ZIF-derived in situ nitrogen-doped porous carbons as efficient metal-free electrocatalysts for oxygen reduction reaction†
Peng
Zhang
a,
Fang
Sun
a,
Zhonghua
Xiang
a,
Zhigang
Shen
a,
Jimmy
Yun
bc and
Dapeng
Cao
*a
aState Key Lab of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, P.R. China. E-mail: caodp@mail.buct.edu.cn; Tel: +8610-64443254
bSchool of Chemical Science and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
cChangzhou Institute of Advanced Materials, Beijing University of Chemical Technology, Changzhou 213164, P.R. China
First published on 7th November 2013
Abstract
We have successfully prepared nanoporous Carbon-L and -S materials by using ZIF-7 as a precursor and glucose as an additional carbon source. Results indicate that Carbon-L and -S show an appropriate nitrogen content, high surface area, robust pore structure and excellent graphitization degree. The addition of an environmentally friendly carbon source – glucose – not only improves the graphitization degree of samples, but also plays a key role in removing residual Zn metal and zinc compound impurities, which makes the resulting materials metal-free in situ nitrogen-doped porous carbons. By further investigating the electrocatalytic performance of these nitrogen-doped porous carbons for oxygen reduction reaction (ORR), we find that Carbon-L, as a metal-free electrocatalyst, shows excellent electrocatalytic activity (the onset and half-wave potentials are 0.86 and 0.70 V vs. RHE, respectively) and nearly four electron selectivity (the electron transfer number is 3.68 at 0.3 V), which is close to commercial 20% Pt/C. Moreover, when methanol was added, the Pt/C catalyst would be poisoned while the Carbon-L and -S would be unaffected. By exploring the current-time chronoamperometric response in 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, we found that the duration stability of Carbon-L is much better than the commercial 20% Pt/C. Thus, both Carbon-L and -S exhibit excellent ability to avoid methanol crossover effects, and long-term operation stability superior to the Pt/C catalyst. This work provides a new strategy for in situ synthesis of N-doped porous carbons as metal-free electrocatalysts for ORR in fuel cells.
000 s, we found that the duration stability of Carbon-L is much better than the commercial 20% Pt/C. Thus, both Carbon-L and -S exhibit excellent ability to avoid methanol crossover effects, and long-term operation stability superior to the Pt/C catalyst. This work provides a new strategy for in situ synthesis of N-doped porous carbons as metal-free electrocatalysts for ORR in fuel cells.
Broader contextCurrently, reducing or replacing Pt-based electrodes in fuel cells has become a hot topic, and it is our urgent task to develop novel highly efficient catalysts for oxygen reduction reaction (ORR). In this work, by using a ZIF as a self-sacrificed precursor and environmentally friendly glucose as an additional carbon source, we have fabricated the ZIF-derived nitrogen-doped porous carbons (marked as Carbon-L and Carbon-S) and found that ZIF-derived nitrogen-doped porous carbons as metal-free electrocatalysts for ORR exhibit excellent electrocatalytic activity and operation stability, which is close to commercial 20% Pt/C catalysts. Moreover, when methanol was added, the Pt/C catalysts would be poisoned while the Carbon-L and -S would be unaffected. By exploring the current–time chronoamperometric response in 25000 s, we found that the duration stability of Carbon-L is much better than that of the commercial 20% Pt/C. Definitely, this work provides a new strategy for in situ synthesis of N-doped porous carbons as metal-free electrocatalysts for ORR in fuel cells. Not only ZIFs can be considered as a self-sacrificed precursor and nitrogen source, but also other nitrogen-containing MOFs may be alternative precursors. Owing to the versatility of MOF structures, the MOF-derived porous carbons would significantly broaden the family of nanoporous carbons with novel structures and multifunctional properties including electrocatalysts, sensors, supercapacitors and batteries.
|
1. Introduction
The catalytic oxygen reduction reaction (ORR) at the cathode is a critical step for fuel cells, metal–air batteries and air-breathing cathodes in industrial electrocatalytic processes. The cathode ORR is six or more orders of magnitude slower than the anode hydrogen oxidation reaction and thus limits the performance of fuel cells.1 Developing suitable catalysts for ORR becomes very important in order to prepare low-cost, high-performance fuel cells. As is well known, the platinum materials have been regarded as the most active catalysts for ORR in fuel cells,2–4 but they suffer from high cost, low tolerance to methanol and limited stability.5 Therefore, most investigations related to the ORR were focused on reducing or replacing Pt-based electrodes in fuel cells, including Pt-based alloys,6,7 transition metal macrocyclic compound-based catalysts,8,9 strongly coupled inorganic nanocarbon hybrid materials,10 nitrogen-doped carbon nanostructures and corresponding composites,11,12 and metal-free N-doped carbon materials (such as graphene-based catalysts,13,14 carbon nanotube based catalysts,15,16 and porous organic framework-templated nitrogen porous carbons17).Besides carbons, in fact, no other materials are better as ideal supports or electrocatalysts for ORR in fuel cells, because the carbon material has essential properties such as high electronic conductivity, good corrosion resistance, excellent surface properties and low cost.11 Currently, investigators usually adopt heteroatoms, especially nitrogen, to dope carbon materials in order to improve the catalytic activity and conductivity of carbon-based materials. Generally, nitrogen-doped carbon materials can be categorized into two types:18 (i) directly doping in the synthesis of porous carbon materials, which is called as “in situ” doping;12,17 (ii) post-treatment of as-synthesized carbon materials with a nitrogen-containing precursor (N2, NH3, etc.), i.e., post-doping.14,19 N-doped carbon materials exhibit unique electronic properties, such as high electrical conductivity and good selectivity toward ORR, which originates from the conjugation between the graphene π system and the nitrogen lone-pair.17 Therefore, N-doped carbon is an excellent candidate for the ORR.
Recently, metal–organic frameworks (MOFs) and covalent-organic materials (COMs) have attracted extensive interest in applications such as clean energy,20–24 catalysis,25 luminescent sensing26,27 and so on. Owing to their diverse structures, high surface area, large pore volume and various pore sizes, MOFs and COMs were also considered as an alternative precursor to construct nanoporous carbons, which broaden the library of nanoporous carbons with novel structures and properties.28,29 So far, several MOFs and COMs have been used as precursors to yield highly nanoporous carbons for clean energy applications such as gas adsorption,30–33 electrochemical capacitance,28,34–38 lithium batteries,39–41 sensing,42,43 environmental pollution treatment44,45 and ORR in fuel cells.46–51 However, the electrocatalysts mentioned above often include metal atoms, which are still unsuitable for ORR because the basic media often used for ORR can make these metals leach from electrocatalyst surfaces over time, thus making the system lose the electrocatalytic activity and shelf life.52 To ensure that the fabricated carbon materials do not contain metal or metallic compounds and other impurities, several additional steps must be performed, such as leaching and washing with a highly corrosive acid (HF)35,42 and aqueous alkali. Therefore, it is necessary to find a green and environmentally friendly method to prepare metal-free nitrogen-doped porous carbon materials with high-performance toward ORR.
Zeolitic imidazolate frameworks (ZIFs), a sub-family of MOFs, consist of transition metal ions (such as Zn2+ and Co2+) and imidazolate linkers, which can form 3D tetrahedral frameworks resembling zeolite topologies.53 Different from their MOF analogues with carboxyl-ligands, a number of ZIFs not only exhibit exceptional thermal and chemical stability, but also contain a rich nitrogen source in imidazolate ligands. All these properties make ZIFs an excellent candidate for construction of N-doped porous carbons with superior performances.31,32,35,48,50,51 In particular, the ZIF-7 framework [Zn (PhIM)2·(H2O)3; PhIM = benzimidazole], which is formed by bridging N-containing benzimidazole ligands and zinc cations resulting in a sodalite (SOD) topology,54 may be a suitable nitrogen precursor to yield in situ N-doped porous carbons.
In previous studies, investigators basically used furfuryl alcohol (FA) as an additional carbon precursor.28,30–32,34 As is well known, FA is a toxic, explosive and irritant reagent, and it is extremely harmful to the human body and the environment. Therefore, we must find an alternative green carbon precursor. Glucose may be an alternative carbon precursor, because it can be infiltrated into the external surface and/or voids of the solid ZIF-7, then polymerized and carbonized to form nanoporous carbons.55
In this work, we successfully fabricate ZIF-derived in situ nitrogen-doped porous carbon materials (ZIF-derived carbons) by carbonizing the ZIF-7/glucose composites, and systematically characterize the synthesized samples. Moreover, we also investigate the electrocatalytic activity of the synthesized porous carbons for ORR in an alkaline electrolyte and make a comparison with the 20% commercial platinum/carbon (20 wt% Pt loading, Johnson Matthey, Pt/C).
2. Experimental section
Solvothermal synthesis of ZIF-7
ZIF-7 was synthesized by solvothermal reaction of zinc nitrate hexahydrate and benzimidazole in solvent of N,N-dimethylformamide (DMF), as reported in the literature.53 The detailed process is presented in the ESI.† Elemental analysis: C14H16N4O3Zn = Zn (IM)2·(H2O)3. Calcd: C, 47.54%; H, 4.56%; N, 15.84%. Found: C, 48.65%; H, 4.12%; N, 16.61%. The results are in good agreement with the one from the literature,53 which better validates that the resulting material is ZIF-7.Preparation of porous carbons
The 0.5 g dry ZIF-7 was soaked in 20 mL 0.5 mol L−1 glucose aqueous solution and stirred for 1 h, and the resulting mixture was allowed to stand overnight, followed by filtration and washing with ethanol. The ZIF-7/glucose composite was then transferred into a quartz boat and placed in a furnace, under flowing Ar for 5 h to exclude air and then heated at 200 °C for 2 h at a rate of 2 °C min−1 under an Ar atmosphere. Subsequently, the composite was carbonized under Ar at 950 °C for 5 h with a heating rate of 3 °C min−1. The resulting sample was labelled as Carbon-L, where L means that ZIF-7 and glucose were mixed in a liquid phase. To simplify the operation process, we also adopted a solvent-free and mechanochemical method to prepare the ZIF-7/glucose co-derived carbon, and specific steps are as follows. 0.5 g ZIF-7 and 0.25 g glucose solids were placed in a quartzose mortar and milled to a fine powder mixture. The resulting powder mixture in an alumina crucible was placed within a quartz tube furnace for carbonization. The heating process is the same as above. The resulting black sample is named as Carbon-S, where S means that ZIF/glucose was mixed in a solid phase. To determine the importance of glucose as a carbon source in the preparation of Carbon-S and Carbon-L materials, we also directly carbonized glucose (the resulting sample is denoted as Carbon-G) and ZIF-7 (the resulting sample is denoted as Carbon-Z) by a similar carbonization procedure, for comparison.Working electrode preparation
5 mg as-prepared carbon was added into 1 mL N,N-dimethylformamide (DMF), and then dispersed by ultrasonic irradiation for at least 30 min. Subsequently, 50 μL 5% Nafion (DuPont) was added into the above dispersion or suspension, and further treated by sonication until forming a homogeneous ink. 4 μL ink (containing ∼20 μg catalyst) was loaded on a glassy rotating disk electrode of 5 mm diameter (Pine Instruments). The catalyst loading is approximately 0.1 mg cm−2. The electrode was dried in air and was used as the working electrode for further electrochemical studies.Electrochemical measurements
All the electrochemical tests were carried out in a standard three-electrode cell with a 1 cm2 Pt plate as the counter electrode, and using a saturated calomel electrode (SCE, 0.99 V vs. RHE in 0.1 M KOH) as the reference electrode. An aqueous solution of 0.1 M KOH was used as an electrolyte for electrochemical studies. Electrochemical properties were investigated by CV and LSV on a PARSTAT 2273 potentiostat from Princeton Applied Research. Prior to the measurement, the electrolyte was saturated with oxygen by bubbling O2. In addition, a flow of O2 was maintained over the electrolyte during the recording of CVs. For comparison, the CV measurement was also performed in a N2-saturated electrolyte. The working electrode was cycled at least five times before the data were recorded at a scan rate of 50 mV s−1. The potential range is cyclically scanned between −1.0 and 0 V. RDE measurements were conducted at a rate of 5 mV s−1 with different rotating speeds from 400 to 2025 rpm by using a Pine Instruments device.3. Results and discussion
The as-prepared ZIF-7 is cubic nanoparticles, about 200 nm, as seen in the scanning electron microscopy (SEM) image in Fig. 1a. Powder X-ray diffraction (PXRD) and Fourier-transform infrared spectroscopy (FT-IR) measurements were carried out to examine the structure and composition of as-prepared ZIF-7 (see Fig. 1b and c). Results indicate that the synthesized crystal shows a very similar XRD and FT-IR to the ones reported previously.53 The corresponding positions and intensities of the FT-IR absorption peaks are shown in the ESI.† Thermogravimetric analysis (TGA) of ZIF-7 was performed under argon flowing at a heating rate of 10 °C min−1. Three steps of weight loss are shown in Fig. 1d. The first two steps of weight loss start at 160 °C and end at 700 °C. The corresponding weight loss is attributed to the removal of free and terminal DMF and H2O molecules.27 The third step of weight loss at 700 °C corresponds to the decomposition of the host frameworks. The TGA indicates that the three-dimensional ZIF-7 framework possesses high thermal stability up to 700 °C, which is an advantage as a precursor because high thermal stability prevents its vaporization at high temperature. Moreover, the high nitrogen content (15.84%) of ZIF-7 makes it suitable as a self-sacrificed precursor to fabricate nitrogen-doped porous carbons. To further improve the textural property and electrocatalytic activity of the ZIF-derived carbons, we also added an additional green and environmentally friendly carbon source – glucose – in the ZIF matrix, which can help form the robust porosity. By carbonizing the ZIF-7/glucose composites, we successfully prepared the porous Carbon-L and -S materials, where L means that the ZIF-7/glucose composite was prepared in a liquid phase and S indicates that the ZIF-7/glucose composite was obtained by milling the mixture in a solid phase. For comparison, we also fabricated Carbon-Z and -G by directly carbonizing the ZIF-7 and glucose, respectively. | ||
| Fig. 1 (a) SEM image, (b) PXRD graph, (c) FT-IR spectra and (d) TGA curve of as-prepared ZIF-7 in this work. | ||
The morphologies of as-prepared porous carbons were characterized via SEM and transmission electron microscopy (TEM). As shown in Fig. 2a and b and S1,† both Carbon-L and Carbon-S show sheet-like or graphene-like structure, containing the clear-cut carbon nanosheets. However, Carbon-Z shows a flower-like structure (see Fig. 2c and d) and Carbon-G shows an amorphous structure (Fig. S2†). The results reveal that the morphology of the porous carbons changes significantly in the cases with and without an additional carbon source (glucose). Actually, the addition of the carbon source glucose is favorable for the formation of graphene-like structures, which is attributed to the pre-melting and polymerization of glucose before the ZIF-7 was carbonized completely. To further observe the microstructure, we also show the high resolution transmission electron microscopy (HRTEM) image in Fig. 3, and detailed analysis was presented in a later section.
 | ||
| Fig. 2 (a) SEM image and (b) TEM image of as-prepared Carbon-L, (c) SEM image and (d) TEM image of as-prepared Carbon-Z. | ||
 | ||
| Fig. 3 HRTEM images of three ZIF-derived carbons: (a) Carbon-L; (b) Carbon-S; (c) Carbon-Z. In (a) and (b), the arrows point to the graphitic sheets; while in (c), the arrow points to the interplanar distance. | ||
To get insight into the pore properties of the synthesized porous carbons, we measured the N2 adsorption–desorption isotherms at 77 K (Fig. 4a) and used density functional theory (DFT) to obtain pore size distribution (PSD) (see Fig. S3†). The isotherms of three ZIF-derived porous carbons exhibit a type-IV curve, which suggests the existence of both micropores and mesopores. The type-I isotherm of Carbon-G demonstrates that this material only contains micropores, which can also be clearly observed through PSD in Fig. S3.† It should be noted that the presence of mesopores and micropores depends on the gasification of the carbons and high evaporation etching of the Zn, while the formation of partial macropores in Carbon-Z may be attributed to the destruction of the cubic structure. With the increase of temperature, the polymerization or fusion of glucose first occurs on the surface of ZIF-7, which prevents the formation of macropores in Carbon-L and -S.
 | ||
| Fig. 4 The structure characterization of as-prepared three ZIF-derived porous carbons and glucose-derived carbon: (a) nitrogen adsorption–desorption isotherms. (b) PXRD graphs. (c) Raman spectra. (d) XPS spectra. Red line (Carbon-L), black line (Carbon-S), blue line (Carbon-Z), and magenta line (Carbon-G). | ||
The feature parameters of four carbons are summarized in Table 1. The BET specific surface area (SSA) and total pore volume of Carbon-L and Carbon-S are 783 m2 g−1, 0.56 cm3 g−1 and 515 m2 g−1, 0.46 cm3 g−1, respectively, which are obviously higher than Carbon-Z (406 m2 g−1, 0.29 cm3 g−1). Combined with their morphologies, it can be deduced that the addition of glucose is critical for the structural evolution of the resulting porous carbons.
| Samples | S BET (m2 g−1) | S Langmuir (m2 g−1) | V t (cm3 g−1) | V micro (cm3 g−1) | V micro/Vt (%) | D HK (nm) |
|---|---|---|---|---|---|---|
| a The specific surface area (SBET) was calculated by the Brunauer–Emmett–Teller (BET) method. SBET calculated in the region of P/P0 = 0.05 to 0.3. b V t represents the total pore volume. Determined at P/P0 = 0.9997. c V micro represents the volume of micropore. d D HK represents the median pore width calculated by Horvath–Kawazoe. | ||||||
| Carbon-L | 783 | 1152 | 0.56 | 0.37 | 66 | 0.50 |
| Carbon-S | 515 | 766 | 0.46 | 0.23 | 50 | 0.53 |
| Carbon-Z | 406 | 607 | 0.29 | 0.22 | 76 | 0.51 |
| Carbon-G | 132 | 198 | 0.09 | 0.08 | 89 | 0.51 |
We also performed PXRD analyses of synthesized porous carbons to validate their graphitic nature and the presence or not of metal Zn impurities in the carbon matrix. The PXRD patterns of four samples (Fig. 4b) display only two broad peaks at around 23° and 44°, corresponding to the carbon (002) and (101) diffractions, respectively. No diffraction peaks of Zn impurities were observed in the ZIF-derived carbons. Furthermore, the carbonization temperature was close to the boiling point of Zn, indicating that the carbon-reduced Zn metal was vaporized away along with the Ar flowing under carbonization conditions.34
During the carbonization process, the carbon source glucose would be carbonized first, because the thermal stability of glucose is poorer than ZIF-7. At the beginning of decomposition of the host ZIF framework, ZnO will be produced, which was further reduced to Zn by glucose-derived carbon (or CO) at T > 800 °C and Zn would be evaporated during carbonization.56 Since the boiling point of the Zn metal is 908 °C, it will be emitted with Ar flowing, yielding metal-free samples at T = 950 °C. The absence of Zn in Carbon-L and -S is further confirmed by the analysis of the EDS and ICP results. As shown in Fig. S4,† no signal of Zn and zinc compounds appears in EDS data of Carbon-L and -S. All the results (including the HRTEM images (Fig. 3a and b)) indicate that Carbon-L and Carbon-S are metal-free porous materials. However, it is observed from the HRTEM images in Fig. 3c and EDS in Fig. S4† that trace amounts of the Zn compound in the Carbon-Z still exist, even though no corresponding metal peak appears in the PXRD graph (see Fig. 4b). The HRTEM image of Carbon-Z in Fig. 3c reveals that the interplanar distance is in the range of 0.345 to 0.348 nm, which is consistent with the lattice spacing of 0.3456 nm for the {220} planes of the facet of Zn3N2 (3.456 Å, PDF#35-0762). The above results prove that high temperature only eliminates zinc oxide rather than other zinc compounds. Moreover, glucose also plays a key role in complete removal of Zn or zinc compounds, which is unlike FA simply as a carbon source.
Raman spectra of all the obtained porous carbons are shown in Fig. 4c, where D and G bands are located at 1353 cm−1 and 1588 cm−1, respectively arising from the disordered carbon structures and the vibration mode to the movement in opposite directions of two carbon atoms in a single graphene sheet.35 The relative ratios of the G band to the D band illustrate the degree of graphitization. The IG/ID of ZIF-derived porous carbons is higher than Carbon-G, but these values are almost the same (approximately 1), which suggests that in all the synthesized porous carbons the graphene sheets were not well developed, and the local carbon structures contain both graphene and disordered carbons. In fact, it can also be observed from the HRTEM images (Fig. 3a and b) that the Carbon-L and -S were fabricated by randomly assembling the nanometer-sized sheets, which coincided with the Raman and XRD results.
X-ray photoelectron spectroscopy (XPS) measurements (Fig. 4d) and elemental analysis (Table S1†) were performed to probe the chemical composition and the content of nitrogen in the ZIF-derived porous carbons. The XPS spectra of four samples distinctly show the presence of N1s, C1s and O1s peaks without any other signals, and high-resolution N1s XPS spectrum details are given in a later section. The C1s peaks for the ZIF-derived porous carbons were centered at approximately 285.0 eV (continuation of sp2 graphitic carbon) and were slightly asymmetric, which is a common characteristic for nitrogen-doped carbon materials.57 The presence of oxygen can be attributed to moisture, atmospheric O2 or CO2 adsorbed on ZIF-derived carbon as well as the residual oxygen-containing groups from ZIF-7 (nitroxyl group) and glucose (hydroxyl group, carbonyl group). The same result can be proved well by their FT-IR spectra (Fig. S5†). All samples share a stretching band at 3437 cm−1, which indicates the existence of the hydroxyl functional group and adsorbed water molecule.
In this work, the ZIF-7 framework was acted as a self-sacrificed precursor and nitrogen source, which resulted in the formation of the nitrogen-doped porous carbon. The addition of glucose not only affects their structure, morphology, composition, graphitization degree, and electrical properties, but also influences the electrocatalytic performance of ZIF-derived porous carbons for ORR. Remarkably, the resulting porous carbons not only possess an appropriate nitrogen content, high surface area, and robust pore structure, but also show enhanced electrical conductivity. Such unique features are favorable for the access of oxygen to the catalyst surface and can facilitate the rapid diffusion of electrons in the electrode during the oxygen reduction process.58
The electrocatalytic activity of ZIF-derived carbons for ORR was evaluated by cyclic voltammetry (CV) scanning in 0.1 M KOH aqueous solution saturated with N2 or O2 gas at room temperature (see Fig. 5a). As shown in Fig. 5b, a featureless voltammetric current within the potential range from 0 to 1.0 V (versus RHE) was observed for Carbon-L in the nitrogen-saturated solution, while a well-defined oxygen reduction peak at 0.73 V emerges in the O2-saturated solution, which is superior to Carbon-S (0.68 V), Carbon-Z (0.50 V), and Carbon-G (0.49 V), and close to commercial 20% Pt/C (0.84 V) (see Fig. 5a), suggesting a pronounced electrocatalytic activity of Carbon-L for ORR.
 | ||
| Fig. 5 (a) CV curves of three ZIF-derived carbons (Carbon-L, Carbon-S, and Carbon-Z), glucose-derived carbon (Carbon-G) and commercial 20% Pt/C in O2-saturated (solid line) or N2-saturated (dashed line) 0.1 M KOH at a sweep rate of 50 mV s−1. (b) CV curve of Carbon-L under the same conditions as (a). (c) Rotating-disk voltammograms of Carbon-L (the catalyst loading was about 0.1 mg cm−2) in O2-saturated 0.1 M KOH with a sweep rate of 5 mV s−1 at the different rotating rates indicated. The inset is Koutecky–Levich (K–L) plots at different potentials (0.3, 0.35, 0.4 and 0.45 V, vs. RHE). Electron transfer number (n) is calculated at the potential of 0.3 V. (d) Linear–sweep voltammograms in 0.1 M KOH under oxygen bubbling at a scan rate of 5 mV s−1 and electrode-rotation speed of 1600 rpm. (e) The K–L plots of different catalysts in the potential range of 0.3 V (vs. RHE). (f) The electron transfer numbers (n) at different porous carbons in the potential range of 0.1 V and 0.6 V on the basis of K–L equations. | ||
The impressive electrocatalytic activity of Carbon-L was further confirmed by recording the linear sweep voltammetry (LSV) curves on a rotating disk electrode (RDE) in Fig. 5c. The LSV curves in O2-saturated 0.1 M KOH electrolyte at a scanning rate of 5 mV s−1 show that the current density increases with the increase of rotation rate from 400 to 2025 rpm. The ORR on the Carbon-L is diffusion-controlled when the potential is less than 0.65 V, and it is the kinetics-diffusion mixing controlled in the potential range from 0.65 V to 0.82 V. For comparison, other carbons were also investigated under the same conditions (see Fig. S6†). The rotating-disk voltammograms in O2-saturated 0.1 M KOH solution at a rotation rate of 1600 rpm are shown in Fig. 5d. Strikingly, the onset potentials of Carbon-L and Carbon-S are more positive than Carbon-Z and Carbon-G. Moreover, the limiting current density (JL) of Carbon-L is up to about −4.6 mA cm−2, which is much higher than other carbons and comparable to 20% Pt/C (−4.9 mA cm−2) in Fig. 5d. These results suggest that the addition of glucose in ZIF-7 significantly affects the electrocatalytic behavior of the carbonized samples and the overpotential of the electrode. To gain further insight into the electrocatalytic performance of Carbon-L, the electrochemical properties of other carbons are also summarized in Table 2. Interestingly, the electron transfer numbers of Carbon-L (3.68 at 0.3 V, the inset of Fig. 5c) and Carbon-S (3.71 at 0.4 V, Fig. S6b†) are comparable to other metal-free catalysts (n = 3.23–3.89, see Table S2 in the ESI†)12,17,59–62 and MOF-derived ORR electrocatalysts reported recently.49,50 Besides the electron transfer number, we also compared the onset potential, half-wave potential and limited current density with other electrocatalysts for ORR, as shown in Table S2.†
| Electrocatalyst | Onset potential [V] (vs. RHE) | Limiting current density [mA cm−2] | Half-wave potential [V] (vs. RHE) | n (number of electron transfers at 0.3 V) |
|---|---|---|---|---|
| Carbon-L | 0.8610 | −4.5918 | 0.6972 | 3.68 |
| Carbon-S | 0.8441 | −4.4345 | 0.6782 | 3.66 |
| Carbon-Z | 0.7434 | −2.5052 | 0.3246 | 2.33 |
| Carbon-G | 0.7630 | −2.7344 | 0.2654 | 1.65 |
| 20% Pt/C | 0.9249 | −4.8679 | 0.7998 | 3.97 |
By using the Koutecky–Levich (K–L) equation, we further analyzed the RDE data. The K–L plots of different electrocatalysts at the potential of 0.3 V are shown in Fig. 5e. The linearity of K–L plots suggests first-order reaction kinetics toward the concentration of dissolved oxygen.12 The slopes of Carbon-L and Carbon-S are much lower Carbon-Z and Carbon-G, and are almost the same as 20% Pt/C. The exact electron transfer numbers of five electrocatalysts at different potentials were calculated by the slopes of K–L plots and are summarized in Fig. 5f. Obviously, n values of Carbon-L and Carbon-S are significantly higher than those of Carbon-G and Carbon-Z, and are close to commercial 20% Pt/C. Obviously, both Carbon-L and Carbon-S are the excellent electrocatalysts for ORR. We also used a rotating ring-disk electrode (RRDE) to measure the voltammograms of Carbon-L for ORR, as shown in Fig. S7.†
Recent investigations16,58,59 indicate that the content of nitrogen in carbon materials, especially the pyridinic or/and graphite nitrogen portion, is crucial for promotion of the electrocatalytic reaction in ORR. Generally speaking, the N-doped carbon materials include several types of N-containing functional groups, which can be identified by the bonding state of the N atom in the composite. The high-resolution N1s spectrum can be deconvoluted into three types: 398.4 ± 0.2, 400.0 ± 0.2 and 401.0 ± 0.2 eV, corresponding to pyridinic N, pyrrolic N, and graphitic N, respectively.63,64 The XPS data in Fig. 6 indicate that the content of graphitic N is nearly kept constant while that of pyridinic N is different significantly. Therefore, it is reasonable to believe that the higher activity of Carbon-L than other three carbon electrocatalysts may be attributed to the higher content of pyridinic N in the Carbon-L. Since the pyridinic nitrogen atoms with strong electron-accepting ability, which possesses one lone pair of electrons in addition to the one electron donated to the conjugated π bond system, can create a net positive charge on the adjacent carbon atoms in the porous carbons, they are favorable for the adsorption of oxygen atoms and can readily attract electrons from the anode, thus facilitating the ORR.16 The content of pyridinic nitrogen of Carbon-Z is higher than Carbon-S, but the electrochemical activity of Carbon-S is much lower than Carbon-Z. In addition to the content of pyridinic N, graphene-like morphology, high BET SSA, and high porosity also contribute to enhanced activity for ORR. Actually, the high BET SSA and high porosity can enhance the mass-transport, benefiting to expose the catalytic site to reactants.50 Lower BET SSA and porosity of Carbon-Z make catalytic sites not well exposed, which prevents the mass-transport between activity sites and oxygen. The lack of glucose in Carbon-Z leads to that the surface of Carbon-Z has no C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, as shown in Fig. S5.† This weakens the hydrophilicity and wettability of the Carbon-Z surface, which affects oxygen access to the electrocatalyst in alkaline solution.
O groups, as shown in Fig. S5.† This weakens the hydrophilicity and wettability of the Carbon-Z surface, which affects oxygen access to the electrocatalyst in alkaline solution.
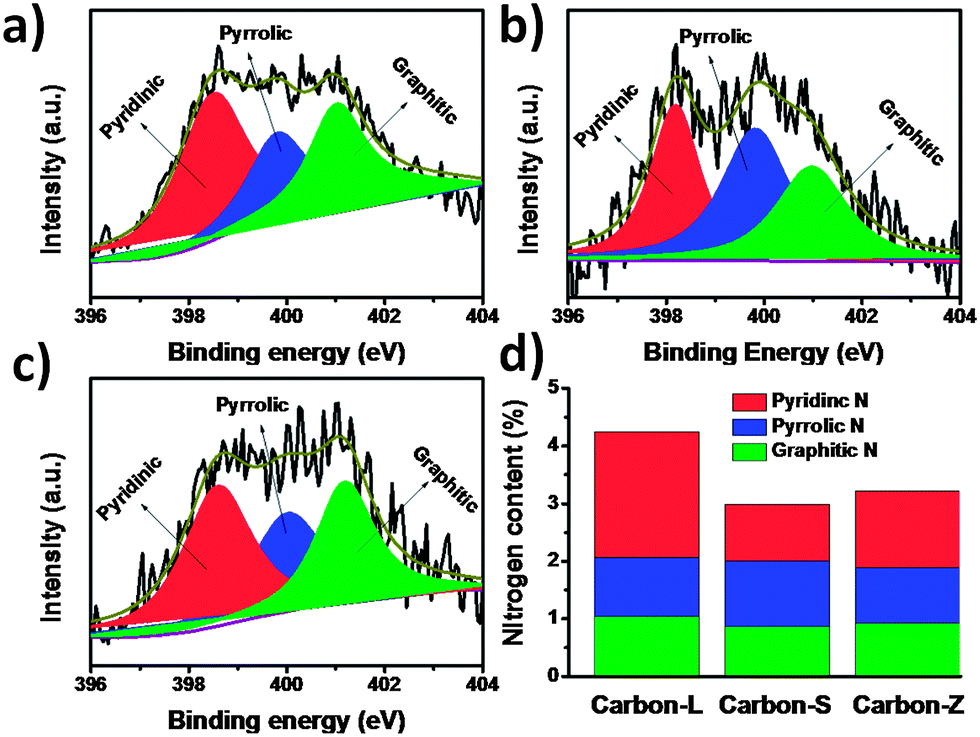 | ||
| Fig. 6 High-resolution N1s XPS spectra of (a) Carbon-S, (b) Carbon-L and (c) Carbon-Z. (d) The content of three types of nitrogen in ZIF-derived nitrogen-doped porous carbons. | ||
The electrical conductivity is vital in achieving high-performance of nitrogen-doped carbon materials for ORR since it significantly affects the electron transportation in the electrode and active sites for oxygen reduction.58 The difference of electrochemical activity for four porous carbons may be attributed to the increase of mesoporosity owing to the introduction of glucose in ZIF-7, which results in enhanced electrical conductivity. It is well known that mesopores are helpful to quick mass transfer and smooth diffusion of electrolyte. This observation can be further confirmed by the electrochemical impedance spectra (EIS). As shown in Fig. S8,† the resistances of Carbon-L and Carbon-S are much lower than Carbon-Z and Carbon-G. Moreover, the increased graphitization degrees of Carbon-L and -S in Raman spectra (Fig. 4c) also improve their electronic conductivity.65 All these results show that the high electrocatalytic activity of Carbon-L and Carbon-S should originate from synergistic effects of their low resistance, high SSA and high content pyridinic nitrogen.66
In addition to the activity, the stability is another key parameter for high-performance electrocatalysts.67 We conducted a 25000 s stability test for Carbon-L toward ORR by chronoamperometry at a constant voltage of −0.4 V in an O2-saturated 0.1 M KOH solution at a rotation rate of 1000 rpm. As shown in Fig. 7a, although the current densities of both Carbon-L and commercial 20% Pt/C decrease with time, the Carbon-L exhibits a very slow attenuation and a high relative current of 75% still persists after 25000 s, while only 53% of the initial catalytic current is maintained for the 20% Pt/C catalyst in the same testing time. This result demonstrates that the Carbon-L electrode holds better duration stability than commercial 20% Pt/C for ORR in an alkaline medium. Besides, we also performed continuous potential cycling to investigate the capacitance stability of Carbon-L. As shown in Fig. 7b, the current does not show significant decrease after 50000 continuous cycles (15 days) between −1.0 and 0 V in oxygen-saturated 0.1 M KOH, which demonstrates the excellent capacitance stability of the ZIF-derived carbon electrode. Hence, they may also serve as electrode materials for supercapacitors.
 | ||
| Fig. 7 (a) Current–time (i–t) chronoamperometric response of Carbon-L and commercial 20% Pt/C electrodes at −0.4 V in O2-saturated 0.1 M KOH at a rotation rate of 1000 rpm. (b) CVs of the Carbon-L electrode in oxygen-saturated 0.1 M KOH before and after a continuous potentiodynamic sweep for 50000 cycles at room temperature with a scan rate of 100 mV s−1; (c) current–time (i–t) chronoamperometric response of Carbon-L (Carbon-S) and 20% Pt/C electrodes by adding 3 mL methanol after about 400 s. | ||
The ORR electrocatalysts should also be robust in the real application and the crossover effect must be considered.58 To examine possible crossover effects, we measured the electrocatalytic selectivity of Carbon-L, Carbon-S and 20% Pt/C against the electrooxidation of methanol (a common fuel molecule) in O2-saturated 0.1 M KOH by adding 3 mL methanol at 400 s. As shown in Fig. 7c, an obvious response was observed for the 20% Pt/C catalyst after methanol was added. In contrast, no noticeable change happens in the oxygen-reduction current for Carbon-L and Carbon-S under the same conditions. This may be ascribed to the increased graphitization degree, which not only increases the electronic conductivity, but also enhances the corrosion resistance.65 Thus, ZIF-derived carbons exhibit a high selectivity for ORR with a remarkably good ability to avoid crossover effects, which is superior to the commercial Pt/C catalyst.
4. Conclusions
In summary, we have proposed an effective approach for in situ synthesis of nitrogen-doped porous carbons from the ZIF-7/glucose composites. Importantly, the addition of an environmentally friendly carbon source (glucose) not only improves the graphitization degree of samples, but also favors removal of Zn metal and zinc compound impurities from ZIFs, which make the resulting materials metal-free nitrogen-doped porous carbons. Compared to other porous carbons, Carbon-L exhibits not only very high electrocatalytic activity which is close to commercial 20% Pt/C, but also better stability and increased tolerance to the methanol crossover effects which is superior to the 20% Pt/C catalyst. Results indicate that both high electrical conductivity and the content of pyridinic N of the prepared Carbon-L play a key role in electrocatalytic activity for ORR, because the two factors significantly affect the electron transportation in electrodes and active sites for oxygen reduction. In addition, the graphene-like morphology, high BET SSA, and high porosity also contribute to enhanced activity for ORR. This work shows that not only ZIFs can be considered as a self-sacrificed precursor and nitrogen source, but also other nitrogen-containing MOFs may be alternative precursors. Owing to the versatility of MOF structures, the MOF-derived porous carbons would significantly broaden the family of nanoporous carbons with novel structures and multifunctional properties including electrocatalysts, sensors, supercapacitors and batteries.Acknowledgements
This work is supported by National 863 Programs (2013AA031901), National 973 Program (2011CB706900), National Scientific Research Funding (ZZ1304) and Outstanding Talent Funding from BUCT. We are thankful to Prof. X. M. Sun at BUCT for the RDE test.References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 145–154 CrossRef CAS.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- R. R. Adzic, J. Zhang, K. Sasaki, M. B. Vukmirovic, M. Shao, J. Wang, A. U. Nilekar, M. Mavrikakis, J. Valerio and F. Uribe, Top. Catal., 2007, 46, 249–262 CrossRef CAS.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- A. Morozan, S. Campidelli, A. Filoramo, B. Jousselme and S. Palacin, Carbon, 2011, 49, 4839–4847 CrossRef CAS.
- L. Wu, Y. Nabae, S. Moriya, K. Matsubayashi, N. M. Islam, S. Kuroki, M.-A. Kakimoto, J.-I. Ozaki and S. Miyata, Chem. Commun., 2010, 46, 6377–6379 RSC.
- H. Wang and H. Dai, Chem. Soc. Rev., 2013, 42, 3088–3133 RSC.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS.
- Z. Liu, G. Zhang, Z. Lu, X. Jin, Z. Chang and X. Sun, Nano Res., 2013, 6, 293–301 CrossRef CAS.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- D. Yu, Q. Zhang and L. Dai, J. Am. Chem. Soc., 2010, 132, 15127–15129 CrossRef CAS PubMed.
- P. Pachfule, V. M. Dhavale, S. Kandambeth, S. Kurungot and R. Banerjee, Chem.–Eur. J., 2013, 19, 974–980 CrossRef CAS PubMed.
- C. Ewels and M. Glerup, J. Nanosci. Nanotechnol., 2005, 5, 1345–1363 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 2691–2718 CAS.
- J. Lan, D. Cao, W. Wang and B. Smit, ACS Nano, 2010, 4, 4225–4237 CrossRef CAS PubMed.
- Z. Xiang, Z. Hu, D. Cao, W. Yang, J. Lu, B. Han and W. Wang, Angew. Chem., Int. Ed., 2011, 50, 491–494 CrossRef CAS PubMed.
- D. P. Cao, J. H. Lan, W. Wang and B. Smit, Angew. Chem., Int. Ed., 2009, 48, 4730–4733 CrossRef CAS PubMed.
- Y. Zhou, Z. Xiang, D. Cao and C.-J. Liu, Chem. Commun., 2013, 49, 5633–5635 RSC.
- Z. Xiang and D. Cao, Macromol. Rapid Commun., 2012, 33, 1184–1190 CrossRef CAS PubMed.
- S. Liu, Z. Xiang, Z. Hu, X. Zheng and D. Cao, J. Mater. Chem., 2011, 21, 6649–6653 RSC.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS.
- W. Chaikittisilp, K. Ariga and Y. Yamauchi, J. Mater. Chem. A, 2013, 1, 14–19 CAS.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- P. Pachfule, B. P. Biswal and R. Banerjee, Chem.–Eur. J., 2012, 18, 11399–11408 CrossRef CAS PubMed.
- A. Almasoudi and R. Mokaya, J. Mater. Chem., 2012, 22, 146–152 RSC.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- W. Chaikittisilp, M. Hu, H. Wang, H.-S. Huang, T. Fujita, K. C.-W. Wu, L. C. Chen, Y. Yamauchi and K. Ariga, Chem. Commun., 2012, 48, 7259–7261 RSC.
- P. Su, L. Jiang, J. Zhao, J. Yan, C. Li and Q. Yang, Chem. Commun., 2012, 48, 8769–8771 RSC.
- D. Yuan, J. Chen, S. Tan, N. Xia and Y. Liu, Electron. Commun., 2009, 11, 1191–1194 CrossRef CAS.
- J. Hu, H. Wang, Q. Gao and H. Guo, Carbon, 2010, 48, 3599–3606 CrossRef CAS.
- S. J. Yang, S. Nam, T. Kim, J. H. Im, H. Jung, J. H. Kang, S. Wi, B. Park and C. R. Park, J. Am. Chem. Soc., 2013, 135, 7394–7397 CrossRef CAS PubMed.
- X. Wang, X. Fang, X. Guo, Z. Wang and L. Chen, Electrochim. Acta, 2013, 97, 238–243 CrossRef CAS.
- G. Xu, B. Ding, L. Shen, P. Nie, J. Han and X. Zhang, J. Mater. Chem. A, 2013, 1, 4490–4496 CAS.
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 2864–2867 CrossRef CAS PubMed.
- N. L. Torad, M. Hu, Y. Kamachi, K. Takai, M. Imura, M. Naito and Y. Yamauchi, Chem. Commun., 2013, 49, 2521–2523 RSC.
- A. Banerjee, R. Gokhale, S. Bhatnagar, J. Jog, M. Bhardwaj, B. Lefez, B. Hannoyer and S. Ogale, J. Mater. Chem., 2012, 22, 19694–19699 RSC.
- J.-D. Xiao, L.-G. Qiu, X. Jiang, Y.-J. Zhu, S. Ye and X. Jiang, Carbon, 2013, 59, 372–382 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- G. Goenaga, S. Ma, S. Yuan and D.-J. Liu, ECS Trans., 2010, 33, 579–586 CAS.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- D. Zhao, J.-L. Shui, C. Chen, X. Chen, B. M. Reprogle, D. Wang and D.-J. Liu, Chem. Sci., 2012, 3, 3200–3205 RSC.
- S. Ma, G. A. Goenaga, A. V. Call and D. J. Liu, Chem.–Eur. J., 2011, 17, 2063–2067 CrossRef CAS PubMed.
- Q. Yang, P. Su, H. Xiao, J. Zhao, Y. Yao, Z.-G. Shao and C. Li, Chem. Sci., 2013, 4, 2941–2946 RSC.
- B. Wang, J. Power Sources, 2005, 152, 1–15 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 10186–10191 CrossRef CAS PubMed.
- Y. Li, F. Liang, H. Bux, W. Yang and J. Caro, J. Membr. Sci., 2010, 354, 48–54 CrossRef CAS.
- S.-L. Jin, H.-G. Deng, L. Zhan, W.-M. Qiao and L.-C. Ling, New Carbon Mater., 2012, 27, 87–92 CrossRef CAS.
- E. A. Fletcher, Ind. Eng. Chem. Res., 1999, 38, 2275–2282 CrossRef CAS.
- G. Lalande, D. Guay, J. Dodelet and G. Denes, J. Electrochem. Soc., 1998, 145, 2411–2418 CrossRef.
- S. Yang, X. Feng, X. Wang and K. Mullen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- C. Zhu and S. Dong, Nanoscale, 2013, 5, 1753–1767 RSC.
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593–5597 CrossRef CAS PubMed.
- Y. Zhao, C. Hu, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371–11375 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, R. Schlögl and D. S. Su, Chem. Commun., 2008, 4891–4893 RSC.
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. V. Dommele, J. H. Bitter, M. Santa, G. Grundmeier, M. Bron and W. Schuhmann, J. Phys. Chem. C, 2009, 113, 14302–14310 CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- H. Tang, H. Yin, J. Wang, N. Yang, D. Wang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 5585–5589 CrossRef CAS PubMed.
- S. Wang, D. Yu, L. Dai, D. W. Chang and J.-B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ee42799d |
| This journal is © The Royal Society of Chemistry 2014 |