 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile approach for the synthesis of monolithic hierarchical porous carbons – high performance materials for amine based CO2 capture and supercapacitor electrode†
Luis
Estevez‡
a,
Rubal
Dua‡
b,
Nidhi
Bhandari
a,
Anirudh
Ramanujapuram
a,
Peng
Wang
*b and
Emmanuel P.
Giannelis
*a
aDepartment of Materials Science and Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: epg2@cornell.edu
bWater Desalination and Reuse Center, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: peng.wang@kaust.edu.sa
First published on 3rd May 2013
Abstract
An ice templating coupled with hard templating and physical activation approach is reported for the synthesis of hierarchically porous carbon monoliths with tunable porosities across all three length scales (macro- meso- and micro), with ultrahigh specific pore volumes ∼11.4 cm3 g−1. The materials function well as amine impregnated supports for CO2 capture and as supercapacitor electrodes.
Broader contextThe move from a fossil fuel based energy economy to a renewable energy based economy is currently underway. In order to realize this goal, it is vital to mitigate the environmental affects of the current fossil fuel infrastructure, while advancing the effectiveness of renewable energy alternatives. Advances in novel hierarchical porous carbon (HPC) materials with high surface area and pore volume can address both goals. By providing a scaffold support for amine impregnation, HPCs can be used to fabricate effective CO2 capture materials; while the open, vascular structure of HPCs can be used to produce effective EDLC supercapacitors for energy storage and high power output. In this work we developed a facile and scalable synthesis technique for producing a family of highly tunable HPC materials. The HPCs showed an excellent capability to be used as effective EDLC electrodes with a maximum charge storage capability of 6 W h kg−1 and a maximum power density of 14 kW kg−1; and an exceptional ability when used as scaffolds for amine based CO2 capture, achieving a maximum CO2 capacity of 4.2 mmol g−1. |
Recently, hierarchical porous carbons (HPCs) possessing well-defined macropores and interconnected meso- and micropores, have attracted much attention.1–18 HPCs can combine in one system: improved mass transport facilitated by the macropores and high surface area and pore volume from micro-/mesopores.19–22 Such hierarchically porous carbons provide better accessibility and active sites for several energy and environmental applications including electrode materials for batteries,9,21 supercapacitors,13,19,20,23,24 fuel cells,25,26 and capacitive deionization,27,28 as well as sorbents for CO2 capture.6
A large number of techniques have been explored for the synthesis of HPCs.29,30 In general, most of the techniques for producing carbons combining macro- and mesoporosity are based on a dual templating strategy, where two templates with dimensions at different length scales are combined to generate the multimodal pores. These techniques either involve two hard templates31,32 or a combination of hard and soft templates.33,34 Removal of the templates, either through decomposition or etching leaves behind a porous scaffold. Other techniques involve template replication of hierarchical inorganic materials,35,36 with hierarchical silica being the most commonly used template. The sol–gel method is another widely used technique for the production of hierarchical carbon, commonly termed as either carbon aerogels29,37 or carbon cryogels.38,39 Ice templating has been also explored for assembling micropore–mesopore dominated carbon materials, such as CNTs, into 3D interconnected hierarchical carbon materials.26,40
A major challenge to date has been the development of HPCs, which can exhibit high surface areas, pore volumes and porosities at all three different length scales: macro-, meso-, and micro, in a simple material platform. Additional challenges with the synthesis techniques include the requirement to synthesize porous inorganic materials or special nanoparticles as hard templates, which involve multiple steps and are thus time consuming and costly. Furthermore, most of the soft templates used are based on surfactants and block-copolymers, which are rather expensive and non-renewable.30 Moreover, the size of the mesopores can also be difficult to tune because of aggregation of the nanoparticles in the polymerizing carbon precursor matrix.41 In addition, techniques like sol–gel suffer from the critical drawback associated with the long synthesis period required for gelation, solvent exchange and supercritical drying in the case of aerogels.29
Here we demonstrate a novel strategy to synthesize HPCs with high surface areas, large pore volumes (up to 2096 m2 g−1 and 11.4 cm3 g−1, respectively), and more importantly, tunable micro/meso/macro porosities. Our approach is based on combining, ice templating alongside a hard template (colloidal silica), and physical activation for generating interconnected macro-, meso-, and microporosity, respectively. We also demonstrate that the newly developed HPCs are promising candidates for amine supported sorbents for CO2 capture and as electrode materials for an electric double-layer capacitor (EDLC). This new, simple, and inexpensive technique provides a suitable platform for producing state of the art HPCs for these applications and beyond. In addition, because of their easy tunability, the new HPCs provide excellent model systems to understand the effect of different types of porosity (e.g. macro, meso and micro) on their performance in a series of applications.
Ice templating42,43 has been used previously to synthesize macroporous44 and even hierarchical materials45,46 due to its simplicity and versatility. However this is the first time ice-templating has been used to synthesize HPCs with tunable porosity by combining it with hard templating and physical activation. Our approach offers several key additional advantages: (1) the range, size and extent of porosity can be easily controlled. For example, the distribution of the mesopores and the extent of mesoporosity can be simply tuned by either using silica of different size or varying the silica to carbon precursor weight ratio. The excellent control of the mesopore size distribution (vide infra) alleviates the need of using costly surfactants,41 fast stirring speeds and/or dilute carbon precursor concentrations.47 In addition, as has been shown in literature,48,49 the macropore size and structure can be controlled by controlling the dipping rate (of the colloidal silica–glucose mixture in liquid nitrogen), concentration of carbon precursor, concentration and size of colloidal particle and by other established techniques. Furthermore the activation reaction conditions can control the microposity. (2) The synthesis is simple, reproducible, greener, and uses inexpensive and widely available starting materials (water/ice for macropores, silica for mesopores, CO2 for micropores), all of which make the process highly scalable.
Scheme 1 shows a graphical representation of the synthesis technique (the details of the synthesis technique are provided in the ESI†). The solidification of water, as a result of plunging the mixture into the liquid nitrogen, expels both the silica particles and glucose molecules away from the growing ice crystals. The ice is then simply removed by sublimation. Care must be taken during the freeze-drying process to prevent the ice templated glucose–silica material from melting. The glucose–silica composite scaffold (shown in Fig. S1†) is then carbonized, resulting in a macroporous carbon–silica structure. The network of interconnected macropores formed after sublimation (Fig. S1a†) remains intact during pyrolysis and after silica etching (Fig. S1b†). Fig. 1(a, c and d) shows the SEM images of the resultant HPC material after the silica etching using NaOH. The SEM images are consistent with an HPC scaffold whose macroporous walls are made up of an interconnected mesoporous carbon. This is further confirmed by TEM images (Fig. 1(e and f) and Fig. S2†) that reveal an extensive network of mesopores. The mesopores seen in the TEM images are in good agreement with the N2 adsorption data (vide infra).
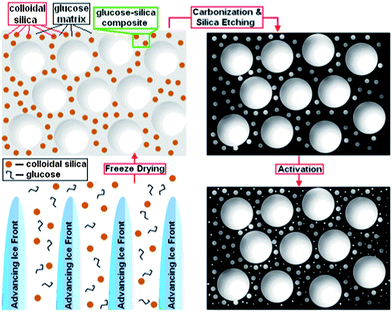 | ||
| Scheme 1 Schematic showing the basic steps and the material thus produced after each step. The four steps shown clockwise from bottom left are: (1) (side view) ice templating – ice front movement through the glucose and colloidal silica aqueous suspension forming ice crystals, (2) (top view) freeze drying – the glucose–silica composite material with macropores after removal of ice, (3) (top view) carbonization and silica etching – macro-mesopore dominated carbon and finally, (4) (top view) physical activation – hierarchical carbon. Note: for simplification, the pores are not shown as interconnected. For convenience the samples are denoted by KCU-C x–y–z, where x represents the average colloidal silica size (nm), y represents the mass ratio of silica to glucose and z represents the time (h) for CO2 activation. | ||
 | ||
| Fig. 1 (a–d) SEM images of HPC scaffolds. Images show: (a) the monolithic character of the HPCs, (b) the interconnected macroporous structure evident before etching of the colloidal silica and (c) a similar interconnected macroporous structure after etching. (d) Higher resolution SEM shows a roughened morphology consistent with mesopores. Representative TEM images of (e) KCU-C 12–1 (f) KCU-C 12–2 (scale bars for TEM images are 200 nm for the larger images and 20 nm for the inset images). | ||
The surface area and pore volume of the macropores measured by mercury porosimetry are 11 m2 g−1 and 9.1 cm3 g−1 respectively for the KCU-C 4–1 sample. Care was taken to vary the pressure applied under mercury porosimetry so that any pores under 50 nm were not analyzed and incorporated into the results. The macropore size distribution obtained via mercury porosimetry measurements (Fig. S3†) is in good agreement with those seen in SEM images in Fig. 1. These results are consistent with expectations from previous work44 that within the range of colloidal silica nanoparticles used, macroporosity is controlled mostly by the speed of ice formation (i.e. freezing speed).
The total BET surface area and pore volume (excluding macropores) for a series of samples is summarized in Table 1. It is important to note that these pore volumes, calculated from N2 sorption, for the KCU-C x–2 samples (from 3.0 to 4.1 cm3 g−1) are among the highest values measured for mesoporous carbon materials.50,51 In contrast to macroporosity, the silica to glucose ratio and the size of silica affects the mesoporosity. For HPCs synthesized using silica to glucose ratio of 1 the surface area and pore volume decrease as the silica particle size increases. No such dependence is seen for the silica to glucose ratio of 2, where the surface area is generally the same regardless of silica size. For a given silica particle size, the surface area and the pore volume increase as the silica to glucose ratio increases from 1 to 2. The percent increase in surface area is much more pronounced for the 12 and 20 nm silica particles (the increase for all particle sizes are 1, 11, 36 and 53% for the 4, 8, 12 and 20 nm silica, respectively). The specific surface area and extent of microporosity can be further increased by CO2 activation. Under CO2 activation, the duration and flow rate of CO2 are important factors in determining the increase in microporosity and surface area. CO2 activation was performed on the KCU-C 4–1 sample, for different time holds at 950 °C, at a constant CO2 flow rate of 50 cm3 min−1. 3–4 hours of activation time was found to be optimal. The thus synthesized KCU-C 4–1–4 sample had a BET surface area of 2096 m2 g−1 and pore volume of 3.0 cm3 g−1, comprising of increased microporous content and broadened mesoporosity (see Fig. S4†).
| Sample name | BET surface area (m2 g−1) | N2 adsorption pore volume (cm3 g−1) |
|---|---|---|
| KCU-C 4–1–4 | 2096 | 3.0 |
| KCU-C 4–1 | 1316 | 2.3 |
| KCU-C 4–2 | 1327 | 4.1 |
| KCU-C 8–1 | 1129 | 2.0 |
| KCU-C 8–2 | 1265 | 3.6 |
| KCU-C 12–1 | 893 | 1.9 |
| KCU-C 12–2 | 1216 | 3.0 |
| KCU-C 20–1 | 841 | 1.9 |
| KCU-C 20–2 | 1289 | 3.7 |
A distinct advantage of our approach is the ability to easily control the pore sizes. For example, the size of the mesopores can be easily tuned by using different size silica particles. Fig. 2 shows the pore size distribution calculated using the BJH model for a series of HPCs, synthesized using different size silica nanoparticles (4, 8, 12 and 20 nm). In the past, special efforts were devoted to ensure that the pore size of the resulting carbons is the same as the size of the starting template.41,47 Such efforts typically focused on preventing aggregation of the hydrophilic silica nanoparticles within the pyrolyzing (and becoming increasingly hydrophobic) carbon precursor matrix. In our approach, the instantaneous locking of the silica nanoparticles within the glucose matrix and subsequent carbonization minimize such aggregation and phase separation. Note the high fidelity of the process in generating mesopores especially for the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of silica to glucose; the size of the resulting mesopores corresponds well with the size of the silica nanoparticles used in the process (Fig. 2). This fidelity holds true for the 20 nm silica particles even for SiO2/glucose ratio of 2. However at this higher silica/glucose ratio of 2, as the particle size decreases, aggregation of the particles during freezing leads to larger pore sizes and this is especially true for the 4 nm silica particles. Note that as the number of silica nanoparticles (relative to the glucose) increases, the amount of glucose present between the nanoparticles decreases, resulting in aggregation at higher ratios.
:1 ratio of silica to glucose; the size of the resulting mesopores corresponds well with the size of the silica nanoparticles used in the process (Fig. 2). This fidelity holds true for the 20 nm silica particles even for SiO2/glucose ratio of 2. However at this higher silica/glucose ratio of 2, as the particle size decreases, aggregation of the particles during freezing leads to larger pore sizes and this is especially true for the 4 nm silica particles. Note that as the number of silica nanoparticles (relative to the glucose) increases, the amount of glucose present between the nanoparticles decreases, resulting in aggregation at higher ratios.
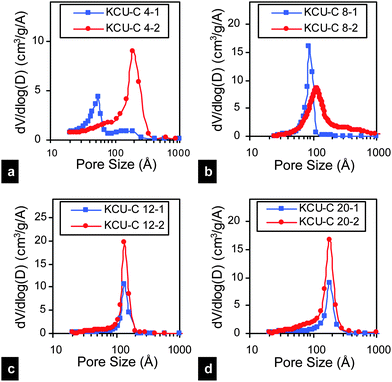 | ||
| Fig. 2 BJH pore size distributions for the KCU-C x–y samples. The BJH pore size distributions of the activated HPCs can be found in the ESI (Fig. S4†). | ||
Our approach also offers the possibility to easily make HPCs with bimodal mesoporosity distribution, by merely starting with two different sized silica nanoparticles. The bimodal nature of the mesoporosity can be clearly seen, in the pore size distribution shown in Fig. S5 in the ESI,† for the sample synthesized using 4 nm and 20 nm silica.
Another advantage of this approach is the flexibility it offers to produce carbon scaffolds either in powder form or as monoliths of desired shape and size, just by altering the mold used (see Fig. S6†). SEM imaging and adsorption measurements confirm that the monoliths possess surface area, specific pore volume and pore size distribution similar to the powder samples. The apparent density of the cylindrical monoliths was found to be approximately 0.09 g cm−3, by measuring the bulk samples' weight and apparent volume, and was confirmed via Hg porosimetry. The modulus of the monoliths evaluated using compression testing was ∼1 MPa, consistent with other porous carbon materials.52 Further discussion of the mechanical properties can be found in Fig. S7.†
Lately there has been much interest in developing multi-modal (macropore and mesopore dominated) support materials (metal oxides53–55) for amine based CO2 capture. However, the synthesis of most of such porous metal oxide based sorbents involve the use of costly non-renewable chemicals such as surfactants or pore expanders, which limits their use for large scale CO2 capture. Our HPCs with their inherent high pore volumes and specifically, the samples with large pore sizes appear to be excellent alternative candidates for amine based CO2 capture. Thus, a KCU-C 4–3 sample, with ultralarge mesopores of ∼50 nm, was synthesized (Fig. S8†), impregnated with polyethyleneimine (PEI), achieving a PEI loading of 73 wt% and its CO2 capture capacity was briefly evaluated. As shown in Fig. 3(a), a sharp weight gain was observed after the sorbent was exposed to CO2 in the first 5 min and 97% of the adsorption (4.1 mmol g−1) was obtained after 10 min. A capacity of up to 4.2 mmol g−1 was achieved after 45 min, which is among the highest reported value for amine-impregnated carbon-based sorbents.56 More importantly, for dilute CO2 (10% CO2–90% N2 gas mixture), the sorption capacity measured under dry and moist conditions was the same, unlike physisorption of moist dilute CO2 in microporous carbons, which is severely effected by the presence of moisture.57
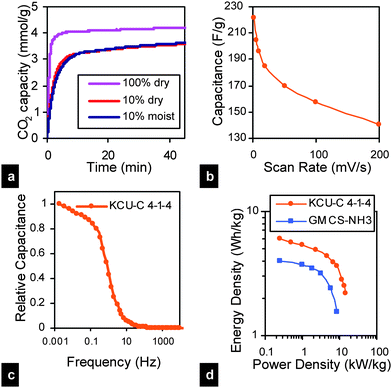 | ||
| Fig. 3 (a) CO2 sorption capacity of PEI–KCU-C 4–3 (73 wt% PEI) composite, (b) variation of specific capacitance with sweep rate, measured from cyclic voltammetry data for KCU-C 4–1–4 in 1 M H2SO4 within the potential range 0–1 V (vs. Ag/AgCl), (c) capacitive frequency response of KCU-C 4–1–4 measured in symmetrical two electrode configuration (d) Ragone plot for KCU-C 4–1–4 and GMCS–NH3 (NH3 treated, hierarchical, graphene mesoporous carbon spheres13) measured in symmetrical two electrode configuration using different current densities in 1 M H2SO4 within the potential range 0–1 V. | ||
The HPCs were also briefly evaluated as electrodes for supercapacitors. The advantage of HPCs for this application is that the presence of mesopores and macropores may provide pathways for fast ionic transport while the micropores provide sites for ion sorption.20,22 For a material to be used successfully as electrode material, it must have good electrical conductivity as well. The electrical conductivity measured using a four point probe was in the order of 1 S cm−1 for the various KCU-C materials. Using the KCU 4–1–4 sample in a three electrode setup with 1 M H2SO4 as the electrolyte, a specific capacitance of 221 F g−1 was obtained at a scan rate of 2 mV s−1, with 64% retention even at a two orders of magnitude higher scan rate of 200 mV s−1.
These values compare favorably well with the values reported in the literature for other hierarchical carbons.1,12 The sample also shows promising capacitive frequency response Fig. 3(c), calculated from electrochemical impedance spectroscopy data58 (Fig. S9(b)†), retaining 50% of its maximum capacitance even when the frequency increases to 0.73 Hz. Symmetric cells prepared from KCU-C 4–1–4 could deliver a specific energy density upto the order of 6 W h kg−1 and a power density in the range of 14 kW kg−1, measured using galvanostatic charge–discharge testing. For comparison, we include the performance of another hierarchical carbon, NH3 treated, graphene mesoporous carbon spheres.13 Further EDLC data for KCU-C 4–1–4 are presented in the ESI (Fig. S9†). Detailed investigations on use of KCU-Cs as support material for amine based CO2 capture and as electrode materials for supercapacitors are currently in progress.
In summary, we report a novel approach to synthesize HPCs, which combine macro-, meso- and microporosity in a simple material platform. Our approach integrates ice templating with hard templating and physical activation to produce HPCs with interconnected porous structure, large surface area, and pore volume. The technique offers tight control and tunability of porosity (macro- meso- and microscale) in terms of both size and extent. It also offers the ability to make HPC monoliths of desired shape and size. The hierarchical carbons show excellent performance as – sorbents for amine based CO2 capture and as electrode materials for supercapacitors.
This work is supported in part by award no. KUS-C1-018-02 made by King Abdullah University of Science and Technology (KAUST), KAUST baseline fund, The National Science Foundation (NSF) Grassroots GK-12 program (award no. DGE 1045513) and by the Energy Materials Center at Cornell (EMC2) – an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001086. This work made use of the Cornell Center for Materials Research Shared Facilities supported through the NSF MRSEC program (DMR-1120296). We also wish to thank Ritu Sahore (Cornell University) and Dr Hua Tan (KAUST Analytical Core Lab) for help with electrical conductivity measurement and microporosity characterization respectively, as well as Joerg Werner and Dr Genggeng Qi for helpful discussions.
Notes and references
- Y. Liang, D. Wu and R. Fu, Sci. Rep., 2013, 3, 1119 Search PubMed.
- D.-C. Guo, J. Mi, G.-P. Hao, W. Dong, G. Xiong, W.-C. Li and A.-H. Lu, Energy Environ. Sci., 2013, 6, 652–659 CAS.
- H. M. Sun, L. Y. Cao and L. H. Lu, Energy Environ. Sci., 2012, 5, 6206–6213 CAS.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- M. Oschatz, L. Borchardt, M. Thommes, K. A. Cychosz, I. Senkovska, N. Klein, R. Frind, M. Leistner, V. Presser, Y. Gogotsi and S. Kaskel, Angew. Chem., Int. Ed., 2012, 51, 7577–7580 CrossRef CAS.
- G. P. Hao, W. C. Li, D. Qian, G. H. Wang, W. P. Zhang, T. Zhang, A. Q. Wang, F. Schüth, H. J. Bongard and A. H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS.
- S. C. Wei, H. Zhang, Y. Q. Huang, W. K. Wang, Y. Z. Xia and Z. B. Yu, Energy Environ. Sci., 2011, 4, 736–740 CAS.
- M. C. Gutierrez, D. Carriazo, C. O. Ania, J. B. Parra, M. L. Ferrer and F. del Monte, Energy Environ. Sci., 2011, 4, 3535–3544 CAS.
- Z. L. Wang, D. Xu, J. J. Xu, L. L. Zhang and X. B. Zhang, Adv. Funct. Mater., 2012, 22, 3699–3705 CrossRef CAS.
- J. C. Lytle, J. M. Wallace, M. B. Sassin, A. J. Barrow, J. W. Long, J. L. Dysart, C. H. Renninger, M. P. Saunders, N. L. Brandell and D. R. Rolison, Energy Environ. Sci., 2011, 4, 1913–1925 CAS.
- C. X. Guo and C. M. Li, Energy Environ. Sci., 2011, 4, 4504–4507 CAS.
- Z. S. Wu, Y. Sun, Y. Z. Tan, S. Yang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 19532–19535 CrossRef CAS.
- Z. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 CAS.
- C. Xue, B. Tu and D. Zhao, Nano Res., 2009, 2, 242–253 CrossRef CAS.
- C. Xue, B. Tu and D. Zhao, Adv. Funct. Mater., 2008, 18, 3914–3921 CrossRef CAS.
- Y. Huang, H. Cai, D. Feng, D. Gu, Y. Deng, B. Tu, H. Wang, P. A. Webley and D. Zhao, Chem. Commun., 2008, 2641–2643 RSC.
- H.-J. Liu, X.-M. Wang, W.-J. Cui, Y.-Q. Dou, D.-Y. Zhao and Y.-Y. Xia, J. Mater. Chem., 2010, 20, 4223–4230 RSC.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373–376 CrossRef CAS.
- F. Xu, R. Cai, Q. Zeng, C. Zou, D. Wu, F. Li, X. Lu, Y. Liang and R. Fu, J. Mater. Chem., 2011, 21, 1970–1976 RSC.
- S. Wenzel, T. Hara, J. Janek and P. Adelhelm, Energy Environ. Sci., 2011, 4, 3342–3345 CAS.
- Z. Chen, J. Wen, C. Yan, L. Rice, H. Sohn, M. Shen, M. Cai, B. Dunn and Y. Lu, Adv. Energy Mater., 2011, 1, 551–556 CrossRef CAS.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 CAS.
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. J. Stephenson and D. Mitlin, Energy Environ. Sci., 2013, 6, 871–878 CAS.
- V. Flexer, N. Brun, O. Courjean, R. Backov and N. Mano, Energy Environ. Sci., 2011, 4, 2097–2106 CAS.
- K. Katuri, M. L. Ferrer, M. C. Gutierrez, R. Jimenez, F. del Monte and D. Leech, Energy Environ. Sci., 2011, 4, 4201–4210 CAS.
- R. T. Mayes, C. Tsouris, J. O. Kiggans Jr, S. M. Mahurin, D. W. DePaoli and S. Dai, J. Mater. Chem., 2010, 20, 8674–8678 RSC.
- C. Tsouris, R. Mayes, J. Kiggans, K. Sharma, S. Yiacoumi, D. DePaoli and S. Dai, Environ. Sci. Technol., 2011, 45, 10243–10249 CrossRef CAS.
- A. H. Lu, G. P. Hao, Q. Sun, X. Q. Zhang and W. C. Li, Macromol. Chem. Phys., 2012, 213, 1107–1131 CrossRef CAS.
- H. Nishihara and T. Kyotani, Adv. Mater., 2012, 24, 4473–4498 CrossRef CAS.
- G. S. Chai, I. S. Shin and J. S. Yu, Adv. Mater., 2004, 16, 2057–2061 CrossRef CAS.
- S. Zhang, L. Chen, S. Zhou, D. Zhao and L. Wu, Chem. Mater., 2010, 22, 3433–3440 CrossRef CAS.
- Y. Deng, C. Liu, T. Yu, F. Liu, F. Zhang, Y. Wan, L. Zhang, C. Wang, B. Tu, P. A. Webley, H. Wang and D. Zhao, Chem. Mater., 2007, 19, 3271–3277 CrossRef CAS.
- Z. Wang and A. Stein, Chem. Mater., 2008, 20, 1029–1040 CrossRef CAS.
- Z. Wang, F. Li, N. S. Ergang and A. Stein, Chem. Mater., 2006, 18, 5543–5553 CrossRef CAS.
- A. Taguchi, J. H. Smått and M. Lindén, Adv. Mater., 2003, 15, 1209–1211 CrossRef CAS.
- J. Biener, M. Stadermann, M. Suss, M. A. Worsley, M. M. Biener, K. A. Rose and T. F. Baumann, Energy Environ. Sci., 2011, 4, 656–667 CAS.
- H. Nishihara, S. R. Mukai and H. Tamon, Carbon, 2004, 42, 899–901 CrossRef CAS.
- S. L. Candelaria, R. Chen, Y. H. Jeong and G. Cao, Energy Environ. Sci., 2012, 5, 5619–5637 CAS.
- M. B. Bryning, D. E. Milkie, M. F. Islam, L. A. Hough, J. M. Kikkawa and A. G. Yodh, Adv. Mater., 2007, 19, 661–664 CrossRef CAS.
- S. Han and T. Hyeon, Chem. Commun., 1999, 1955–1956 RSC.
- S. Deville, E. Saiz, R. K. Nalla and A. P. Tomsia, Science, 2006, 311, 515–518 CrossRef CAS.
- M. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Mater., 2008, 20, 634–648 CrossRef.
- L. Estevez, A. Kelarakis, Q. Gong, E. H. Da'as and E. P. Giannelis, J. Am. Chem. Soc., 2011, 133, 6122–6125 CrossRef CAS.
- M. C. Gutiérrez, M. Jobbágy, N. Rapún, M. L. Ferrer and F. delMonte, Adv. Mater., 2006, 18, 1137–1140 CrossRef.
- M. Klotz, I. Amirouche, C. Guizard, C. Viazzi and S. Deville, Adv. Eng. Mater., 2012, 14, 1123–1127 CrossRef CAS.
- H. I. Lee, G. D. Stucky, J. H. Kim, C. Pak, H. Chang and J. M. Kim, Adv. Mater., 2011, 23, 2357–2361 CrossRef CAS.
- M. C. Gutiérrez, Z. Y. García-Carvajal, M. Jobbágy, F. Rubio, L. Yuste, F. Rojo, M. L. Ferrer and F. delMonte, Adv. Funct. Mater., 2007, 17, 3505–3513 CrossRef.
- S. Deville, C. Viazzi and C. Guizard, Langmuir, 2012, 28, 14892–14898 CrossRef CAS.
- A. H. Lu, W. Schmidt, B. Spliethoff and F. Schüth, Adv. Mater., 2003, 15, 1602–1606 CrossRef CAS.
- Y. Xia, Z. Yang and R. Mokaya, Nanoscale, 2010, 2, 639–659 RSC.
- L. Qiu, J. Z. Liu, S. L. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef.
- G. Qi, L. Fu, B. H. Choi and E. P. Giannelis, Energy Environ. Sci., 2012, 5, 7368–7375 CAS.
- G. Qi, Y. Wang, L. Estevez, X. Duan, N. Anako, A.-H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444–452 CAS.
- C. Chen, S.-T. Yang, W.-S. Ahn and R. Ryoo, Chem. Commun., 2009, 3627–3629 RSC.
- C. C. Hwang, Z. Jin, W. Lu, Z. Sun, L. B. Alemany, J. R. Lomeda and J. M. Tour, ACS Appl. Mater. Interfaces, 2011, 3, 4782–4786 CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS.
- C.-W. Huang, C.-T. Hsieh, P.-L. Kuo and H. Teng, J. Mater. Chem., 2012, 22, 7314–7322 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional SEM and optical images, data for Hg and N2 porosimetry, mechanical testing, CO2 sorption, EDLC testing. See DOI: 10.1039/c3ee40549d |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |