32 × 106 particles cm−3) | Inhalation exposure: 6 h per day, 5 times per week, 4 weeks | No significant changes in body weight, hematology and blood biochemical values for both male and female rats | 105 |
| 25 | Adult-male C57BL/6N mice | 100, 500, 1000 mg kg−1 | Intraperitoneal injection for 24 h | Free radical induced oxidative stress, gene expression alteration and neurotoxicity | 106 |
| 20, 80, 110 | Male Wistar rats | 23.8, 26.4, 27.6 μg mL−1 | Intravenous injection: once per day, 5 consecutive days | Size dependent tissue distribution | 102 |
| Colloidal silver | Weaned piglets | 25, 50 and 100 μg per g diet | Ingestion exposure: mixed with diet for 5 weeks | No lactobacilli proportion observed, no AgNPs accumulation in skeletal muscles or kidneys, and only small contents found in liver | 110 |
| 60 | Fischer 344 rats | 10 mL kg−1 | Ingestion exposure: 90 days | Gender-related differences in accumulation of AgNPs in kidneys | 108 |
| 60 | Sprague–Dawley rats | Low-dose group (30 mg kg−1); medium-dose group (300 mg kg−1); high-dose group (1000 mg kg−1) | Ingestion exposure: mixed with diet for 28 weeks | Significant dose-dependent changes in the alkaline phosphatase, cholesterol values; dose-dependent accumulation of silver content in all the tissues examined; gender-related differences in accumulation of AgNPs in kidneys | 109 |
When rats were exposed to an atmosphere containing ultrafine AgNPs at a concentration of 133 μg m−3, AgNPs were detected in lung and liver immediately after the exposure, and afterward significant contents of Ag were also found in heart, brain, blood and other organs. The Ag concentration in the lungs decreased rapidly after inhalation, and the authors speculated that AgNPs existed in the alveolae wall might enter the blood capillaries.101 Lankveld and co-workers also demonstrated that when three different sized AgNPs (20, 80, 110 nm) were intravenously injected into rats at a concentration of 23.8 μg mL−1 for 5 days, the concentration of Ag was reduced rapidly in the blood, and AgNPs redistributed to liver, lung, brain, heart and all other organs, showing a systemic distribution.102
In another inhalation experiment with Sprague–Dawley rats, no remarkable changes were found in nasal cavity and lungs at a high dose of 1.32 × 106 particles cm−3 AgNPs in an inhalation chamber for 28 days; however, the size and number of goblet cells containing neutral mucins increased, suggesting that AgNPs may affect the neutral mucins in the respiratory mucosa.103 Lung function related damage was also reported in the literature. After 90 days inhalation exposure to AgNPs at a dose of 2.9 × 106 particles cm−3, lung inflammation appeared in rats, and the lung function test displayed that the tidal volume and minute volume decreased remarkably, indicating AgNPs may cause lung lesions and affect their normal function.104 However, in another inhalation toxicity study, no significant changes were found in body weight, hematology and blood biochemical values for both male and female rats after 28 days exposure at a high dose of 1.32 × 106 particles cm−3, implying that AgNPs at a concentration near silver dust limit (100 μg m−3) did not produce any significant health effects.105
Additionally, it is also proven that AgNP exposure could influence the gene expression in mouse brains. Data showed that the mice's gene expression in the caudate nucleus, frontal cortex and hippocampus all changed after treatment with 1000 mg kg−1 AgNPs, revealing that AgNPs may create neurotoxicity and apoptosis by altering gene expression and producing ROS-related oxidative stress.106 Lee et al. evaluated the effects of AgNPs on gene expression in mouse brain by exposing C57BL/6 mice to 22 nm AgNPs (1.91 × 107 particles cm−3) for two weeks, and found 468 genes in the cerebrum and 952 genes in the cerebellum were AgNP responsive. These altered genes were associated with motor neuron disorders, neurodegenerative disease, and immune cell function, suggesting AgNPs might produce potential neurotoxicity and immunotoxicity.107
Size dependent distribution of AgNPs was reported. A study showed that when AgNPs with different sizes were intravenously injected into rats, 20 nm particles deposited mainly in liver, followed by kidney and spleen, whereas larger particles (80 and 110 nm) mainly distributed in spleen followed by liver and lung.102 Gender related differences in AgNPs accumulation were also identified. Kim et al.108 reported a twofold higher concentration of AgNPs were detected in female rat kidneys compared to males after 90 days exposure at a dose of 10 mL kg−1 AgNPs via oral ingestion, which agreed with another study.109
The oral toxicity study demonstrated that alkaline phosphatase and cholesterol values changed notably in both male and female rats after 28 days exposure at a dose of 30 mg kg−1, and the variation was dose dependent. However, no genetic toxicity in rat bone marrow was observed.109 In another study, it was reported that AgNPs showed no effect on the gastrointestinal tract. When weaned piglets were treated with colloidal silver for a short time at 100 μg Ag per g diet, the ileal concentration of coliforms or lactobacilli proportion was not disturbed. After being fed with AgNPs at 20 mg Ag per g diet for 5 weeks, no AgNPs accumulation was found in skeletal muscles or kidneys, and only small Ag content of was observed in liver.110
5.3 Toxicity to non-mammals
A number of non-mammals have also been used to test the adverse effect of AgNPs. However, most of these studies are based on aquatic organisms, and the literature about the non-aquatic species related toxicity is very limited (Table 2). Considering that a large quantity of AgNPs released from fabrics and textiles would flow into aquatic system, this is not surprising.| Ag NP size/nm | Organism | Dose concentration | Exposure time | Effect measured | Ref. |
|---|---|---|---|---|---|
| 11.6 ± 3.5 | Zebrafish embryos | 0.04–0.71 nM | 120 h | Dose-dependent mortality and developmental abnormality | 112 |
| 3, 10, 50, 100 | Zebrafish embryos | 0.25, 2.5, 25, 100, 250 μM | 24–120 h | Almost 100% mortality at 120 h post-fertilization, generation of a variety of embryonic morphological malformations | 113 |
| 41.6 ± 9.1 | Zebrafish embryos | 0.02–0.7 nM | 120 h | Dose-dependent mortality and developmental abnormality | 114 |
| 49.6 | Japanese medaka (Oryzias latipes) | 1, 25 μg mL−1 | 1, 2, 4 days | Cellular and DNA damage, carcinogenic and oxidative stresses, induction of genes related metal detoxification/metabolism regulation and radical scavenging action | 115 |
| 25 | Japanese medaka (Oryzias latipes) | 100–1000 μg mL−1 | 70 days | Retarded development, reduced pigmentation and morphological malformations in embryos | 116 |
| 3–40 | Rainbow trout (Oncorhynchus mykiss) | Cells grown: 10–20 mg L−1. cytotoxicity: 0.1–10 mg L−1 | 48 h | Cytotoxicity: membrane integrity showing reduction in viability, higher levels of oxidative stress | 117 |
| 35 nm for PVP-AgNPs 40 nm for Citrate-AgNPs | Daphnia magna | PVP-AgNPs: 3.1–50 μg L−1; citrate-AgNPs: 0.625–5.0 μg L−1; AgNO3: 0.16–2.5 μg L−1 | 24 h | AgNPs disrupt protein metabolism and signal transduction, induce metal responsive and DNA damage repair genes | 118 |
| AgNO3 caused a downregulation of developmental processes, particularly in sensory development | |||||
| 30 ± 5 | Estuarine polychaete (Nereis diversicolor) | Expected final concentrations: 250 ng Ag per g sediment | Ingestion exposure: sediment mixed with AgNPs as diet, 10 days | Direct internalization of AgNPs into gut epithelium | 119 |
| 20 | Soil nematode (Caenorhabditis elegans) | 0.05, 0.1, 0.5 mg L−1 AgNPs and AgNO3 | Ingestion exposure, 24–72 h | Increased expression of the superoxide dismutases-3 (sod-3) and abnormal dauer formation protein (daf-12) genes, concurrently with significant decreases in reproduction ability | 120 |
| 10, 80 | Earthworm (Eisenia fetida) | 20, 100, 500 mg kg−1 | 14 days | Dose-dependent inhibition of the activities of AP and Na+, K+-ATPase | 121 |
| Solid dispersion, 3 μm | Fruit fly (Drosophila melanogaster) | Acute toxicity: 10–100 mg L−1 Ag; chronic toxicity: 5 mg L−1 Ag | Ingestion exposure: AgNPs prepared in solid dispersion were added into culture medium, 10 days | Acute toxicity: 50% of the tested flies unable to leave the pupae, did not finish developmental cycle | 122 |
| Chronic toxicity: influence the fertility of Drosophila during the first three filial generations | |||||
| 10 | Fruit fly (Drosophila melanogaster) | 50, 100 μg mL | Ingestion exposure: 24, 48 h | Upregulation of the expression of heat shock protein 70 and induction of oxidative stress | 123 |
Zebrafish, as a correlative and predictive model, has been used in many studies to evaluate the effects of AgNPs.111 Earlier in vivo study112 demonstrated that single AgNPs (11.6 ± 3.5 nm) could transport into zebrafish embryos through the chorion pore canal, and AgNPs were detected inside embryos at each developmental stage. At a critical concentration of 0.19 nM, developmental abnormalities could be triggered. In another study, four different sized AgNPs were synthesized to test their toxicity to zebrafish embryos, and only a few differences were observed between them. It is reported that AgNPs induced almost 100% mortality after exposure for 120 h at 250 μM, and a variety of embryonic morphological malformations were observed at a dose of 100 μM.113 A recent article also demonstrated dose-dependent mortality and developmental abnormalities in zebrafish embryos. Striking size-dependent toxicity was also reported. However, different from previous studies that smaller particles were more toxic, the authors found that larger AgNPs (41.6 ± 9.1 nm) produced higher toxic impacts and more severely deformed zebrafish than the smaller ones (11.6 ± 3.5 nm) at the same concentration.114
Chae et al.115 used Japanese medaka (Oryzias latipes) as a model animal to assess the toxic effects of AgNPs. Real time RT-PCR analysis was utilized to monitor the variation of stress-related gene expression after exposure to 1 and 25 μg mL−1 AgNPs. Results demonstrated that AgNPs could cause cellular dysfunction, DNA damage, as well as carcinogenic and oxidative stress. Developmental toxicity of AgNPs was also reported by using medaka at early life stages. High dose AgNPs (≥400 μg L−1) could induce retarded development and reduced pigmentation in the treated embryos, and dose-dependent decrease of the maximum width of the optic tectum (an indicator of midbrain development) was also observed. Furthermore, different kinds of morphological malformations such as edema, spinal abnormalities, finfold abnormalities, heart malformations and eye defects emerged after long time exposure, indicating the developmental toxicity of AgNPs.116 Toxicity of AgNPs in rainbow trout gill cells showed that nanoparticles were taken up into cells and lead to cytotoxicity related with membrane integrity disruption and oxidative stress.117
Poynton et al.118 developed a 15k oligonucleotide microarray to distinguish the toxicity from AgNPs and Ag+ for Daphnia magna. It is revealed that AgNPs disrupted protein metabolism and signal transduction, and metal responsive and DNA damage repair genes were induced as well. On the other hand, a downregulation of developmental process, especially in sensory development was caused by AgNO3, suggesting different mechanisms of toxicity between AgNPs and Ag+. An estuarine polychaete Nereis diversicolor was fed with 250 ng AgNPs per g sediment for 10 days, and obvious bioaccumulation occurred in the body. TEM images showed that AgNPs were directly internalized into the gut epithelium, and small AgNPs connected with the gut epithelial apical membrane were also present in areas with high endocytotic activity denoted by a great many endosomes and small vesicles near the cellular membrane, predicting that cellular uptake of AgNPs followed an endocytic pathway.119
Ecotoxicity study on the soil nematode Caenorhabditis elegans showed that AgNPs (0.1, 0.5 mg L−1) led to the increased expression of superoxide dismutases-3 (sod-3), abnormal dauer formation protein (daf-12) genes and reduced reproductive potential.120 Ho et al.121 reported the impact of AgNPs on earthworm Eisenia fetida at 20, 100 and 500 mg kg−1 dosages, and dose-dependent inhibition of the activity of acid phosphatase, Na+, K+-ATPase was observed after 14 days' exposure.
Due to the easy manipulation and cultivation, as well as the possibility of inducing mutations of fruit fly (Drosophila melanogaster), it has been used as a model organism in a great number of toxicity tests. Panacek et al.122 reported the acute and chronic toxicity effects of AgNPs on Drosophila. Acute toxicity assay indicated that half of the tested flies failed to finish their developmental cycle, and could not leave the pupae at a Ag concentration of 20 mg L−1. After long-time exposure to 5 mg L−1 AgNPs, the fertility of Drosophila during the first three filial generations was significantly decreased, nevertheless the fecundity of subsequent generations reached back to the normal level of the control group due to adaptation. Another study123 also found that AgNPs could induce oxidative stress and up-related the expression of heat shock protein 70 in third instar larvae of Drosophila at exposure concentrations of 50 and 100 μg mL−1, and DNA damage and apoptosis related toxicity was also observed.
5.4 Toxicity source: AgNPs or Ag+
There is a hot debate on the source of AgNP toxicity. Though great effort has been made, it is still challenging to elucidate whether the toxicity is related to nanoparticles or ions because of the uncontrollable release of Ag+ and their co-occurrence with AgNPs. There are only a handful studies which have tried to discern the two species. In an early article124 on the study of the AgNP toxicity to Chlamydomonas reinhardtii, cysteine was used to complex with Ag+ to decrease the Ag+ concentration and thus remove the contribution of Ag+ to the overall toxicity. It seemed that the toxicity of AgNPs could not be fully explained by the Ag+ in AgNPs solutions, and the particles served as sources of Ag+. Kawata et al.125 also found both the nanoparticles and Ag+ contributed to the toxicity of AgNPs to human hepatoma HepG2 cells. In another study126 evaluating the effect of AgNPs on a common grass Lolium multiflorum, AgNPs exposed seedlings showed abnormal growth with highly vacuolated and collapsed cortical cells and broken epidermis and root cap at a exposure concentration up to 40 mg L−1 AgNPs, whereas no such abnormalities were found in seedlings exposed to the same concentration of AgNO3, suggesting the toxicity of AgNPs was higher than of Ag+. Ag+ based toxicity has also been studied intensively recently. The toxicity of AgNPs was compared to that of Ag+ in Caenorhabditis elegans.127 A linear correlation between AgNP toxicity and Ag+ was found, indicating the dissolved silver ions were the key parameter that determined the toxicity of AgNPs. Xiu et al.128 tried to discern the toxicity of AgNPs and Ag+ under anaerobic conditions that prevented the oxidation of AgNPs. Results showed that Ag+ was 20 times more toxic to E. coli than AgNPs (EC50: 0.10 ± 0.01 mg L−1 for Ag+vs. 2.04 ± 0.07 mg L−1 for AgNPs), while the presence of common ligands such as Cl−, S2− and PO43− could largely decrease the Ag+ toxicity, which might explain the higher toxicity of AgNPs than Ag+ reported in previous studies. Their further study129 revealed that AgNPs (5 nm and 11 nm PEG–AgNPs) did not inhibit the growth of E. coli under strictly anaerobic conditions, whereas the AgNP toxicity greatly increased after exposure to air for 6 h and prolonged air exposure led to higher antibacterial activity, suggesting that Ag+ was the genuine molecular toxicant.In conclusion, adverse effects can be caused by AgNPs, involving developmental abnormality, DNA damage, gene expression variation and metabolic disturbance, which is dependent on concentration. Levels of AgNP toxicity vary significantly, depending on the size, shape and capping agents, as well as the exposure pathway. Dissolved Ag+ plays a vital role in the AgNP toxicity, while AgNPs acted as the source of Ag+ and the carrier to deliver NPs to organisms.
6. Summary and outlook
AgNPs are widely used in a growing number of applications due to their unique properties. However, with the accelerating introduction of AgNPs into commercial products, there is likelihood of their release into the environment, which gives rise to health and environmental concerns. This article focuses on a variety of aspects of AgNPs in the environment, involving the analysis, source, fate and transport, and potential risks to organisms. Although great effort has been made in studying each of these aspects, the information is still limited and results are uncertain and even controversial. As a result, long-term exploratory research is needed before definitive answers can be found.It is well known that AgNPs are introduced into various products. As it is reported that the type of incorporation greatly affects the nanoparticle release,30 additional research should be performed to carefully investigate the strength of the bonds between products and AgNPs. On the other hand, the majority of AgNPs containing products do not provide information of size, shape, capping agent, concentration and type of incorporation, and policy makers should regulate the market to reduce the potential adverse effects of AgNPs.
Though great progress has been made in recent years in the monitoring of AgNPs, and there are a great number of techniques available to characterize and quantify AgNPs, it is still hard to track AgNPs in the environment. As the amount of Ag in the environment is extremely small, it is beyond the detection ability of most instruments. Furthermore, the majority of these techniques are designed to characterize newly synthesized AgNP suspensions; complex environmental media hinder their application. Thus, in order to fully investigate the fate and transport of AgNPs in the environment, appropriate methods for the preconcentration, separation and speciation of AgNPs should be developed, and analytical tools for the characterization and detection of AgNPs in complicated environmental and biological samples are also urgently needed.
Due to the large surface area and relatively high surface energy, once released into the environment, AgNPs could be highly dynamic and different transformations such as oxidation, aggregation, sulfurization and chlorination will readily occur, which would greatly affect the behavior of the AgNPs. Also, environmental transformation related AgNP toxicity and stability should be investigated. Because environmental systems are always variable and stochastic, and AgNPs have limited stability and propensity of being easily oxidized and releasing Ag+, it is hard to predict the fate and transport of AgNPs. Furthermore, in the presence of DOM, dissolved Ag+ can also be reduced to AgNPs. Previous study has demonstrated that Ag+ release is mediated by dissolved oxygen and protons, however, our recent study revealed that the dissolved oxygen would generate superoxide anion in natural waters under sunlight and significantly promote the reformation of AgNPs. Given their complicated behavior in the environment, we must make great effort to broaden our knowledge of the transformation of AgNPs so as to correctly forecast their environmental and human health risks.
Several different mechanisms on the toxicity of AgNPs have been proposed, but no universal conclusion has been drawn. An important question as to whether the toxicity of AgNPs is from the nanoparticles or is related to Ag+ remains unanswered. Ideally further effort should be made to try to discern the toxicity of NPs and silver ions. Furthermore, in most toxicity studies the researchers only consider the pristine size or shape of AgNPs, but during the exposure process the morphology of AgNPs may change significantly, which would greatly affect their toxicity. Thus, the stability of AgNPs in different exposure media or environmental relevant conditions should be thoroughly investigated. The toxicity data available is mainly focused on aquatic organisms, and more research is needed to better understand the potential adverse impacts on terrestrial animals, and to predict the health effect in humans.
Acknowledgements
This work was supported by the National Basic Research Program of China (2010CB933502) and the National Natural Science Foundation of China (20977101, 21025729, 21227012).References
- H. J. Klasen, Burns, 2000, 26, 117–130 CrossRef CAS.
- A. D. Russell and W. B. Hugo, Prog. Med. Chem., 1994, 31, 351–370 CrossRef CAS.
- H. J. Klasen, Burns, 2000, 26, 131–138 CrossRef CAS.
- P. L. Drake and K. J. Hazelwood, Ann. Occup. Hyg., 2005, 49, 575–585 CrossRef CAS.
- S. N. Luoma, Silver Nanotechnologies and the Environment: Old Problems or New Challenges?, Woodrow Wilson International Center for Scholars, Washington, DC, 2008 Search PubMed.
- M. C. Lea, Am. J. Sci., 1889, 37, 476–491 Search PubMed.
- B. Nowack, H. Krug and M. Height, Environ. Sci. Technol., 2011, 45, 3189 CrossRef CAS.
- J. Tate, J. A. Rogers, C. D. W. Jones, B. Vyas, D. W. Murphy, W. J. Li, Z. A. Bao, R. E. Slusher, A. Dodabalapur and H. E. Katz, Langmuir, 2000, 16, 6054–6060 CrossRef CAS.
- Y. N. Li, Y. L. Wu and B. S. Ong, J. Am. Chem. Soc., 2005, 127, 3266–3267 CrossRef CAS.
- T. M. Tolaymat, A. M. El Badawy, A. Genaidy, K. G. Scheckel, T. P. Luxton and M. Suidan, Sci. Total Environ., 2010, 408, 999–1006 CrossRef CAS.
- Q. Ye, J. S. Zhao, F. F. Huo, J. Wang, S. Y. Cheng, T. F. Kang and H. X. Dai, Catal. Today, 2011, 175, 603–609 CrossRef CAS.
- K. M. Manesh, A. I. Gopalan, K. P. Lee and S. Komathi, Catal. Commun., 2010, 11, 913–918 CrossRef CAS.
- L. H. Ai, C. M. Zeng and Q. M. Wang, Catal. Commun., 2011, 14, 68–73 CrossRef CAS.
- B. Naik, S. Hazra, V. S. Prasad and N. N. Ghosh, Catal. Commun., 2011, 12, 1104–1108 CrossRef CAS.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- M. M. Harper, J. A. Dougan, N. C. Shand, D. Graham and K. Faulds, Analyst, 2012, 137, 2063–2068 RSC.
- A. Ravindran, V. Mani, N. Chandrasekaran and A. Mukherjee, Talanta, 2011, 85, 533–540 CrossRef CAS.
- Y. Cao, J. Wang, Y. Y. Xu and G. X. Li, Biosens. Bioelectron., 2010, 25, 1032–1036 CrossRef CAS.
- B. Roy, P. Bairi and A. K. Nandi, Analyst, 2011, 136, 3605–3607 RSC.
- S. S. Birla, V. V. Tiwari, A. K. Gade, A. P. Ingle, A. P. Yadav and M. K. Rai, Lett. Appl. Microbiol., 2009, 48, 173–179 CrossRef CAS.
- W. R. Li, X. B. Xie, Q. S. Shi, S. S. Duan, Y. S. Ouyang and Y. B. Chen, BioMetals, 2010, 24, 135–141 CrossRef.
- H. H. Lara, N. V. Ayala-Nunez, L. D. I. Turrent and C. R. Padilla, World J. Microbiol. Biotechnol., 2009, 26, 615–621 CrossRef.
- A. R. Shahverdi, A. Fakhimi, H. R. Shahverdi and S. Minaian, Nanomed.: Nanotechnol., Biol. Med., 2007, 3, 168–171 CrossRef CAS.
- J. B. Wright, K. Lam, D. Hansen and R. E. Burrell, Am. J. Infect. Control, 1999, 27, 344–350 CrossRef CAS.
- R. W. Y. Sun, R. Chen, N. P. Y. Chung, C. M. Ho, C. L. S. Lin and C. M. Che, Chem. Commun., 2005, 5059–5061 RSC.
- L. Lu, R. W. Y. Sun, R. Chen, C. K. Hui, C. M. Ho, J. M. Luk, G. K. K. Lau and C. M. Che, Antiviral Ther., 2008, 13, 253–262 CAS.
- D. Baram-Pinto, S. Shukla, N. Perkas, A. Gedanken and R. Sarid, Bioconjugate Chem., 2009, 20, 1497–1502 CrossRef CAS.
- http://www.nanotechproject.org/inventories/consumer/analysis_draft/ .
- T. M. Benn and P. Westerhoff, Environ. Sci. Technol., 2008, 42, 4133–4139 CrossRef CAS.
- L. Geranio, M. Heuberger and B. Nowack, Environ. Sci. Technol., 2009, 43, 8113–8118 CrossRef CAS.
- K. Kulthong, S. Srisung, K. Boonpavanitchakul, W. Kangwansupamonkon and R. Maniratanachote, Part. Fibre Toxicol., 2010, 7, 8–17 CrossRef.
- C. Lorenz, L. Windler, N. von Goetz, R. P. Lehmann, M. Schuppler, K. Hungerbuhler, M. Heuberger and B. Nowack, Chemosphere, 2012, 89, 817–824 CrossRef CAS.
- Y. Yan, H. F. Yang, J. F. Li, X. J. Lu and C. Wang, Text. Res. J., 2012, 82, 1422–1429 CrossRef.
- R. Kaegi, B. Sinnet, S. Zuleeg, H. Hagendorfer, E. Mueller, R. Vonbank, M. Boller and M. Burkhardt, Environ. Pollut., 2010, 158, 2900–2905 CrossRef CAS.
- J. Farkas, H. Peter, P. Christian, J. A. G. Urrea, M. Hassellov, J. Tuoriniemi, S. Gustafsson, E. Olsson, K. Hylland and K. V. Thomas, Environ. Int., 2011, 37, 1057–1062 CrossRef CAS.
- D. Cleveland, S. E. Long, P. L. Pennington, E. Cooper, M. H. Fulton, G. I. Scott, T. Brewer, J. Davis, E. J. Petersen and L. Wood, Sci. Total Environ., 2012, 421–422, 267–272 CrossRef CAS.
- A. Dudkiewicz, K. Tiede, K. Loeschner, L. H. S. Jensen, E. Jensen, R. Wierzbicki, A. B. A. Boxall and K. Molhave, TrAC, Trends Anal. Chem., 2011, 30, 28–43 CrossRef CAS.
- J. F. Liu, S. J. Yu, Y. G. Yin and J. B. Chao, TrAC, Trends Anal. Chem., 2012, 33, 95–106 CrossRef CAS.
- K. Tiede, A. B. A. Boxall, S. P. Tear, J. Lewis, H. David and M. Hassellov, Food Addit. Contam., Part A, 2008, 25, 795–821 CrossRef CAS.
- J. F. Liu, J. B. Chao, R. Liu, Z. Q. Tan, Y. G. Yin, Y. Wu and G. B. Jiang, Anal. Chem., 2009, 81, 6496–6502 CrossRef CAS.
- K. Tiede, A. B. A. Boxall, D. Tiede, S. P. Tear, H. David and J. Lewis, J. Anal. At. Spectrom., 2009, 24, 964–972 RSC.
- K. Tiede, A. B. A. Boxall, X. M. Wang, D. Gore, D. Tiede, M. Baxter, H. David, S. P. Tear and J. Lewis, J. Anal. At. Spectrom., 2010, 25, 1149–1154 RSC.
- A. R. Poda, A. J. Bednar, A. J. Kennedy, A. Harmon, M. Hull, D. M. Mitrano, J. F. Ranville and J. Steevens, J. Chromatogr., A, 2011, 1218, 4219–4225 CrossRef CAS.
- M. E. Hoque, K. Khosravi, K. Newman and C. D. Metcalfe, J. Chromatogr., A, 2012, 1233, 109–115 CrossRef CAS.
- M. E. Quadros and L. C. Marr, Environ. Sci. Technol., 2011, 45, 10713–10719 CrossRef CAS.
- J. B. Chao, J. F. Liu, S. J. Yu, Y. D. Feng, Z. Q. Tan and R. Liu, Anal. Chem., 2011, 83, 6875–6882 CrossRef CAS.
- F. Laborda, J. Jimenez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2011, 26, 1362–1371 RSC.
- J. Tuoriniemi, G. Cornelis and M. Hassellov, Anal. Chem., 2012, 84, 3965–3972 CrossRef CAS.
- J. A. Gomez-Caballero, M. G. Villasenor-Cabral, P. Santiago-Jacinto and F. Ponce-Abad, Can. Mineral., 2010, 48, 1237–1253 CrossRef CAS.
- L. S. Wen, P. H. Santschi, G. A. Gill, C. L. Paternostro and R. D. Lehman, Environ. Sci. Technol., 1997, 31, 723–731 CrossRef CAS.
- D. S. Sal'nikov, A. S. Pogorelova, S. V. Makarov and I. Y. Vashurina, Russ. J. Appl. Chem., 2009, 82, 545–548 CrossRef CAS.
- N. Akaighe, R. I. MacCuspie, D. A. Navarro, D. S. Aga, S. Banerjee, M. Sohn and V. K. Sharma, Environ. Sci. Technol., 2011, 45, 3895–3901 CrossRef CAS.
- Y. G. Yin, J. F. Liu and G. B. Jiang, ACS Nano, 2012, 6, 7910–7919 CrossRef CAS.
- R. D. Glover, J. M. Miller and J. E. Hutchison, ACS Nano, 2011, 5, 8950–8957 CrossRef CAS.
- J. L. Gardea-Torresdey, E. Gomez, J. R. Peralta-Videa, J. G. Parsons, H. Troiani and M. Jose-Yacaman, Langmuir, 2003, 19, 1357–1361 CrossRef CAS.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS.
- P. Raveendran, J. Fu and S. L. Wallen, J. Am. Chem. Soc., 2003, 125, 13940–13941 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2008, 10, 859–862 RSC.
- J. P. Xie, J. Y. Lee, D. I. C. Wang and Y. P. Ting, ACS Nano, 2007, 1, 429–439 CrossRef CAS.
- D. P. Yang, S. H. Chen, P. Huang, X. S. Wang, W. Q. Jiang, O. Pandoli and D. X. Cui, Green Chem., 2010, 12, 2038–2042 RSC.
- N. C. Mueller and B. Nowack, Environ. Sci. Technol., 2008, 42, 4447–4453 CrossRef CAS.
- F. Gottschalk and B. Nowack, J. Environ. Monit., 2011, 13, 1145–1155 RSC.
- M. Tejamaya, I. Roemer, R. C. Merrifield and J. R. Lead, Environ. Sci. Technol., 2012, 46, 7011–7017 CrossRef CAS.
- X. Jin, M. H. Li, J. W. Wang, C. Marambio-Jones, F. B. Peng, X. F. Huang, R. Damoiseaux and E. M. V. Hoek, Environ. Sci. Technol., 2010, 44, 7321–7328 CrossRef CAS.
- K. A. D. Guzman, M. P. Finnegan and J. F. Banfield, Environ. Sci. Technol., 2006, 40, 7688–7693 CrossRef.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS.
- J. Park, B. K. Kwak, E. Bae, J. Lee, Y. Kim, K. Choi and J. Yi, J. Nanopart. Res., 2009, 11, 1705–1712 CrossRef CAS.
- A. R. VandeVoort and Y. Arai, Environmental Chemistry of Silver in Soils: Current and Historic Perspective, Adv. Agron., 2012, 114, 59–90 CrossRef CAS.
- T. Hofmann and F. von der Kammer, Environ. Pollut., 2009, 157, 1117–1126 CrossRef CAS.
- J. A. Nason, S. A. McDowell and T. W. Callahan, J. Environ. Monit., 2012, 14, 1885–1892 RSC.
- J. Y. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS.
- J. L. Barriada, A. D. Tappin, E. H. Evans and E. P. Achterberg, TrAC, Trends Anal. Chem., 2007, 26, 809–817 CrossRef CAS.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2012, 46, 6900–6914 CrossRef CAS.
- W. P. Cai, H. C. Zhong and L. D. Zhang, J. Appl. Phys., 1998, 83, 1705–1710 CrossRef CAS.
- W. Zhang, Y. Yao, N. Sullivan and Y. S. Chen, Environ. Sci. Technol., 2011, 45, 4422–4428 CrossRef CAS.
- J. Y. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903–6913 CrossRef CAS.
- A. M. El Badawy, T. P. Luxton, R. G. Silva, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef CAS.
- X. Li, J. J. Lenhari and H. W. Walker, Langmuir, 2012, 28, 1095–1104 CrossRef CAS.
- F. Piccapietra, L. Sigg and R. Behra, Environ. Sci. Technol., 2012, 46, 818–825 CrossRef CAS.
- Y. W. Cheng, L. Y. Yin, S. H. Lin, M. Wiesner, E. Bernhardt and J. Liu, J. Phys. Chem. C, 2011, 115, 4425–4432 CAS.
- W. Zhang, Y. Yao, K. G. Li, Y. Huang and Y. S. Chen, Environ. Pollut., 2011, 159, 3757–3762 CrossRef CAS.
- J. L. Elechiguerra, L. Larios-Lopez, C. Liu, D. Garcia-Gutierrez, A. Camacho-Bragado and M. J. Yacaman, Chem. Mater., 2005, 17, 6042–6052 CrossRef CAS.
- M. McMahon, R. Lopez, H. M. Meyer, L. C. Feldman and R. F. Haglund, Appl. Phys. B: Lasers Opt., 2005, 80, 915–921 CrossRef CAS.
- B. Kim, C. S. Park, M. Murayama and M. F. Hochella, Environ. Sci. Technol., 2010, 44, 7509–7514 CrossRef CAS.
- R. Kaegi, A. Voegelin, B. Sinnet, S. Zuleeg, H. Hagendorfer, M. Burkhardt and H. Siegrist, Environ. Sci. Technol., 2011, 45, 3902–3908 CrossRef CAS.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2011, 45, 5260–5266 CrossRef CAS.
- J. Y. Liu, K. G. Pennell and R. H. Hurt, Environ. Sci. Technol., 2011, 45, 7345–7353 CrossRef CAS.
- B. C. Reinsch, C. Levard, Z. Li, R. Ma, A. Wise, K. B. Gregory, G. E. Brown, Jr and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 6992–7000 CrossRef CAS.
- T. E. Graedel, J. Electrochem. Soc., 1992, 139, 1963–1970 CrossRef CAS.
- B. V. Andryushechkin, K. N. Eltsov, V. M. Shevlyuga and V. Y. Yurov, Surf. Sci., 1999, 431, 96–108 CrossRef CAS.
- C. A. Impellitteri, T. M. Tolaymat and K. G. Scheckel, J. Environ. Qual., 2009, 38, 1528–1530 CrossRef CAS.
- O. Choi and Z. Q. Hu, Environ. Sci. Technol., 2008, 42, 4583–4588 CrossRef CAS.
- E. T. Hwang, J. H. Lee, Y. J. Chae, Y. S. Kim, B. C. Kim, B. I. Sang and M. B. Gu, Small, 2008, 4, 746–750 CrossRef CAS.
- S. Kim, J. E. Choi, J. Choi, K.-H. Chung, K. Park, J. Yi and D.-Y. Ryu, Toxicol. in Vitro, 2009, 23, 1076–1084 CrossRef CAS.
- I. Sondi and B. Salopek-Sondi, J. Colloid Interface Sci., 2004, 275, 177–182 CrossRef CAS.
- K. J. Kim, W. S. Sung, B. K. Suh, S. K. Moon, J. S. Choi, J. Kim and D. G. Lee, BioMetals, 2008, 22, 235–242 CrossRef.
- J. Farkas, P. Christian, J. A. G. Urrea, N. Roos, M. Hassellov, K. E. Tollefsen and K. V. Thomas, Aquat. Toxicol., 2010, 96, 44–52 CrossRef CAS.
- C. N. Lok, C. M. Ho, R. Chen, Q. Y. He, W. Y. Yu, H. Z. Sun, P. K. H. Tam, J. F. Chiu and C. M. Che, J. Proteome Res., 2006, 5, 916–924 CrossRef CAS.
- P. V. AshaRani, G. L. K. Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS.
- C. Marambio-Jones and E. M. V. Hoek, J. Nanopart. Res., 2010, 12, 1531–1551 CrossRef CAS.
- S. Takenaka, E. Karg, C. Roth, H. Schulz, A. Ziesenis, U. Heinzmann, P. Schramel and J. Heyder, Environ. Health Perspect., 2001, 109, 547–551 CAS.
- D. P. K. Lankveld, A. G. Oomen, P. Krystek, A. Neigh, A. Troost-de Jong, C. W. Noorlander, J. C. H. Van Eijkeren, R. E. Geertsma and W. H. De Jong, Biomaterials, 2010, 31, 8350–8361 CrossRef CAS.
- J. S. Hyun, B. S. Lee, H. Y. Ryu, J. H. Sung, K. H. Chung and I. J. Yu, Toxicol. Lett., 2008, 182, 24–28 CrossRef CAS.
- J. H. Sung, J. H. Ji, J. U. Yoon, D. S. Kim, M. Y. Song, J. Jeong, B. S. Han, J. H. Han, Y. H. Chung, J. Kim, T. S. Kim, H. K. Chang, E. J. Lee, J. H. Lee and I. J. Yu, Inhalation Toxicol., 2008, 20, 567–574 CrossRef CAS.
- J. H. Ji, J. H. Jung, S. S. Kim, J. U. Yoon, J. D. Park, B. S. Choi, Y. H. Chung, I. H. Kwon, J. Jeong, B. S. Han, J. H. Shin, J. H. Sung, K. S. Song and I. J. Yu, Inhalation Toxicol., 2007, 19, 857–871 CrossRef CAS.
- M. F. Rahman, J. Wang, T. A. Patterson, U. T. Saini, B. L. Robinson, G. D. Newport, R. C. Murdock, J. J. Schlager, S. M. Hussain and S. F. Ali, Toxicol. Lett., 2009, 187, 15–21 CrossRef CAS.
- H. Y. Lee, Y. J. Choi, E. J. Jung, H. Q. Yin, J. T. Kwon, J. E. Kim, H. T. Im, M. H. Cho, J. H. Kim, H. Y. Kim and B. H. Lee, J. Nanopart. Res., 2009, 12, 1567–1578 CrossRef.
- W. Y. Kim, J. Kim, J. D. Park, H. Y. Ryu and I. J. Yu, J. Toxicol. Environ. Health, Part A, 2009, 72, 1279–1284 CrossRef CAS.
- Y. S. Kim, J. S. Kim, H. S. Cho, D. S. Rha, J. M. Kim, J. D. Park, B. S. Choi, R. Lim, H. K. Chang, Y. H. Chung, I. H. Kwon, J. Jeong, B. S. Han and I. J. Yu, Inhalation Toxicol., 2008, 20, 575–583 CrossRef CAS.
- M. Fondevila, R. Herrer, M. C. Casallas, L. Abecia and J. J. Ducha, Anim. Feed Sci. Technol., 2009, 150, 259–269 CrossRef CAS.
- M. Ahamed, M. S. AlSalhi and M. K. J. Siddiqui, Clin. Chim. Acta, 2010, 411, 1841–1848 CrossRef CAS.
- K. J. Lee, P. D. Nallathamby, L. M. Browning, C. J. Osgood and X. H. N. Xu, ACS Nano, 2007, 1, 133–143 CrossRef CAS.
- O. Bar-Ilan, R. M. Albrecht, V. E. Fako and D. Y. Furgeson, Small, 2009, 5, 1897–1910 CrossRef CAS.
- K. J. Lee, L. M. Browning, P. D. Nallathamby, T. Desai, P. K. Cherukuri and X. H. N. Xu, Chem. Res. Toxicol., 2012, 25, 1029–1046 CrossRef CAS.
- Y. J. Chae, C. H. Pham, J. Lee, E. Bae, J. Yi and M. B. Gu, Aquat. Toxicol., 2009, 94, 320–327 CrossRef CAS.
- Y. A. Wu, Q. F. Zhou, H. C. Li, W. Liu, T. Wang and G. B. Jiang, Aquat. Toxicol., 2010, 100, 160–167 CrossRef CAS.
- J. Farkas, P. Christian, J. A. Gallego-Urrea, N. Roos, M. Hassellov, K. E. Tollefsen and K. V. Thomas, Aquat. Toxicol., 2011, 101, 117–125 CrossRef CAS.
- H. C. Poynton, J. M. Lazorchak, C. A. Impellitteri, B. J. Blalock, K. Rogers, H. J. Allen, A. Loguinov, J. L. Heckman and S. Govindasmawy, Environ. Sci. Technol., 2012, 46, 6288–6296 CrossRef CAS.
- J. Garcia-Aonso, F. R. Khan, S. K. Misra, M. Turmaine, B. D. Smith, P. S. Rainbow, S. N. Luoma and E. Valsami-Jones, Environ. Sci. Technol., 2011, 45, 4630–4636 CrossRef.
- J. Roh, S. J. Sim, J. Yi, K. Park, K. H. Chung, D. Ryu and J. Choi, Environ. Sci. Technol., 2009, 43, 3933–3940 CrossRef CAS.
- C. Hu, M. Li, W. Wang, Y. Cui, J. Chen and L. Yang, Toxicol. Environ. Chem., 2012, 94, 732–741 CrossRef CAS.
- A. Panacek, R. Prucek, D. Safarova, M. Dittrich, J. Richtrova, K. Benickova, R. Zboril and L. Kvitek, Environ. Sci. Technol., 2011, 45, 4974–4979 CrossRef CAS.
- M. Ahamed, R. Posgai, T. J. Gorey, M. Nielsen, S. M. Hussain and J. J. Rowe, Toxicol. Appl. Pharmacol., 2010, 242, 263–269 CrossRef CAS.
- E. Navarro, F. Piccapietra, B. Wagner, F. Marconi, R. Kaegi, N. Odzak, L. Sigg and R. Behra, Environ. Sci. Technol., 2008, 42, 8959–8964 CrossRef CAS.
- K. Kawata, M. Osawa and S. Okabe, Environ. Sci. Technol., 2009, 43, 6046–6051 CrossRef CAS.
- L. Y. Yin, Y. W. Cheng, B. Espinasse, B. P. Colman, M. Auffan, M. Wiesner, J. Rose, J. Liu and E. S. Bernhardt, Environ. Sci. Technol., 2011, 45, 2360–2367 CrossRef CAS.
- X. Y. Yang, A. P. Gondikas, S. M. Marinakos, M. Auffan, J. Liu, H. Hsu-Kim and J. N. Meyer, Environ. Sci. Technol., 2012, 46, 1119–1127 CrossRef CAS.
- Z. M. Xiu, J. Ma and P. J. J. Alvarez, Environ. Sci. Technol., 2011, 45, 9003–9008 CrossRef CAS.
- Z. M. Xiu, Q. B. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |