Improved cathode materials for microbial electrosynthesis
Tian
Zhang†
a,
Huarong
Nie†
b,
Timothy S.
Bain
a,
Haiyun
Lu
b,
Mengmeng
Cui
b,
Oona L.
Snoeyenbos-West
a,
Ashley E.
Franks
a,
Kelly P.
Nevin
a,
Thomas P.
Russell
*b and
Derek R.
Lovley
*a
aDepartment of Microbiology, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: dlovley@microbio.umass.edu; Fax: +1 413-545-1578; Tel: +1 413-545-9651
bDepartment of Polymer Science and Engineering, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: russell@mail.pse.umass.edu; Fax: +1 413-577-1510; Tel: +1 413-545-2680
First published on 1st November 2012
Abstract
Microbial electrosynthesis is a promising strategy for the microbial conversion of carbon dioxide to transportation fuels and other organic commodities, but optimization of this process is required for commercialization. Cathodes which enhance electrode–microbe electron transfer might improve rates of product formation. To evaluate this possibility, biofilms of Sporomusa ovata, which are effective in acetate electrosynthesis, were grown on a range of cathode materials and acetate production was monitored over time. Modifications of carbon cloth that resulted in a positive-charge enhanced microbial electrosynthesis. Functionalization with chitosan or cyanuric chloride increased acetate production rates 6–7 fold and modification with 3-aminopropyltriethoxysilane gave rates 3-fold higher than untreated controls. A 3-fold increase in electrosynthesis over untreated carbon cloth cathodes was also achieved with polyaniline cathodes. However, not all strategies to provide positively charged surfaces were successful, as treatment of carbon cloth with melamine or ammonia gas did not stimulate acetate electrosynthesis. Treating carbon cloth with metal, in particular gold, palladium, or nickel nanoparticles, also promoted electrosynthesis, yielding electrosynthesis rates that were 6-, 4.7- or 4.5-fold faster than the untreated control, respectively. Cathodes comprised of cotton or polyester fabric treated with carbon nanotubes yielded cathodes that supported acetate electrosynthesis rates that were ∼3-fold higher than carbon cloth controls. Recovery of electrons consumed in acetate was ∼80% for all materials. The results demonstrate that one approach to increase rates of carbon dioxide reduction in microbial electrosynthesis is to modify cathode surfaces to improve microbe-electrode interactions.
Broader contextMicrobial electrosynthesis is a recently conceived bioenergy strategy in which microorganisms use electrons derived from electrodes to reduce carbon dioxide to organic products that are excreted from the cells. Any form of electrical energy can power microbial electrosynthesis, but when electricity is obtained from solar technologies and water is the source of electrons, microbial electrosynthesis is an artificial form of photosynthesis with many potential advantages over biomass-based energy strategies. This study demonstrates that there are several strategies for modifying cathode surface properties that can enhance rates of microbial electrosynthesis. |
Introduction
Microbial electrosynthesis is a novel bioenergy strategy in which electricity serves as the energy source for microbial reduction of carbon dioxide to multi-carbon organic molecules that can serve as transportation fuels or other useful organic commodities.1–4 The conversion of electrical energy to extracellular, multi-carbon products represents an attractive option for energy storage and distribution.5 When the electricity for microbial electrosynthesis is derived from solar sources, microbial electrosynthesis represents an artificial form of photosynthesis with many potential advantages over bioenergy strategies that rely on biological photosynthesis.3,4,6–8 Initial proof-of-concept studies demonstrated that acetogenic bacteria, such as Sporomusa and Clostridium species, could accept electrons from negatively charged graphite electrodes as the electron donor for the reduction of carbon dioxide to acetate that was released extracellularly.3,4Clostridium ljungdahlii, one of the strains capable of electrosynthesis, can be genetically manipulated,9 offering the promise of generating products with higher value than acetate.3Commercialization of microbial electrosynthesis will require optimization and scaling. One key feature is enhancing electron exchange at the cathode surface while maintaining low costs. Although there have been substantial improvements in understanding how microorganisms transfer electrons to electrodes, the mechanisms for electron transfer from electrodes to microbes are still poorly understood.1,2,8,10,11 Thus, initial approaches to improve cathode design are likely to be largely empirical, but still potentially productive. For example, cathode-driven anaerobic respiration by Geobacter sulfurreducens12 was enhanced by switching from graphite to different forms of stainless steel and modifying surface roughness.13–15
A number of approaches that can improve microbe–electrode electron exchange (Scheme 1) have been identified in studies of anode material studies for biosensors and microbial fuel cells.16–22 For example, a positive charge at the electrode surface, established with ammonia gas treatment,23 chitosan,24–29 cyanuric chloride (CC),30–33 3-aminopropyltriethoxysilane (APTES),34–37 melamine38,39 or polyaniline (PANi),40–46 has the potential of leading to better electron transfer. Thin layers of metal catalysts, such as Au,17,47–50 Pd17,47,50–52 or Ni,17,53–56 can reduce the activation energy threshold of electron transfer from electrodes to bacteria. Fabrics coated with carbon nanotubes offer an open, three-dimensional, conductive matrix for microbial growth.57–61
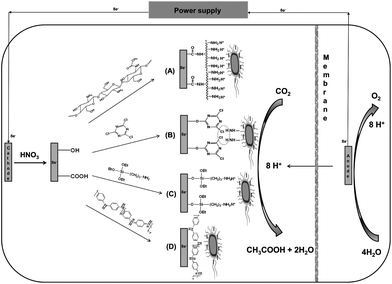 | ||
| Scheme 1 Schematic of the cathode configuration and electron-consumption between S. ovata and electrode for the electrosynthesis of acetate. (A) Carbon cloth cathode coated by chitosan. (B) Carbon cloth cathode coated with cyanuric chloride. (C) Carbon cloth cathode coated with 3-aminopropyltriethoxysilane. (D) Carbon cloth cathode coated with PANi. | ||
Here, we report on a study of the performance of diversity of cathode materials for microbial electrosynthesis by Sporomusa ovata. These results suggest that several modifications that provide a positive charge at the cathode surface can effectively enhance microbial electrosynthesis rates.
Results and discussion
Carbon cloth was a suitable cathode material for microbial electrosynthesis with Sporomusa ovata (Fig. 1 and Table 1). There was a steady consumption of current over time with the concomitant production of acetate (Fig. 1B). Recovery of electrons consumed in acetate was high (74 ± 14%; mean ± standard deviation, n = 3). Acetate production could be attributed to cells attached to the cloth fibers (Fig. 1C) which live/dead staining indicated were metabolically active (Fig. 1D).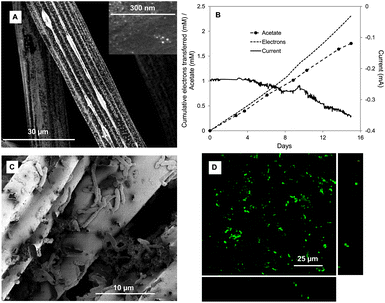 | ||
| Fig. 1 S. ovata electrosynthesis of acetate with untreated carbon cloth cathode. (A) SEM image of the untreated carbon cloth. (B) Electron consumption, acetate and current production over time (C) SEM image of S. ovata on the cathode. (D) Confocal scanning laser microscopic image of S. ovata on the cathode. Results shown are from a representative example of three replicate cultures. | ||
| Carbon cloth cathode treatment | Average current consumption densitya (mA m−2) | Acetatea (mM m−2 day−1) | Coulombic efficiencya |
|---|---|---|---|
| a Each value is the mean and standard deviation of three replicates. | |||
| Carbon cloth | −71 ± 11 | 30 ± 7 | 76 ± 14 |
| Chitosan | −475 ± 18 | 229 ± 56 | 86 ± 12 |
| Cyanuric chloride | −451 ± 79 | 205 ± 50 | 81 ± 16 |
| 3-Aminopropyltriethoxysilane | −206 ± 11 | 95 ± 20 | 82 ± 11 |
| Polyaniline | −189 ± 18 | 90 ± 22 | 85 ± 7 |
| Melamine | −69 ± 9 | 31 ± 8 | 80 ± 15 |
| Ammonia | −60 ± 21 | 28 ± 14 | 82 ± 8 |
| Au | −388 ± 43 | 181 ± 44 | 83 ± 14 |
| Pd | −320 ± 64 | 141 ± 35 | 79 ± 16 |
| Ni | −302 ± 48 | 136 ± 33 | 80 ± 15 |
| CNT–cotton | −220 ± 1 | 102 ± 25 | 83 ± 10 |
| CNT–polyester | −210 ± 13 | 96 ± 24 | 82 ± 8 |
Cathode modifications to confer positive surface charge
Gram negative microorganisms like S. ovata typically have a negative outer-surface charge.62,63 The surface charge of untreated carbon cloth is neutral.64 Therefore, strategies for generating a positively charged cathode surface were evaluated to determine if this approach would promote better electronic interaction between the cells and the cathode.Chitosan, an amino- and hydroxyl-group rich polysaccharide, is one of the most commonly used natural biopolymers for enzyme immobilization65,66 or the dispersion of nanoparticles67 in biosensors or microbial fuel cells27,28 due to its biocompatibility, nontoxicity, film-forming ability, high water permeability, excellent mechanical strength and low cost.26,29 Chitosan was bound to the carbon cloth via the reaction between –COOH groups on the electrode surface and –NH2 groups on the chitosan (Scheme 1A). Scanning electron microscopy revealed that a thin layer of chitosan covered the entire electrode surface, with pore sizes suitable for microbial access (Fig. 2A). The rate of acetate production via microbial electrosynthesis (Fig. 2B and Table 1) was 7.6-fold higher than with the unmodified carbon cloth electrode (Fig. 1B). Electron recovery in acetate remained high with 86% ± 12% of the electrons consumed recovered in acetate (Fig. 2B). Confocal laser-scanning fluorescence microscopy (Fig. 2C) revealed a more than 9-fold higher cell density on the chitosan modified cathode 3.02 ± 1.95 × 107 cells per cm−2 than the untreated cloth 3.98 ± 1.24 × 106 cells per cm−2, which may account for the higher rates of electrosynthesis.
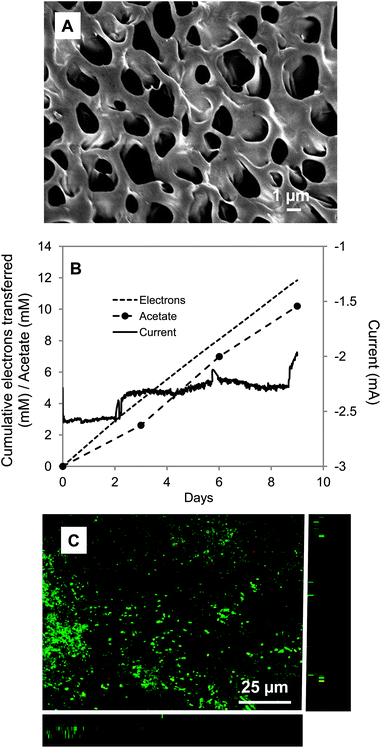 | ||
| Fig. 2 S. ovata electrosynthesis of acetate with chitosan-coated carbon cloth cathode. (A) SEM image of the chitosan-coated carbon cloth. (B) Electron consumption, acetate and current production over time. (C) Confocal scanning laser microscopic image of S. ovata on the cathode. Results shown are from a representative example of three replicate cultures. | ||
Cyanuric chloride (2,4,6-trichloro-1,3,5-triazine) has been widely used to modify graphite electrodes to promote the attachment of enzymes.30–33 In addition to providing an overall positive charge, there is the possibility that chlorines that have not reacted with the carbon cloth will react with functional groups on the cell surface, such as amino groups of surface-exposed proteins, to promote absorption of bacteria onto the electrode surface.68 X-ray photoelectron spectroscopy confirmed the binding of cyanuric chloride to carbon cloth (Fig. 3A). Edges corresponding to N 1s (binding energy, 400 eV), Cl 2s (binding energy, 269.6 eV) and Cl 2p (binding energy, 197.6 eV) were apparent in the profiles of the cyanuric chloride-modified carbon cloth, whereas only carbon and oxygen edges, corresponding to C 1s (binding energy, 284.6 eV) and O 1s (binding energy. 532 eV), were observed in the spectrum of the untreated carbon cloth (Fig. 3A). The rate of acetate production via electrosynthesis with the cyanuric chloride-treated cloth was 6.8-fold higher than for untreated cloth with a recovery of 81% ± 16% (n = 3) of the electrons consumed recovered in acetate (Fig. 3B and Table 1).
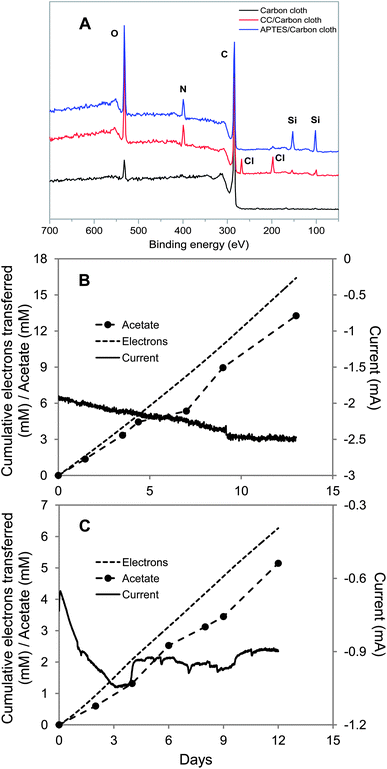 | ||
| Fig. 3 S. ovata electrosynthesis of acetate with carbon cloth cathodes coated with cyanuric chloride or 3-aminopropyltriethoxysilane. (A) XPS spectra untreated and treated carbon cloth. (B) Electron consumption, acetate and current production over time with the cyanuric chloride-coated carbon cloth. (C) Electron consumption, acetate and current production over time with the 3-aminopropyltriethoxysilane-coated carbon cloth. Results shown are from a representative example of three replicate cultures. | ||
3-Aminopropyltriethoxysilane (APTES) is commonly used for surface functionalization in biosensors because the silane group can covalently bind to the silicon oxide substrate and amine functionality can promote the adsorption of negatively charged proteins or other biomolecules.34–36,69 Hydroxyl groups exposed on the surface of HNO3-pretreated carbon cloth electrode are expected to covalently bind APTES to the electrode surface (Scheme 1C). Evidence for the attachment of APTES to the carbon cloth was provided by the appearance of two edges at 192.8 and 101.6 eV arising from the Si 2s and Si 2p, as well as N 1s edge (400 eV), in the XPS spectra after surface modification (Fig. 3A). Acetate electrosynthesis rates were 3-fold higher than those with the untreated carbon cloth (Fig. 3C and Table 1) with a recovery of electrons consumed in acetate of 82% ± 11%.
Polyaniline (PANi), an organic conducting polymer, has been used to modify anodes in microbial fuel cells to improve performance, due to its high electrical conductivity, ease of synthesis, and chemical stability.40,41,43 Electrospinning is a straightforward technique for fabricating three-dimensional scaffolds with high surface to volume ratios, significant fiber interconnectivity, and microscale porosity.45,70,71 Polymer fibers, obtained by using the electrospinning technique, afford a nanofibrous scaffold with strong adsorbability and abundant space for biomacromolecules.70 PANi–PAN was prepared by electrospinning with coaxial polymer nanofibers of PANi and polyacrylonitrile (PAN). Microporous composite mats of PANi–PAN were obtained (Fig. 4A). The rate of acetate production with PANi–PAN cathodes was 3-fold higher than for the control carbon cloth (Fig. 4B and Table 1) with a recovery of electrons consumed in acetate production of 85% ± 7%.
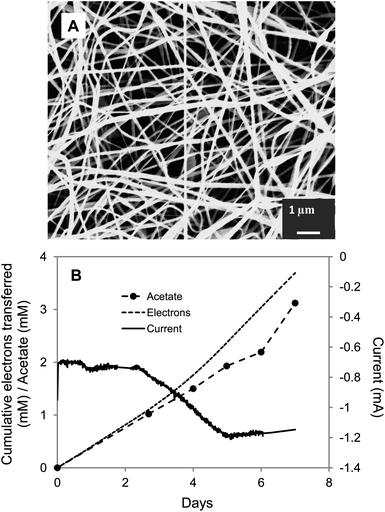 | ||
| Fig. 4 S. ovata electrosynthesis of acetate with carbon cloth cathode coated with PANi–PAN. (A) SEM image of the PANi–PAN coated carbon cloth. (B) Electron consumption, acetate and current production over time. Results shown are from a representative example of three replicate cultures. | ||
In contrast to the enhancement in acetate electrosynthesis with the cathode modifications summarized above, two other modifications designed to generate a positively charged cathode surface were not successful. Treating carbon cloth with melamine or ammonia is expected to yield a positive surface charge due to the presence of nitrogen-containing surface functional groups.23,72 However, neither of these treatments enhanced microbial electrosynthesis of acetate over that in untreated controls.
These results demonstrated that it is possible to enhance the rate of microbial electrosynthesis by modifications that provide a positive charge at the cathode surface. However, a positive charge is not sufficient and other features of the cathode modifications designed to provide a positive charge may be important.
Cathode modification with metal nanoparticles
Metal nanoparticles are attractive possibilities for enhancing electrode–microbe electron exchange because they have excellent catalytic activity, biocompatibility, high active surface area, and chemical stability, and their particle sizes can readily be controlled.50,53 Enhanced performance of biosensors and microbial fuel cells has been achieved with metal nanoparticles,17,47,54 which can facilitate electron transfer between the cells and electrode due to the low charge-transfer resistances and high conductivities of the nanoparticles.50,52In order to evaluate their potential for improving electrosynthesis, thin layers of Au, Pd or Ni nanoparticles were homogeneously coated onto the carbon cloth by physical deposition. Characteristic (111) X-ray diffraction (XRD) peaks of Au, Pd, or Ni were observed at 38.4°, 40.2° or 45.2°, respectively, confirming nanoparticles deposition (Fig. 5A). Each of the treatments promoted acetate electrosynthesis with rates 6-, 4.7- or 4.5-fold faster than the untreated control for Au, Pd, and Ni, respectively (Fig. 5B–D and Table 1). In each case electron recovery in acetate was comparable to other cathode modification strategies.
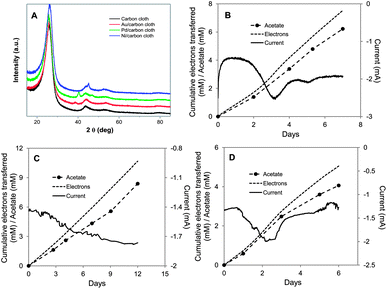 | ||
| Fig. 5 S. ovata electrosynthesis of acetate with carbon cloth cathodes coated with metal nanoparticles. (A) X-ray diffraction patterns of plain carbon cloth or cloth coated with Au, Pd, or Ni nanoparticles. (B) Electron consumption, acetate and current production over time with Au nanoparticle coated carbon cloth. (C) Electron consumption, acetate and current production over time with Pd nanoparticle coated carbon cloth. (D) Electron consumption, acetate and current production over time with Ni nanoparticle coated carbon cloth. Results shown are from a representative example of three replicate cultures. | ||
Carbon nanotube–textile composite cathode
Electrodes modified with carbon nanotubes (CNTs) have been shown to enhance the performance of biosensing systems61,73–75 and microbial fuel cells,57,58,60 due to their high aspect ratio, high conductivity and excellent biocompatibility. It is possible to incorporate CNTs into lightweight, stretchable and flexible textiles, like cotton58,76,77 or polyester fabric77 by a simple, yet scalable, dip-coating process. Polyester or carbon fabric were successfully coated with single-walled carbon nanotubes (SWNTs) as described in the literature,58 with stronger and more efficient adsorption on polyester or cotton sheets than on carbon fiber, as reported previously.76 After 8 repetitive dipping and drying steps, the polyester and cotton fabric became conductive with a low resistivity of 50 Ω cm−1. The SWNTs were well-distributed on the cotton (Fig. 6A) and polyester textile fibers (Fig. 6D) and cells readily attached to the materials (Fig. 6C and F). The rate of acetate production by S. ovata was comparable with both materials with rates that were 3.4- and 3.2-fold higher than the control (Fig. 6B and E and Table 1). Electron recoveries were comparable with other materials.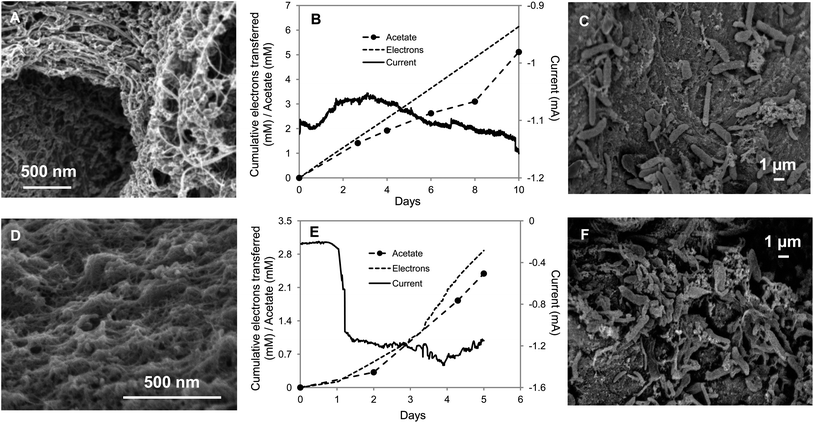 | ||
| Fig. 6 S. ovata electrosynthesis of acetate with carbon nanotube–textile composite. (A) SEM image of the cotton fabric coated with carbon nanotubes (CNTs). (B) Electron consumption, acetate and current production over time with CNT–cotton cathode. (C) SEM image of S. ovata on the CNT–cotton. (D) SEM image of the CNT–polyester. (E) Electron consumption, acetate and current production over time with CNT–polyester cathode. (F) SEM image of S. ovata on the CNT–polyester. Results shown are from a representative example of three replicate cultures. | ||
Experimental
Organism source and culture conditions
Sporomusa ovata (DSM 2662) was obtained from Deutsche Sammlung Mikroorganismen und Zellkulturen and routinely grown in DSM medium 311 (omitting betaine, fructose, casitone, and resazurin) with hydrogen as the electron donor (H2–CO2 [80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20]) at 30 °C under strict anaerobic conditions as previously described.3,4,78
:20]) at 30 °C under strict anaerobic conditions as previously described.3,4,78
Chemicals
Chitosan, cyanuric chloride (CC, 2,4,6-trichloro-1,3,5-triazine), 3-aminopropyltriethoxysilane (APTES), polyacrylonitrile (PAN, Mw, 150k), polyaniline (PANi, emeraldine base, Mw, 6.4k), N-hydroxysuccinimide melamine, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, camphor-10-sulfonic acid and single-walled carbon nanotubes (SWNTs) were purchased from Sigma-Aldrich.Acetate production by microbial electrosynthesis with an electrode as the electron donor
Each cathode material was tested at 25 °C in a three-electrode, dual-chambered system, with S. ovata grown in the cathode chamber as described previously.3,4 The carbon cloth cathode (47 cm2; GC-14, Electrolytica, Amherst, NY) and graphite stick anode (65 cm2; Mersen, Greenville, MI) were suspended in 200 ml of media in two chambers which are separated by a Nafion 117 cation-exchange membrane (Electrolytica, Amherst, NY). The anode chamber was continually bubbled with N2–CO2 (80:20). The cathode was equipped with a potentiostat (ECM8, Gamry Instruments, PA, USA) at −600 mV (versus Ag/AgCl). Hydrogen-grown cultures of S. ovata were established in the cathode chamber with a hydrogen-containing gas mix N2–CO2–H2 (83:10:7). The cathode gas mix was switched to N2–CO2 (80:20) after several fresh medium swaps. As previously described,4 there was no significant H2 production with any of the cathode materials and although some of the cathode materials were organic, they did not serve as a carbon source for acetate production as evidenced by a lack of acetate production when cathodes were not connected to anodes, as well as the correspondence between electron consumption and electrons appearing in products during electrosynthesis.
Electrode modification procedure
:1, v/v) coupling medium containing 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide (50 mM/50 mM) at room temperature overnight, then carefully washed with ethanol and dried by vacuum at room temperature overnight. Cyanuric chloride was anchored onto the electrode surface by immersing carbon cloth in 50 mM cyanuric chloride toluene solution for 24 h as previously described.32 To improve the density of cyanuric chloride on the electrode surface, the reaction was performed at 0 °C rather than room temperature. 3-Aminopropyltriethoxysilane functionalized electrode surface were obtained by immersing HNO3-pretreated carbon cloth in a 5% 3-aminopropyltriethoxysilane solution in an anhydrous toluene for 30 min. After carefully washing with toluene and acetone to remove the non-specific 3-aminopropyltriethoxysilane, the carbon cloth electrodes were dried at 110 °C for 1 h. Melamine treated electrodes were obtained by immersion of carbon cloth in methanol–water solution (1:1, v/v) containing 50 mM of melamine, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide (50 mM/50 mM) at room temperature overnight. Then they were washed with methanol several times to remove the unabsorbed melamine.
Carbon cloth was ammonia-treated with a gas mix of 5% NH3 and 95% helium as previously described.23
Electrospun mats of PANi–PAN
Nanofibrous mats of PANi–PAN were fabricated by electrospinning as previously described.79 2% of PANi solution was obtained by doping PANi base with camphor-10-sulfonic acid, filtered, and then washed with dimethylformamide. By dissolving PAN in the 2% of PANi solution, polymer solution was obtained and used for electrospinning at 25 kV. The feeding rate was 20 μl min−1 and the collecting distance was 15 cm. The composite fibers were collected on the carbon cloth. The electrospinning apparatus included a high voltage power supply (AU-120P0.5, Matsusada Precision Inc., Kusatsu, Shiga, Japan), a syringe pump (KDS 101, KD Scientific Inc., Holliston, MA) and a grounded cylinder target.Deposition of metal nanoparticles
Au and Ni layers were homogeneously sputtered on the carbon cloth for 100 s. The thin Pd layer was obtained through vapor-deposition.Carbon nanotube–textile composite
Carbon nanotube–textile composite electrode was synthesized through a simple process by dipping–drying textile cloth in aqueous CNT ink, which was prepared by dispersing 0.16% of single-walled CNTs in water by weight and 1% of sodium dodecylbenzene sulfonate as a surfactant as previously described.58 Two types of textile were used in this procedure, a piece of intertwined polyester fiber (1 mm in thickness) and a cotton sheet (1–2 mm in thickness) from JoAnn Fabric.Analytical methods
Acetate was measured via high performance liquid chromatography (HPLC) as previously described.3,80 The biofilms of S. ovata on the cathodes were stained with the LIVE/DEAD BacLight Viability Kit and imaged with confocal laser microscopy as previously described.3,80 The average cell number was calculated by examining at least five fields of view. For examination with scanning electron microscopy (SEM), samples of cathode materials were collected and fixed overnight in a buffer solution (0.1 M phosphate buffer, pH 7) containing 2.5% glutaraldehyde at room temperature. Then, samples were washed using a phosphate buffer solution (pH 7) and immersed successively in different aqueous solutions with increasing ethanol content (30, 50, 60, 70, 80, 90 and 100% ethanol), and was washed a second time with increasing acetonitrile content (50, 60, 70, 80, 90 and 100% acetonitrile). Vacuum dried samples were finally coated with Au before SEM observation. The microscopic features of the samples were investigated using a JEOL 6320 model scanning electron microscope (SEM) at an accelerating voltage of 5 kV.X-ray photoelectron spectroscopy (XPS) was performed on a Physical Electronics Quantum 2000 Scanning ESCA Microprobe. Depth profiling was done by collecting spectra at 15° and 75° take-off angles with respect to the plane of the sample surface. The analysis at 15° has a penetration depth of ∼10 Å and that at 75° corresponds to a penetration depth of ∼40 Å.
X-ray diffraction (XRD) experiments were performed in a Shimadzu XRD-6000 X-ray powder diffractometer with Cu Kα (λ = 0.154 nm) radiation at a generator voltage of 40 kV and a current of 40 mA.
Conclusions
These results demonstrate that there are several surface modifications of cathodes that can significantly increase the rate of microbial electrosynthesis. Modifying carbon cloth with chitosan or cyanuric chloride, which is relatively inexpensive, increased microbial electrosynthesis of acetate 6–7 fold. Modification with metal nanoparticles moderately increased microbial electrosynthesis rates, and although nickel is relatively inexpensive, the cost of gold or palladium would make large-scale microbial electrosynthesis reactors with such cathodes economically infeasible.17,81 The carbon nanotube–textile composite cathodes are the least cost effective materials due to the high cost of carbon nanotubes81,82 with only 3-fold increase in rates of microbial electrosynthesis compared to the untreated control.One factor limiting the design of cathode materials is a lack of understanding of the mechanisms by which electrons are transferred from cathodes to cells.6,10,11 Rates of microbial electron transfer to anodes as high as 30 A m−2 have been reported22 and cathode biofilms of Geobacter sulfurreducens consumed up to 20 A m−2 when reducing fumarate.83 If similar rates of electron transfer could be achieved with microorganisms reducing carbon dioxide then rates of microbial electrosynthesis could be increased 40–60 fold higher than the highest rates reported here. Preliminary mechanistic studies have been conducted on electron transfer into cells of fumarate-reducing Geobacter sulfurreducens,84 but even in this instance the cell components required for cell–cathode electrical connections have not be definitively identified and G. sulfurreducens does not effectively reduce carbon dioxide to organic products. Further research in this area is expected to make it possible to tune materials and cathode potentials to best interact with the appropriate electron carriers in microorganisms capable of electrosynthesis and further optimize this process.
Acknowledgements
This research was supported by Advanced Research Projects Agency – Energy (ARPA-E), U.S. Department of Energy, under Award Number DE-AR0000159 to DRL and the U.S. Department of Energy, Office of Basic Energy Sciences under contract DOE-DE-FG02-45612 to TPR.References
- D. R. Lovley, Energy Environ. Sci., 2011, 4, 4896–4906 CAS
.
- D. R. Lovley, Annu. Rev. Microbiol., 2012, 66, 391–409 CrossRef CAS
- K. P. Nevin, S. A. Hensley, A. E. Franks, Z. M. Summers, J. Ou, T. L. Woodard, O. L. Snoeyenbos-West and D. R. Lovley, Appl. Environ. Microbiol., 2011, 77, 2882–2886 CrossRef CAS
- K. P. Nevin, T. L. Woodard, A. E. Franks, Z. M. Summers and D. R. Lovley, mBio, 2010, 1, e00103 CrossRef
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS
- D. R. Lovley, Environ. Microbiol. Rep., 2011, 3, 27–35 CrossRef CAS
- D. R. Lovley and K. P. Nevin, Curr. Opin. Biotechnol., 2011, 22, 441–448 CrossRef CAS
- K. Rabaey and R. A. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS
- M. Köpke, C. Held, S. Hujer, H. Liesegang, A. Wiezer, A. Wollherr, A. Ehrenreich, W. Liebl, G. Gottschalk and P. Durre, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13087–13092 CrossRef
- L. Huang, J. M. Regan and X. Quan, Bioresour. Technol., 2011, 102, 316–323 CrossRef CAS
- M. Rosenbaum, F. Aulenta, M. Villano and L. T. Angenent, Bioresour. Technol., 2011, 102, 324–333 CrossRef CAS
- K. B. Gregory, D. R. Bond and D. R. Lovley, Environ. Microbiol., 2004, 6, 596–604 CrossRef CAS
- C. Dumas, A. Mollica, D. Feron, R. Basseguy, L. Etcheverry and A. Bergel, Electrochim. Acta, 2007, 53, 468–473 CrossRef CAS
- L. Pons, M. L. Delia, R. Basseguy and A. Bergel, Electrochim. Acta, 2011, 56, 2682–2688 CrossRef CAS
- L. Pons, M. L. Delia and A. Bergel, Bioresour. Technol., 2011, 102, 2678–2683 CrossRef CAS
- T. Nöll and G. Nöll, Chem. Soc. Rev., 2011, 40, 3564–3576 RSC
- J. Wei, P. Liang and X. Huang, Bioresour. Technol., 2011, 102, 9335–9344 CrossRef CAS
- K. Watanabe, J. Biosci. Bioeng., 2008, 106, 528–536 CrossRef CAS
- B. Willner, E. Katz and I. Willner, Curr. Opin. Biotechnol., 2006, 17, 589–596 CrossRef CAS
- B. E. Logan, Appl. Microbiol. Biotechnol., 2010, 85, 1665–1671 CrossRef CAS
- Z. Du, H. Li and T. Gu, Biotechnol. Adv., 2007, 25, 464–482 CrossRef CAS
- S. L. Chen, H. Q. Hou, F. Harnisch, S. A. Patil, A. A. Carmona-Martinez, S. Agarwal, Y. Y. Zhang, S. Sinha-Ray, A. L. Yarin, A. Greiner and U. Schroder, Energy Environ. Sci., 2011, 4, 1417–1421 CAS
- S. A. Cheng and B. E. Logan, Electrochem. Commun., 2007, 9, 492–496 CrossRef
- M. N. V. R. Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef CAS
- V. K. Mourya and N. N. Inamdar, React. Funct. Polym., 2008, 68, 1013–1051 CrossRef CAS
- I. T. Cavalcanti, B. V. Silva, N. G. Peres, P. Moura, M. D. Sotomayor, M. I. Guedes and R. F. Dutra, Talanta, 2012, 91, 41–46 CrossRef CAS
- S. R. Higgins, D. Foerster, A. Cheung, C. Lau, O. Bretschger, S. D. Minteer, K. Nealson, P. Atanassov and M. J. Cooney, Enzyme Microb. Technol., 2011, 48, 458–465 CrossRef CAS
- X. W. Liu, X. F. Sun, Y. X. Huang, G. P. Sheng, S. G. Wang and H. Q. Yu, Energy Environ. Sci., 2011, 4, 1422–1427 CAS
- F. Kuralay, T. Vural, C. Bayram, E. B. Denkbas and S. Abaci, Colloids Surf., B, 2011, 87, 18–22 CrossRef CAS
- G. Blotny, Tetrahedron, 2006, 62, 9507–9522 CrossRef CAS
- A. C. Franzoi, I. C. Vieira, J. Dupont, C. W. Scheeren and L. F. de Oliveira, Analyst, 2009, 134, 2320–2328 RSC
- Y. Wang and Y. Hasebe, Sens. Actuators, A, 2011, 155, 722–729 Search PubMed
- D. Quan and W. S. Shin, Mater. Sci. Eng., C, 2004, 24, 113–115 CrossRef
- H. Li, J. Zhang, X. Zhou, G. Lu, Z. Yin, G. Li, T. Wu, F. Boey, S. S. Venkatraman and H. Zhang, Langmuir, 2010, 26, 5603–5609 CrossRef CAS
- J. J. Lin, P. Y. Hsu, Y. L. Wu and J. J. Jhuang, Sensors, 2011, 11, 2796–2808 CrossRef CAS
- V. K. S. Hsiao, J. R. Waldeisen, Y. B. Zheng, P. F. Lloyd, T. J. Bunning and T. J. Huang, J. Mater. Chem., 2007, 17, 4896–4901 RSC
- Y. Wang, W. Qian, Y. Tan and S. Ding, Biosens. Bioelectron., 2008, 23, 1166–1170 CrossRef CAS
- A. Pietrzyk, W. Kutner, R. Chitta, M. E. Zandler, F. D'Souza, F. Sannicolo and P. R. Mussini, Anal. Chem., 2009, 81, 10061–10070 CrossRef CAS
- Z. H. Sheng, X. Q. Zheng, J. Y. Xu, W. J. Bao, F. B. Wang and X. H. Xia, Biosens. Bioelectron., 2012, 34, 125–131 CrossRef CAS
- B. Lai, X. Tang, H. Li, Z. Du, X. Liu and Q. Zhang, Biosens. Bioelectron., 2011, 28, 373–377 CrossRef CAS
- Y. Qiao, S. J. Bao, C. M. Li, X. Q. Cui, Z. S. Lu and J. Guo, ACS Nano, 2008, 2, 113–119 CrossRef CAS
- Y. Qiao, C. M. Li, S. J. Bao and Q. L. Bao, J. Power Sources, 2007, 170, 79–84 CrossRef CAS
- K. Gurunathan, A. V. Murugan, R. Marimuthu, U. P. Mulik and D. P. Amalnerkar, Mater. Chem. Phys., 1999, 61, 173–191 CrossRef CAS
- K. Scott, G. A. Rimbu, K. P. Katuri, K. K. Prasad and I. M. Head, Process Saf. Environ. Prot., 2007, 85, 481–488 CrossRef CAS
- H. Antaya, M. Richard-Lacroix and C. Pellerin, Macromolecules, 2010, 43, 4986–4990 CrossRef CAS
- J. Niessen, F. Harnisch, M. Rosenbaum, U. Schroder and F. Scholz, Electrochem. Commun., 2006, 8, 869–873 CrossRef CAS
- Y. Fan, S. Xu, R. Schaller, J. Jiao, F. Chaplen and H. Liu, Biosens. Bioelectron., 2010, 26, 1908–1912 CrossRef
- D. Brondani, E. Zapp, I. C. Vieira, J. Dupont and C. W. Scheeren, Analyst, 2011, 136, 2495–2505 RSC
- B. K. Jena, S. Ghosh, R. Bera, R. S. Dey, A. K. Das and C. R. Raj, Recent Pat. Nanotechnol., 2010, 4, 41–52 CrossRef CAS
- M. Oyama, Anal. Sci., 2010, 26, 1–12 CrossRef CAS
- S. Thiagarajan, R. F. Yang and S. M. Chen, Bioelectrochemistry, 2009, 75, 163–169 CrossRef CAS
- X. Wu, F. Zhao, N. Rahunen, J. R. Varcoe, C. Avignone-Rossa, A. E. Thumser and R. C. Slade, Angew. Chem., Int. Ed., 2011, 50, 427–430 CrossRef CAS
- C. W. Welch and R. G. Compton, Anal. Bioanal. Chem., 2006, 384, 601–619 CrossRef CAS
- D. A. Lowy, L. M. Tender, J. G. Zeikus, D. H. Park and D. R. Lovley, Biosens. Bioelectron., 2006, 21, 2058–2063 CrossRef CAS
- M. Ganesana, G. Istarnboulie, J. L. Marty, T. Noguer and S. Andreescu, Biosens. Bioelectron., 2011, 30, 43–48 CrossRef CAS
- R. Ojani, J. B. Raoof and S. Zamani, Talanta, 2010, 81, 1522–1528 CrossRef CAS
- L. Peng, S. J. You and J. Y. Wang, Biosens. Bioelectron., 2010, 25, 1248–1251 CrossRef CAS
- X. Xie, L. B. Hu, M. Pasta, G. F. Wells, D. S. Kong, C. S. Criddle and Y. Cui, Nano Lett., 2011, 11, 291–296 CrossRef CAS
- J. E. Mink, J. P. Rojas, B. E. Logan and M. M. Hussain, Nano Lett., 2012, 12, 791–795 CrossRef CAS
- H. Y. Tsai, C. C. Wu, C. Y. Lee and E. P. Shih, J. Power Sources, 2009, 194, 199–205 CrossRef CAS
- J. Wang, Electroanalysis, 2005, 17, 7–14 CrossRef CAS
- T. J. Beveridge, J. Bacteriol., 1999, 181, 4725–4733 CAS
- M. Sára and U. B. Sleytr, J. Bacteriol., 2000, 182, 859–868 CrossRef
- B. Avasarala, R. Moore and P. Haldar, Electrochim. Acta, 2010, 55, 4765–4771 CrossRef CAS
- H. Dai, Y. W. Chi, X. P. Wu, Y. M. Wang, M. D. Wei and G. N. Chen, Biosens. Bioelectron., 2010, 25, 1414–1419 CrossRef CAS
- X. H. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. H. Lin, Biosens. Bioelectron., 2009, 25, 901–905 CrossRef CAS
- S. Yadav, R. Devi, P. Bhar, S. Singhla and C. S. Pundir, Enzyme Microb. Technol., 2012, 50, 247–254 CrossRef CAS
- G. Kay and E. M. Crook, Nature, 1967, 216, 514–515 CrossRef CAS
- T. Y. Cheang, B. Tang, A. W. Xu, G. Q. Chang, Z. J. Hu, W. L. He, Z. H. Xing, J. B. Xu, M. Wang and S. M. Wang, Int. J. Nanomed., 2012, 7, 1061–1067 CAS
- J. Xie, M. R. MacEwan, A. G. Schwartz and Y. Xia, Nanoscale, 2010, 2, 35–44 RSC
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS
- M. Seredych, D. Hulicova-Jurcakova, G. Q. Lu and T. J. Bandosz, Carbon, 2008, 46, 1475–1488 CrossRef CAS
- M. E. G. Lyons and G. P. Keeley, Int. J. Electrochem. Sci., 2008, 3, 819–853 CAS
- W. Jia, C. Jin, W. Xia, M. Muhler, W. Schuhmann and L. Stoica, Chemistry, 2012, 18, 2783–2786 CrossRef CAS
- J. Zhao, W. Zhang, P. Sherrell, J. M. Razal, X. F. Huang, A. I. Minett and J. Chen, ACS Appl. Mater. Interfaces, 2011, 4, 44–48 Search PubMed
- B. S. Shim, W. Chen, C. Doty, C. Xu and N. A. Kotov, Nano Lett., 2008, 8, 4151–4157 CrossRef CAS
- L. Hu, W. Chen, X. Xie, N. Liu, Y. Yang, H. Wu, Y. Yao, M. Pasta, H. N. Alshareef and Y. Cui, ACS Nano, 2011, 5, 8904–8913 CrossRef CAS
- B. Moller, R. Ossmer, B. H. Howard, G. Gottschalk and H. Hippe, Arch. Microbiol., 1984, 139, 388–396 CrossRef
- Y. Zhu, L. Feng, F. Xia, J. Zhai, M. X. Wan and L. Jiang, Macromol. Rapid Commun., 2007, 28, 1135–1141 CrossRef CAS
- T. Zhang, S. M. Gannon, K. P. Nevin, A. E. Franks and D. R. Lovley, Environ. Microbiol., 2010, 12, 1011–1020 CrossRef CAS
- M. H. Zhou, M. L. Chi, J. M. Luo, H. H. He and T. Jin, J. Power Sources, 2011, 196, 4427–4435 CrossRef CAS
- X. Xie, G. H. Yu, N. Liu, Z. N. Bao, C. S. Criddle and Y. Cui, Energy Environ. Sci., 2012, 5, 6862–6866 CAS
- C. Dumas, R. Basseguy and A. Bergel, Electrochim. Acta, 2008, 53, 2494–2500 CrossRef CAS
- S. M. Strycharz, R. H. Glaven, M. V. Coppi, S. M. Gannon, L. A. Perpetua, A. Liu, K. P. Nevin and D. R. Lovley, Bioelectrochemistry, 2011, 80, 142–150 CrossRef CAS
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |