Rapid prediction of hydrogen permeation through amorphous metal membranes: an efficient computational screening approach†
Shiqiang
Hao‡
and
David S.
Sholl
*
School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332-0100, USA. E-mail: david.sholl@chbe.gatech.edu
First published on 6th November 2012
Abstract
Efficient purification of hydrogen from high temperature mixed gas streams using dense metal membranes can potentially play a critical role in the large-scale production of hydrogen from gasification of coal or biomaterials. We use first-principles calculations together with statistical methods to systematically predict hydrogen permeability through amorphous ternary Zr–Cu–T films (T = 17 elements) and other selected amorphous materials. These results greatly expand the range of amorphous materials that have been considered as hydrogen purification membranes. More importantly, we demonstrate that relatively simple descriptions of the site binding energies in these amorphous materials can account for the key observations from our detailed first-principles calculations. This outcome significantly reduces the computational effort required in future screening of materials in this application and also places bounds on the ultimate performance of these materials.
1 Introduction
Hydrogen is a promising fuel source that is attractive as an alternative to fossil fuels. Pure hydrogen does not exist in nature, so it must come from other resources. Currently, hydrogen is most economically obtained by reforming of hydrocarbon sources with steam or partial oxidation of hydrocarbons from fossil fuels. When hydrogen is produced from fossil fuels, it is necessary to purify hydrogen from mixed gas streams containing components such as CO, CO2, CH4, and H2S.1 One important challenge related to hydrogen purification is the development of membranes that can operate at elevated temperatures and pressures. Membranes should provide high fluxes, resistance to poisoning, and long operational lifetime with effective cost. Metal membranes are well suited for high-temperature applications.2 Pd-based metal membranes have received much attention for H2 purification because, in principle, they have perfect selectivity for H2 over other gas species. A useful benchmark for considering the permeability of new membrane materials is that the permeability of pure Pd when used with pure H2 as the feed gas is ∼10−8 mol m−1 s−1 Pa−0.5 at 600 K. By separating H2 from a CO2-rich stream, Pd membranes could be helpful in carbon sequestration from gasification processes.3 Pure Pd membranes, however, are prone to H2-induced embrittlement at temperatures below 300 °C and to sulfur poisoning. To improve the performance of pure metal membranes, metal alloys can be considered. Alloying Pd with other metals such as Ag, Au, or Cu reduces the embrittlement problem and in some cases improves resistance to poisoning.4 PdCu alloys have attracted particular attention as sulfur tolerant alloys.5 Even though a large number of binary and multi-component crystalline alloys have been tested experimentally as metal membranes,6,7 the search for durable and cost effective alloys that show high permeability for H2 continues to be an active area.Using thin films of amorphous metals, that is, metals without long-range structural order, offers significant opportunities for fabrication of membranes that outperform existing Pd-based crystalline alloy membranes. In general, concerns about hydrogen embrittlement and sintering are reduced in amorphous materials when compared to crystalline materials.8 The cost of the membrane materials in amorphous films can potentially be far less than in Pd-based alloys. Dolan et al. have recently provided a detailed review of hydrogen-selective amorphous alloy membranes,8 and Ockwig and Nenoff highlighted recent progress with these materials as part of their comprehensive review of membranes for hydrogen production.9 Perhaps the single most important observation from these two reviews is that amorphous metal membranes have been tested by multiple groups, so the methods required for creating pinhole-free membranes now exist. Ockwig and Nenoff described development of these membranes is “still an entirely open field”, in large part due to the huge number of materials that could be used.9 Among the limited number of materials that have been tested as membranes, a number have shown promising permeabilities relative to well-known crystalline membranes. Inoue and coworkers have reported several films involving Zr, Ni, and other metals that have H2 permeabilities comparable to pure Pd.10,11
Theoretical predictions have provided an effective complement to experiments in the development of practical metal membranes for H2 purification. In order for these approaches to be useful, one must provide quantitatively reliable information without requiring parameterization from experimental data. Density Functional Theory (DFT) provides a useful basis for calculations of this kind, and has been shown to be useful for predicting H diffusion and permeability in numerous dense materials, including pure metals,5 metal alloys,6,12,13 and metal sulfides.14,15 Although the properties of interstitial H in amorphous metals have been studied for at least 30 years, most theoretical work on these materials has been phenomenological rather than quantitative.16,17
We have previously developed an approach based on quantum chemistry calculations that gives quantitative information about the flux of hydrogen through amorphous metal films.18 Critically, this approach does not require any input from experimental data. We have verified the accuracy of this DFT-based computational strategy by comparison with experimental results for amorphous Zr36Ni64 and Zr30(Ni0.6Nb0.4)70.19 We have also applied this computational strategy to a series of amorphous materials with relatively high crystallization temperatures.20 Materials with high crystallization temperatures are potentially suitable for H2 purification at 300–500 °C, which are typical for H2 production from hydrocarbons. Our calculations identified Zr54Cu46 and Zr30Cu60Ti10 as the most promising materials among the 36 amorphous alloys we examined.
A key challenge in developing new materials as membranes is the need to efficiently screen a variety of candidate materials to find promising examples for more detailed testing. Even though we have been successful in applying quantitative methods to predict the performance of amorphous membranes, the DFT calculations underlying this approach are time consuming. In this paper, we demonstrate a simple method that can use input from a limited number of detailed DFT calculations to efficiently optimize the membrane performance of multicomponent amorphous materials with respect to their alloy composition.
2 Calculations
During H2 purification, hydrogen permeates through a metal film by dissociation of H2, diffusion of atomic H through interstitial sites in the metal, and recombination of atomic H as H2 on the other side. A schematic illustration of site to site hopping in a metal is shown in Fig. S1.† By considering the common situation where H2 dissociation on the surface of the film is not rate limiting, surface dissociation processes can be neglected, and the net permeability of a film can be predicted if the solubility and diffusivity of H in the bulk material is known.3,5 We used the DFT-calculations described in our earlier work to predict the solubility and diffusivity of interstitial H in amorphous metal alloys.18,19,21 These calculations show that the interstitial sites and transition states that define site to site hopping are well defined and localized. Our calculations were performed with the Vienna ab initio Simulation Package (VASP) using plane wave DFT with the PW91 GGA functional.22,23 Supercells approximating amorphous samples were prepared using ab initio Molecular Dynamics (MD) at 3000 K. These liquid-like samples were then quenched using conjugate gradient relaxation. The liquid state volumes are ∼7% greater than the volume of the quenched amorphous samples.24Binding sites for interstitial H were optimized in calculations using one H atom per supercell. We used the heuristic methods described elsewhere to efficiently locate all of the interstitial sites and transition states between adjacent interstitial sites for H in the computational volume.18 In our previous calculations, we examined the convergence of the properties of Zr55Co25Al20 with supercells containing 32, 64, and 108 atoms.20 The permeability predicted with the 32 atom calculations were consistent with (although not identical to) the larger calculations; in a typical example the permeability was 6.5 × 10−10 mol m−1 s−1 Pa−0.5 for a 32 atom cell, 8.9 × 10−10 mol m−1 s−1 Pa−0.5 for a 64 atom cell, and 1.1 × 10−9 mol m−1 s−1 Pa−0.5 for a 108 atom cell at feed (permeate) pressure of 3 (0.01) atm at 600 K. Motivated by this observation, we used 32 atom supercells to examine all candidate materials. Calculations using 32 atom supercells sampled reciprocal space with 3 × 3 × 3 k-points. Our calculations typically located 85–95 distinct interstitial sites and 240–290 transition states in each 32 atom supercell. The number of interstitial sites is similar to the situation for a crystalline fcc metal such as Pd, which has 3 interstitial sites and 6 distinct transition states per metal atom. Energies of interstitial sites and transition states were found from DFT calculations that allowed all atomic degrees of freedom in the supercell to relax.
Because the solubility of H in amorphous metals can be significant under practical conditions, H–H repulsion plays an important role in determining the overall solubility. We used the Westlake criterion to characterize H–H interactions. This criterion predicts that two interstitial sites separated by less than 0.21 nm cannot be simultaneously occupied by H atoms. We have shown previously that this approach gives results in close agreement with more detailed DFT characterization of H–H interactions in amorphous Fe3B and ZrNi.25 With this definition of the binding energy for each interstitial site and description of H–H repulsion, the net solubility of H in our amorphous samples can be calculated as a function of temperature and H2 pressure using Grand Canonical Monte Carlo (GCMC) simulations. These GCMC calculations equate the chemical potential of interstitial H and the gaseous phase, which was treated as an ideal gas. Unless stated otherwise, all of our calculations for H solubility include lattice expansion effects, because a high concentration of interstitial H can induce lattice expansion, and this expansion can in turn affect H binding energies and therefore solubility. We accounted for this effect using an iterative method introduced in earlier calculations with ZrNiNb.20 This method does not require any input from experimental data.
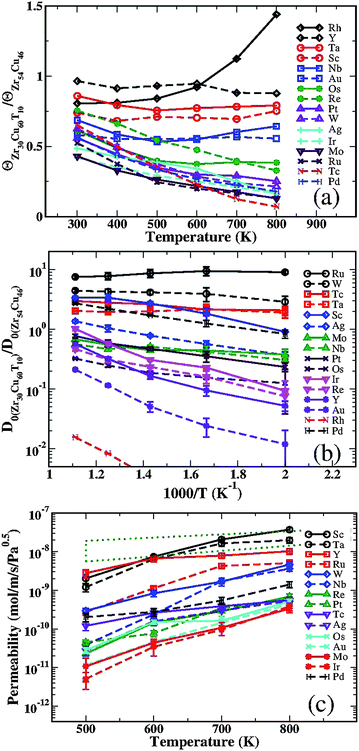
We used Fick's law to describe net transport of hydrogen through a membrane, so the flux of H, J, is related to the transport diffusion coefficient, Dt, and the gradient in the H concentration gradient, c, by J = −Dt(c)∇c. The high solubilities of H in amorphous metals mean that the concentration dependence of Dt must be considered to reliably describe H diffusion. As we have shown in earlier work, this concentration dependence can be described without approximation by 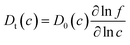 , where D0(c) is the concentration-dependent corrected diffusivity.21,26 Here, f is the fugacity of H in the gas phase in equilibrium with the solid and the derivative is known as the thermodynamic correction factor. The advantage of expressing the transport diffusivity using this equation is that the corrected diffusivity and the thermodynamic correction factor can be computed independently from the model of interstitial H we have defined above.
, where D0(c) is the concentration-dependent corrected diffusivity.21,26 Here, f is the fugacity of H in the gas phase in equilibrium with the solid and the derivative is known as the thermodynamic correction factor. The advantage of expressing the transport diffusivity using this equation is that the corrected diffusivity and the thermodynamic correction factor can be computed independently from the model of interstitial H we have defined above.
The corrected diffusivity of H in each amorphous material is calculated by Kinetic Monte Carlo (KMC) simulations.21 In these simulations, site to site hopping rates are determined by quantum corrected transition state theory, where the DFT-calculated energies and vibrational frequencies of H at each interstitial site and transition state were used to identify the diffusion barriers and prefactors. Corrected diffusivities were computed by averaging over 20 independent KMC simulations at each state point of interest. Obtaining corrected diffusivities involves averaging over the center of mass of a set of atoms in a material as they diffuse, not only the diffusive motion of individual H atoms.26,27 H–H interactions were defined using the Westlake criterion. Here, the effects of H-induced expansion are not explicitly considered, although in situations where this expansion is considerable it could alter the local hopping rates by changing the activation energy for individual site to site hops.19 Other details of the KMC methods are available in our previous reports.21
3 Results for ZrCu ternary amorphous materials
DFT calculations were initially performed for 17 ternary amorphous alloys with composition Zr30Cu60T10 (T = Sc, Ti, Y, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Ta, W, Re, Os, Ir, Pt, Au). The selection of the ternary elements was based on considerations that have been suggested to maintain good glass forming ability.8 Our previous calculations indicated that Zr54Cu46 was a promising amorphous membrane, with a predicted permeability of 5.14 × 10−9 mol m−1 s−1 Pa−0.5 at 600 K.20 This result is similar to pure Pd at the same conditions. We therefore describe the properties of the ternary alloys by comparison with Zr54Cu46. All of our DFT calculations were averaged over two independent calculations using 32 atom supercells. Additional calculations are performed for the top candidates (Zr54Cu46, Zr30Cu60Ti10) with 108 atom supercells.The solubility of H in the ternary materials at a H2 pressure of 1 atm normalized by the result in Zr54Cu46 is shown in Fig. 1(a). Only one material, ZrCuRh, is predicted to have higher solubility than the ZrCu binary material. The other ternary materials have solubility that is smaller than in the ZrCu materials by a factor of up to 20.
 | ||
| Fig. 1 DFT-based results for (a) H solubility at a pressure of 1 atm, (b) H corrected diffusivity at a fixed interstitial concentration of H/M = 0.1 and (c) H permeability in amorphous Zr30Cu60T10 (T = Sc, Ti, Y, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Ta, W, Re, Os, Ir, Pt, Au). The dotted region indicates the permeability of crystalline Pd. In (a) and (b), the results are normalized by the result for Zr54Cu46 at the same conditions. Lines are to guide the eye. | ||
The calculated corrected diffusion coefficients at an interstitial H concentration of H/M = 0.1 (M = metal atoms) are shown in Fig. 1(b). The most obvious feature of Fig. 1(b) is that the corrected diffusivity of H in these materials varies widely depending on the composition of the ternary alloy. At 600 K, for example, D0 increases by almost three orders of magnitude if the ternary element changed from Au to Ru.
The thermodynamic correction factors (TCF) that appear in the relation between the corrected and transport diffusivity can be computed from the same GCMC simulation we used to determine the solubility data.19 These results included the effects of H-induced expansion as described above. After finding the corrected diffusivities and thermodynamic correction factors (TCFs), we can calculate the transport diffusivities for each material. The corrected diffusivities were specified by fitting continuous curves to the results obtained as a function of the interstitial concentration. The TCF for each concentration of interest were evaluated using GCMC by varying the bulk H2 pressure until the observed value of H/M matched the value used in the KMC simulation within a small tolerance.19
Once the solubility and transport diffusivity of H in a metal film are known, it is straightforward to calculate the material's permeability to evaluate the performance of the film when it used as a membrane.5,28 The permeability is 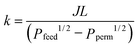 , where Pfeed (Ppermeate) is the H2 pressure on the feed side (permeate side) of the membrane, and L is the membrane thickness. For amorphous metals, the net hydrogen flux can be written as
, where Pfeed (Ppermeate) is the H2 pressure on the feed side (permeate side) of the membrane, and L is the membrane thickness. For amorphous metals, the net hydrogen flux can be written as 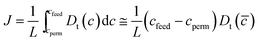 when transport of H through the bulk of the membrane is the dominant transport resistance. Here, cfeed (cperm) is the concentration of interstitial H on the feed (permeate) side, and
when transport of H through the bulk of the membrane is the dominant transport resistance. Here, cfeed (cperm) is the concentration of interstitial H on the feed (permeate) side, and ![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif) = (cfeed + cpermeate)/2.
= (cfeed + cpermeate)/2.
The predicted H2 permeabilities for all the amorphous materials we considered using a feed (permeate) pressure of 3 atm (0.01) atm are shown as symbols connected by lines in Fig. 1(c). The dotted region shows a range of permeabilities that have been reported for crystalline Pd.10,11 Among the ternary alloys we examined, the three with highest predicted permeability, Zr30Cu60T10 with T = Sc, Ta, Y, have permeability similar to Pd for temperatures above ∼600 K. The other ternary elements (Ru, W, Nb, Re, etc.) lead to considerably lower permeability than crystalline Pd. For example, Zr30Cu60Mo10 and Zr30Cu60Ir10 are predicted to have permeabilities that are 2–4 orders of magnitude lower than Pd over the temperature range we considered.
The results above examined a series of materials with composition Zr30Cu60T10. As well as varying the elements used in the amorphous material, it is also possible to vary the composition with a fixed set of alloying elements. DFT calculations can also be used to assess the performance of candidate materials with respect to their composition. Here, we use ZrxCuyTi100−x−y as an example to illustrate this idea. We performed detailed DFT calculations for 9 distinct materials, specifically Zr20Cu80, Zr54Cu46, Zr80Cu20, Zr30Cu60Ti10, Zr50Cu40Ti10, Zr70Cu20Ti10, Zr10Cu40Ti50, Zr25Cu25Ti50, and Zr40Cu10Ti50. We then used one dimensional polynomial interpolation and extrapolation techniques followed by a two dimensional least square surface fitting method to describe H solubility and diffusivity as a function of the ternary alloy composition. This calculation assumes ZrxCuyTi100−x−y is amorphous at all compositions that are considered and the possibility of precipitate formation is neglected. This clearly cannot be correct for compositions close to pure Zr, Cu, or Ti. Caution should be used, therefore, in interpreting these results for compositions that differ greatly from compositions where amorphous phases are known experimentally to exist.
The fitted H solubility and diffusivity for ZrxCuyTi100−x−y at 600 K are plotted in Fig. 2(a) and (b). The results in these plots are normalized by the results for Zr54Cu46 at the same conditions. The solubility monotonically increases with Zr or Ti composition and decreases with Cu content. The behavior of the H diffusivity is more complex. The highest diffusivities are predicted for compositions with roughly equal amounts of Zr and Cu and moderate amounts of Ti. The fitted results, which are tabulated in the ESI,† are in good agreement with the DFT-based solubility and diffusivity data. Based on the fitted results for solubility and diffusivity, we can calculate the corresponding permeability as a function of the alloy compositions. This calculation, which uses the averaged solubility between H2 pressures of 3 and 0.01 atm at 600 K, is shown in Fig. 2(c). The composition with the highest predicted permeability has a predicted permeability about 2.5 times higher than Zr54Cu46. The set of DFT-calculations used to determine the fitted functions shown in Fig. 2(a) and (b) includes a range of values surrounding the high permeability region in Fig. 2(c), so the fitting methods used to make this prediction are reliable.
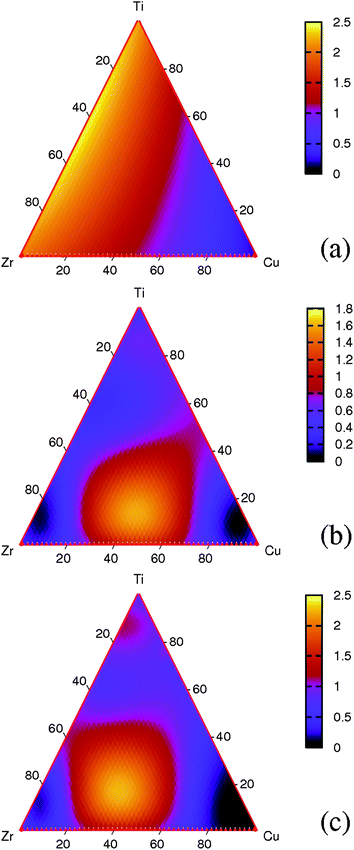 | ||
| Fig. 2 Phase diagram of ternary (a) solubility, (b) diffusivity, and (c) permeability of ZrxCuyTi100−x−y at 600 K. The solubilities are averaged by those at 3 and 0.01 atm. The transport diffusivities are at corresponding hydrogen concentrations and the permeabilities are thus related to feed and permeate pressure 3 and 0.01 atm. All results are normalized with the values for Zr54Cu46 at the same conditions. | ||
4 Discussion
The calculations reported above are useful for predicting the performance of individual materials, but in their current form they do not reveal a great deal about why the materials behave as they do. Understanding how the addition of ternary elements changes membrane properties would assist future efforts to screen materials. To this end, it is helpful to determine if the variation between materials is primarily associated with solubility or with diffusion. If the variations between materials were primarily associated with, for example, solubility, then it would be possible to develop an approximate description of membrane performance that focused only on this quantity. This issue can be addressed by considering the H solubility and diffusivity in a large set of materials normalized to a single reference material. Fig. 3 shows the normalized transport diffusivity as a function of normalized solubility using Zr54Cu46 as the reference material. Here, the solubilities are the average of the solubilities at 600 K of 3 and 0.01 atm and the diffusivities were evaluated at these average concentrations. These conditions correspond to the center of the membrane when it is operated at 600 K with a feed (permeate) pressure of 3 (0.01) atm. Fig. 3 includes data from a collection of binary and ternary materials. It is clear from these results that neither the solubility nor the diffusivity alone is suitable for capturing even the overall trends among this set of materials. For both quantities, there are examples where one quantity is relatively constant among a subset of materials while the other quantity is found to vary by up to two orders of magnitude. Any approximate description of H transport through these amorphous materials must therefore characterize both solubility and diffusivity in order to be useful.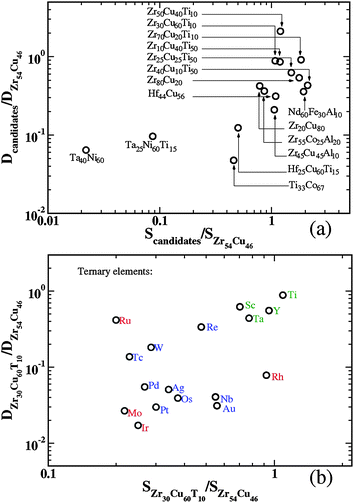 | ||
| Fig. 3 The normalized transport diffusivity as a function of the normalized solubility for (a) selected amorphous materials, and (b) amorphous Zr30Cu60T10 (T = Sc, Ti, Y, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Ta, W, Re, Os, Ir, Pt, Au). The solubility and diffusivity was calculated at the conditions described in the text. The reference system was Zr54Cu46. | ||
4.1 Correlated saddle point model
In this section, we investigate a phenomenological model of hydrogen binding and diffusion properties in amorphous metals16 that can be adapted to aid the interpretation of our results. In this model, the hydrogen binding site energies and saddle point energies are assumed to be distributed by Gaussian functions. The binding energy (saddle point energy) distribution is defined by a mean value μb (μs) and standard deviation σb (σs). During a jump from one site to an adjacent site the hydrogen atom has to move through a saddle point near the center of the polyhedron plane shared by the starting and ending points. This implies that the diffusion energy barrier should be correlated with the binding site energies of the two sites the barrier connects.16 For a jumping process from site i to site k, the diffusion barrier can be expressed (approximately) as Qik = Q0 + (Ei + Ek)/2.16 Here Ei and Ek are binding energies of hydrogen at site i and site k, Q0 = μs − μb is the energy difference between the means of the two Gaussian distributions, and (Ei + Ek)/2 is the average binding energy of the two sites. We will refer to this description as the correlated saddle point model.To use this model to qualitatively analyze H diffusion in the materials we have studied, we have to calculate Q0. This can be done by fitting a Gaussian function to the distribution of binding site and transition state energies from our DFT calculations. We analyzed four materials that represent the full range of behaviors seen in Fig. 2: low solubility/high diffusivity (Zr30Cu60Ru10), high solubility/low diffusivity (Zr30Cu60Rh10), low solubility/low diffusivity (Ta25Ni60Ti15), and high solubility/high diffusivity (Zr54Cu46). All other cases around or within this range are thus understandable with their intermediate characteristics. The fitted Gaussian distributions for each of these four materials are shown in Fig. 4. A more detailed depiction of this data is given in the ESI.†
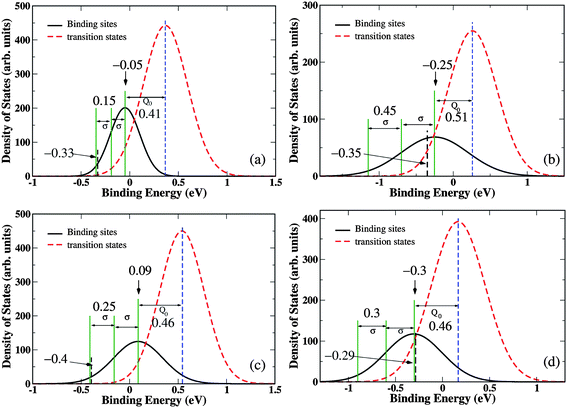 | ||
| Fig. 4 Binding energy and diffusion barrier distributions for (a) Zr30Cu60Ru10, (b) Zr30Cu60Rh10, (c) Ta25Ni60Ti15, and (d) Zr54Cu46. The mean value difference Q0 and the binding energy width are also listed. In each case, the ranges of H occupation are labeled with black dashed line, which are calculated by GCMC at 600 K and 1 atm. | ||
Above, we predicted membrane permeability under conditions where the H2 pressure varied from 3 atm and 0.01 atm across the membrane. The average solubility in these membranes is very close to the solubility with a H2 pressure of 1 atm, so below we focus on this condition to analyze the site occupancy and diffusion behavior at 600 K. We first consider hydrogen solubility in Zr30Cu60Ru10 as shown in Fig. 4(a). At 600 K and 1 atm, GCMC calculations indicate that the sites with binding energy lower than roughly −0.33 eV are occupied. These sites account for only about 2.1% of all hydrogen binding sites as characterized by the Gaussian distribution of binding sites. Because only a small portion of sites are occupied under these conditions, the H solubility in this material is low. If we consider H atoms in these sites, the Gaussian model predicts that a typical hopping event from these sites will move an H atom to a neighboring site with a binding energy similar to the mean value of all binding sites. The energy difference between these two sites is ∼2σ = 0.30 eV, so the correlated saddle point model predicts the diffusion activation energy for this event is ∼ Q0 + (Ei + Ek)/2 = Q0 + σ = 0.56 eV. Starting from those sites at tail positions of Gaussian function, hydrogen atoms can readily diffuse to their neighbor sites with binding energies similar to the mean value. If we take this energy barrier to be characteristic of diffusion in the material, then this simplified model predicts that this is a material with low solubility and relatively high diffusivity. Thus, this model makes it possible to rationalize the performance of this material based on several simple characteristics of the site energy distributions.
We now turn to Zr30Cu60Rh10. The fitted energy distributions for this material are shown in Fig. 4(b). GCMC calculations at 600 K and 1 atm show that only the sites with binding energy lower than roughly −0.35 eV are occupied. These sites make up approximately 45% of the possible binding sites, leading to a high H solubility in this material. Because of the high solubility, it is useful to consider several subpopulations of H atoms separately. For sites with energies more than 2σ lower than the mean, the same analysis described above with the correlated saddle point model predicts a characteristic diffusion activation energy of 0.96 eV. For the second range of hydrogen atoms within the binding energy −1.1 < Ei < −0.7 eV, the diffusion barrier calculated in a similar way is about 0.74 eV. The third group of H atoms have binding energies between −0.7 and −0.35 eV. The characteristic diffusion barrier predicted by the correlated saddle point model for this group is roughly 0.61 eV. For all of these groups, the typical diffusion activation energies are larger than in Zr30Cu60Ru10, so qualitatively this analysis accounts for the observation that this material shows high solubility but relatively low diffusivity.
We can now analyze the remaining materials in Fig. 4 in the same way. For Ta25Ni60Ti15, shown in Fig. 4(c), H occupies only a small number of the overall set of binding sites, and the characteristic diffusion activation energy for these occupied sites is estimated to be ∼0.7 eV. This description predicts that this is a material with low solubility and low diffusivity, in agreement with our detailed DFT calculations. Similar to Zr30Cu60Rh10, the solubility of H in Zr54Cu46 is quite high because of the large number of binding sites with very favorable binding energies (see Fig. 4(d)). We consider the diffusion of these H atoms by splitting them into three energy ranges. The H atoms with binding energy less than −0.9 eV are predicted to have a characteristic diffusion barrier of ∼0.75 eV. For the atoms with binding energies between −0.9 and −0.6 eV, the predicted characteristic diffusion barrier is ∼0.6 eV. The third group of H atoms, and the group that contains the most sites, has binding energies between −0.6 and −0.3 eV. The characteristic diffusion barrier for these atoms is 0.46 eV, which is quite low relative to the other materials considered above. Thus, the correlated saddle point model predicts that Zr54Cu56 has high solubility and high diffusivity, in agreement with our detailed DFT calculations.
4.2 Simple Gaussian model
Although the correlated saddle point model elegantly explains the solubility and diffusion of H in amorphous materials, it requires a large number of DFT calculations before any conclusions can be drawn. Moreover, once these calculations are complete, it is possible to make accurate predictions about a material's performance without needing to resort to a phenomenological model. This model can therefore be thought of a useful way to rationalize the behavior of different materials, but it is not suitable for predicting the properties of new materials. If we want to seek better materials from a large number of possible ternary, quaternary, and more complex multicomponent amorphous alloys,2,8,9 an alternative approach is needed. In this section, we show that a simplified model based only on the distribution of binding site energies (that does not use information related to transition states) can be used to make useful qualitative predictions about H permeation through amorphous materials.Fig. 5(a) shows that fitted site energy distributions for four materials. We argued above that a typical hopping event from a favorable site moves an H atom from a site in the lower part of the site energy distribution into a site near the mean of the distribution. This implies that a favorable membrane material will have a significant number of sites in the energy regime that is occupied under the conditions (temperature and pressure) of interest, and, just as importantly, have a relatively narrow distribution of site binding energies. A site binding energy distribution with a large variance will typically be associated with larger local diffusion barriers than an energy distribution with a smaller variance. This description is only appropriate for materials where the binding energy distribution can be described by a Gaussian distribution, so it would not be useful, for example, for simple crystalline materials where site binding energies take on a small number of discrete values.
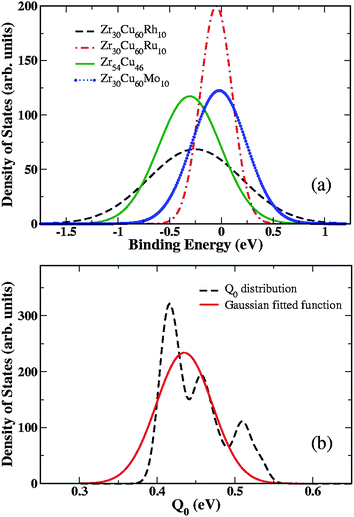 | ||
| Fig. 5 (a) Gaussian distributions of binding energies in four typical cases: Zr30Cu60Rh10, Zr30Cu60Ru10, Zr30Cu60Mo10, and Zr54Cu46. (b) Distribution of Q0 (dashed line) for all available DFT calculated amorphous materials, and fitted Gaussian distribution (solid line) with a mean value of 0.435 eV and a standard deviation of 0.038 eV. | ||
To apply the correlated saddle point model, it is necessary to know not only the variance and mean of the binding site energy distribution, but also the difference in the means of the site energy distribution and the transition state energy distribution, Q0. If Q0 varied widely among different materials, it would not be possible to use this model without explicitly determining the transition state energy distribution. To understand whether this description applies to real materials, we determined Q0 from our DFT calculations for the 16 materials shown in Fig. 2(a) and Zr30Cu60T, with T = Sc, Ti, Y, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Ta, W, Re, Os, Ir, Pt, Au) is shown in Fig. 5(b). Although Q0 varies among the materials, these values can be described by a Gaussian function with a mean value of 0.435 eV and a standard deviation of only 0.038 eV. The small standard deviation of this distribution implies that Q0 is approximately constant across the full set of amorphous materials we have considered. This is a useful observation, since it implies that qualitative predictions about the permeation of H through amorphous metals can now be made using only the binding site energy distribution. Determining the site energy distribution with DFT is considerably less demanding than calculations in which this distribution and the full distribution of transition state energies are determined. It also implies that information about diffusion can be extracted from experimental solubility data provided that the temperature and pressure dependence of the observed solubility in a material can be analyzed to give information about the underlying distribution of binding site energies.
4.3 Optimal parameters for Gaussian model
We have shown above that membrane performance can be qualitatively estimated by the shape and position of the Gaussian distribution of site binding energies. It is useful to consider what values of the parameters (μ, σ) of the Gaussian distribution yield the best membrane performance. In Section 2, we used our detailed DFT-based results to predict the permeability of ZrxCuyTi100−x−y as a function of composition. We also fitted the Gaussian model parameters, μ and σ, as a function of the ZrxCuyTi100−x−y composition. By comparing these results with the predictions from Section 2, we can determine what values of the Gaussian model parameters correspond to desirable membrane performance. Fig. 6 illustrates this approach. In Fig. 6, the black circles are parameters for which the permeabilities are >2 times higher than Zr54Cu46 at 600 K using a feed (permeate) pressure of 3 (0.01) atm. If amorphous alloys made up from any combination of elements can be found with parameters similar to the black circles in Fig. 6, their membrane performance is predicted to exceed that of Zr54Cu46 at 600 K. The Gaussian model parameters for all of the other materials we considered with detailed DFT calculations are also shown on Fig. 6. The alloys with composition Zr30Cu60T10 (T = Sc, Ta, Y, and Ti) are closer than other materials to the black circles in the figure, indicating good permeability. This prediction is in good agreement with our detailed DFT results.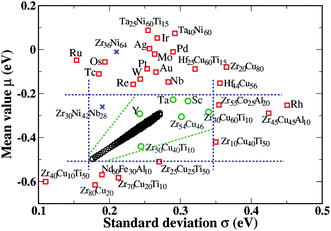 | ||
| Fig. 6 Gaussian model parameters for H binding energy from all materials we examined using DFT (blue, red, and green symbols) and optimized parameters from fitted results for ZrxCuyTi100−x−y (black circles). The element labels beside the data indicate the samples of Zr30Cu60T10 (T = Sc, Ti, Y, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Ta, W, Re, Os, Ir, Pt, Au). Our previous DFT results19 for Zr36Ni64 and Zr30Ni42Nb28 are indicated with blue crosses. | ||
The rectangle indicated in Fig. 6 is drawn to exclude the materials with highly unfavorable membrane performance. Looking at the materials shows there are different mechanisms governing their poor permeabilities. Materials where the mean of the Gaussian distribution is higher than −0.2 eV have low solubility because of their relatively unfavorable binding energies. Examples in this group include Ta25Ni60Ti15, Ta40Ni60, and Zr30Cu60Ir10, whose solubilities are 0.032, 0.008, and 0.099 at 600 K and a H2 pressure of 1 atm. Materials with a mean value lower than −0.5 eV have very high solubility. In these cases, blocking effects decrease the hydrogen diffusivity and as a result suppress the overall permeability. Zr80Cu20, Nd60Fe30Al10, and Zr70Cu20Ti10 belong in this category. For materials with mean values in the range −0.2 < μ < −0.5 eV, the standard deviation of the binding site energy distribution is important. If the standard deviation is too large, high diffusion barriers lead to sluggish diffusivity and low permeability.
We also included the DFT-derived Gaussian parameters of Zr36Ni64 and Zr30Ni42Nb28 in Fig. 6 to make a connection with experimental data. We have showed previously that DFT-based calculations for Zr36Ni64 and Zr30Ni42Nb28 using the same methods we have used here19 make predictions in excellent agreement with experimental observations.10 The permeability of Zr36Ni64 is much lower than that of Zr30Ni42Nb28, while the permeability of Zr30Ni42Nb28 is similar to pure Pd.19 The Gaussian parameters of Zr36Ni64 are located well outside the rectangle in Fig. 6, suggesting in agreement with experiments that this material has fairly poor permeability. Zr30Ni42Nb28, in contrast, is closer to the optimal region in Fig. 6, in agreement with the better performance found with this material experimentally.
Fig. 6 raises the possibility that we can make a general statement about the largest permeability that can be achieved with any amorphous material. Considering the full range of Gaussian parameters represented in Fig. 6, our qualitative model suggests that the highest permeability materials will have permeability ∼3 times larger than pure Pd. This implies that there are potentially amorphous compositions that have higher permeability than materials such as Zr30Ni42Nb28 and Zr54Cu46, but there is little scope for finding materials whose permeability is an order of magnitude or more higher than pure Pd. It is conceivable that materials exist that cannot be reasonably described using the heuristic framework implied by Fig. 6 and our use of the correlated saddle point model, but we have not observed any materials like this among the large number of amorphous materials we have characterized with detailed DFT calculations.
All of the discussions above were based on membranes at 600 K with fixed feed and permeate pressures 3 and 0.01 atm. It is therefore useful to determine if the optimal parameters determined at these conditions are also able to make meaningful predictions at other conditions. We examined this issue by repeating our calculations at 500 K, finding optimal Gaussian parameters as before. The difference between the parameters found in this way at 500 K and 600 K was small. This indicates that the optimal parameters are relatively insensitive to temperature. This is reasonable since the permeabilities of amorphous materials are relatively insensitive to temperature.19,20
5 Summary
We have used first-principles calculations together with statistical methods to systematically evaluate hydrogen solubility, diffusion, and permeability in amorphous Zr30Cu60T10 (T = Sc, Ti, Y, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Ta, W, Re, Os, Ir, Pt, Au) and other selected amorphous materials. These calculations greatly expand the number of amorphous metals for which quantitative information on hydrogen permeability is available. We showed how first-principles calculations can be used to optimize the permeability of an amorphous material with respect to its composition for the ternary material ZrxCuyTi100−x−y.Although the first-principles methods we used to examine the materials listed above give quantitative results, they demand significant computational resources. Much of this computational effort is associated with developing a complete description of the transition states for H atom hopping between interstitial sites, which is a prerequisite for developing a full description of interstitial diffusion. To determine whether this approach can be performed more efficiently, we examined the applicability of several phenomenological models to our detailed first-principles data. This analysis showed that the distribution of binding site energies in amorphous metal films can reliably be described using a Gaussian model and, more significantly, a correlated saddle point model adequately describes the distribution of transition state energies of interstitial hopping.
Once the parameters for the Gaussian distribution of site binding energies are known, only a single parameter is needed to describe the correlated saddle point model. Our calculations indicate that this parameter varies little among the wide variety of amorphous metals we examine. This observation implies that it is now possible to accurately predict the permeability of H through amorphous metal films based only on determining the distribution of site binding energies. This creates possibilities for screening large numbers of materials computationally. It is also possible that this method could be used to make predictions about membrane performance based on experimental measurements of H solubility, since systematically determining the pressure dependence of H solubility provides information on the distribution of binding site energies. This approach could be useful in efforts to screen materials for membrane applications experimentally since making solubility measurements is considerably easier in many cases than performing high quality membrane experiments.
The caveats that must be used when making predictions about membrane performance from theoretical calculations have been carefully discussed in our earlier work.18–21,25 In situations where membrane performance is dominated by the bulk properties of the film rather than by surface effects, the methods we have used here accurately describe net permeation of H through metal films. Our methods cannot give information on effects such as clustering that may occur on time scales of hours or longer in quenching of amorphous materials. The phenomenological models motivated by our detailed calculations place bounds on the overall permeability that can be achieved with amorphous metal films. These models indicate that a range of amorphous materials are possible with permeability exceeding pure Pd by factors of 2–3. The methods we have introduced in this paper should be useful in finding additional alloy compositions that fall in this class and in motivating experimental studies to assess the viability of these materials in practical applications.
Acknowledgements
Financial support for this work came from the US DOE Office of Basic Energy Sciences and the US DOE National Energy Technology Laboratory. Discussions with K. Coulter were greatly appreciated.References
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS.
- D. S. Sholl and Y. H. Ma, MRS Bull., 2006, 31, 770 CrossRef CAS.
- S. N. Paglieri and J. D. Way, Sep. Purif. Methods, 2002, 31, 1 CrossRef CAS.
- C. G. Sonwane, J. Wilcox and Y. H. Ma, J. Phys. Chem. B, 2006, 110, 24549 CrossRef CAS.
- P. Kamakoti, B. D. Morreale, M. V. Ciocco, B. H. Howard, R. P. Killmeyer, A. V. Cugini and D. S. Sholl, Science, 2005, 307, 569 CrossRef CAS.
- S. G. Kang, K. E. Coulter, S. K. Gade, J. D. Way and D. S. Sholl, J. Phys. Chem. Lett., 2011, 2, 3040 CrossRef CAS.
- K. E. Coulter, J. D. Way, S. K. Gade, S. Chaudhari and D. S. Sholl, J. Phys. Chem. C, 2010, 114, 17173 CAS.
- M. D. Dolan, N. C. Dave, A. Y. Ilyushechkin, L. D. Morpeth and K. G. McLennan, J. Membr. Sci., 2006, 285, 30 CrossRef CAS.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078 CrossRef CAS.
- S. Hara, N. Hatakeyama, N. Itoh, H. M. Kimura and A. Inoue, J. Membr. Sci., 2003, 211, 149 CrossRef CAS.
- S. I. Yamaura, M. Sakurai, M. Hasegawa, K. Wakoh, Y. Shimpo, M. Nishida, H. Kimura, E. Matsubara and A. Inoue, Acta Mater., 2005, 53, 3703 CrossRef CAS.
- P. Kamakoti and D. S. Sholl, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 014301 CrossRef.
- L. Semidey-Flecha and D. S. Sholl, J. Chem. Phys., 2008, 128, 144701 CrossRef.
- B. D. Morreale, B. H. Howard, O. Iyoha, R. M. Enick, C. Ling and D. S. Sholl, J. Membr. Sci., 2007, 46, 6313 CAS.
- C. Ling and D. S. Sholl, J. Membr. Sci., 2009, 329, 153 CrossRef CAS.
- R. Kirchheim, Prog. Mater. Sci., 1988, 32, 261 CrossRef CAS.
- R. Kirchheim, T. M. Otschele, W. Kiennger, H. Gleiter, R. Birringer and T. D. Koble, Mater. Sci. Eng., 1988, 99, 457 CrossRef CAS.
- S. Q. Hao and D. S. Sholl, Energy Environ. Sci., 2008, 1, 175 CAS.
- S. Q. Hao and D. S. Sholl, J. Membr. Sci., 2010, 350, 402 CrossRef CAS.
- S. Q. Hao and D. S. Sholl, J. Membr. Sci., 2011, 381, 192 CrossRef CAS.
- S. Q. Hao and D. S. Sholl, J. Chem. Phys., 2009, 130, 11106 CrossRef.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- D. S. Sholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, John Wiley & Sons, Inc., 2009 Search PubMed.
- M. Mihalkovič and M. Widom, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 144107 CrossRef.
- S. Q. Hao, M. Widom and D. S. Sholl, J. Phys.: Condens. Matter, 2009, 21, 115402 CrossRef.
- D. S. Sholl, Acc. Chem. Res., 2006, 39, 403 CrossRef CAS.
- A. I. Skoulidas and D. S. Sholl, J. Phys. Chem. B, 2001, 105, 3151 CrossRef CAS.
- T. L. Ward and T. Dao, J. Membr. Sci., 1999, 153, 211 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ee23180h |
| ‡ Current address: Department of Materials Science and Engineering, Northwestern University, Evanston, IL 60208. |
| This journal is © The Royal Society of Chemistry 2013 |