A high performance oxygen storage material for chemical looping processes with CO2 capture†
Qilei
Song
*ab,
Wen
Liu
a,
Christopher D.
Bohn
a,
Ryan N.
Harper
c,
Easan
Sivaniah
b,
Stuart A.
Scott
c and
John S.
Dennis
*a
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge CB2 3RA, UK. E-mail: jsd3@cam.ac.uk; Fax: +44 (0)1223334796; Tel: +44 (0)1223 334787
bCavendish Laboratory, University of Cambridge, Cambridge CB3 0HE, UK. E-mail: qs209@cam.ac.uk; Fax: +44 (0)1223 337000; Tel: +44 (0)1223 337010
cDepartment of Engineering, University of Cambridge, Cambridge CB2 1PZ, UK
First published on 22nd November 2012
Abstract
Chemical looping combustion (CLC) is a novel combustion technology that involves cyclic reduction and oxidation of oxygen storage materials to provide oxygen for the combustion of fuels to CO2 and H2O, whilst giving a pure stream of CO2 suitable for sequestration or utilisation. Here, we report a method for preparing of oxygen storage materials from layered double hydroxides (LDHs) precursors and demonstrate their applications in the CLC process. The LDHs precursor enables homogeneous mixing of elements at the molecular level, giving a high degree of dispersion and high-loading of active metal oxide in the support after calcination. Using a Cu–Al LDH precursor as a prototype, we demonstrate that rational design of oxygen storage materials by material chemistry significantly improved the reactivity and stability in the high temperature redox cycles. We discovered that the presence of sodium-containing species were effective in inhibiting the formation of copper aluminates (CuAl2O4 or CuAlO2) and stabilising the copper phase in an amorphous support over multiple redox cycles. A representative nanostructured Cu-based oxygen storage material derived from the LDH precursor showed stable gaseous O2 release capacity (∼5 wt%), stable oxygen storage capacity (∼12 wt%), and stable reaction rates during reversible phase changes between CuO–Cu2O–Cu at high temperatures (800–1000 °C). We anticipate that the strategy can be extended to manufacture a variety of metal oxide composites for applications in novel high temperature looping cycles for clean energy production and CO2 capture.
Broader contextThe principal means of controlling emissions of CO2 from the combustion of fossil fuels for electrical power will be to capture it from flue gases followed by sequestration or utilisation. Chemical-looping combustion (CLC) offers the inherent feature of isolating CO2, avoiding the need for costly separation processes. This process requires an oxygen storage material that is reactive, cheap to prepare and durable over many cycles of reduction and oxidation at high temperatures, typically 800–1000 °C. One of the key issues is the sintering of the oxygen storage material occurring during the redox cycles and its deleterious effect on the capacity of the material to transport and release oxygen. This paper demonstrates a simple method of preparing a copper-based oxygen storage material, which has excellent cyclic stability and storage capacity. The oxygen storage material was synthesised by preparing a Cu–Al layered double hydroxide (LDH) precursor, to ensure dispersion of Cu and Al at the molecular level. The LDH precursor was then calcined, giving a fine dispersion of copper phase in the resulting composite of metal oxides. The work shows how it is possible to control dispersion, structure, and the stability of the resulting structures to sintering, by tuning the precursor chemistry. This represents a significant advance over current empirical techniques of preparation oxygen carrier materials and could be extended to other metal oxide composites, e.g. for application in catalysis or novel solid looping cycles for clean energy production and CO2 capture. |
1 Introduction
To capture CO2 from coal-fired power plants, the current commercial, or near-commercial, technologies includes solvent scrubbing of the flue gases or combustion of the coal in mixtures of O2 and CO2 (i.e. oxy-fuel). The major drawback of these methods is the energy penalty associated with gas separation and the extra capital cost of the associated equipment.1 Chemical-looping combustion (CLC) has been proposed as an alternative for CO2 capture from stationary sources of CO2 emission. In contrast to the conventional combustion of fuels in air, the CLC process uses an oxygen storage material, also known as oxygen carrier, usually a transition metal oxide, to provide gaseous or solid state oxygen for combustion and gives an inherent separation of CO2 from nitrogen and other components in air, without significant energy penalty. The principle of CLC was proposed by Lewis and Gilliland in the 1950s.2,3 In the current context of urgent need for clean energy and CO2 capture, the CLC process has regained attention as a promising alternative to traditional fossil fuel conversion technologies.4–13Typically, oxygen carriers make use of transition metal oxide couples, such as NiO–Ni, CuO–Cu, Mn2O3–MnO, and Fe2O3–Fe3O4, supported on various materials.7–12,14,15 Low-cost natural materials, e.g. ilmenite, FeTiO3/FeTiO5,16–19 iron ore,20,21 and calcium-based oxygen carriers, CaSO4/CaS,22–25 have also attracted significant attention. Some metal oxides can release gaseous O2 at a high temperature, which can be used for combustion of solid fuel, and can subsequently be regenerated using air; this process has been termed “chemical-looping with oxygen uncoupling” (CLOU) by Mattisson and coworkers.26,27 Good candidate materials for the CLOU process include CuO–Cu2O, Mn2O3–Mn3O4 and Co3O4–CoO. Perovskite and spinel type mixed oxides are also promising, such as CaMn0.875Ti0.125O3,28,29 FexMn(1−x)O3 and Mg2MnO4.30,31 The CuO–Cu2O couple appears to be the most suitable because of its high oxygen release capacity and suitable range of operation temperature (800–1000 °C).6,26,27,32–35 Particularly, Cu-based oxygen carriers have been demonstrated for coal combustion in a laboratory batch unit by Mattisson, Leion and coworkers,26,27,32 and in a continuously operating unit by Abad et al.36 Chemical looping requires the reversible phase change between CuO–Cu2O–Cu, either from CuO to Cu by reduction with a gaseous fuel, or with the gaseous O2 released from 2CuO(s) = Cu2O(s) + 0.5O2(g), followed by regeneration in air back to CuO. In CLOU, only the CuO–Cu2O cycle is used, since the equilibrium partial pressure of gaseous O2 release for Cu2O–Cu is too low for practical use.
Over the last decade, various types of oxygen carriers have been reported, as summarised in a recent comprehensive review by Adánez and coworkers.6 However, there remain many challenges to produce an oxygen carrier that is stable over many chemical-looping redox cycles. One of the key challenges for stable operation of these materials is overcoming the sintering of the oxygen carrier at high temperatures required for CLC and CLOU. These cycles must operate at a high temperature to give a reasonable efficiency, especially if used in a pressurised cycle with gaseous fuels so as to compete with e.g. gas turbine combined cycles (GTCC) with post-combustion carbon capture; thus the materials may be subjected to extreme conditions which are detrimental to their longevity.20,37 The susceptibility to sintering is related to the melting point of the bulk solid (Tm, K) and characterised by the so-called Hüttig temperature (at which point atoms at defects become mobile, defined empirically as 0.3Tm) and Tammann temperature (at which point atoms from the bulk exhibit mobility, defined empirically as 0.5Tm). The Tammann temperatures for CuO, Cu2O and Cu are 526 °C, 481 °C and 405 °C, respectively. The Hüttig temperature for copper is particularly low (134 °C) owing to its low melting point (1083 °C). Therefore, severe sintering of copper or copper oxide crystals is inevitable at the high operating temperatures (800–1000 °C).
In order to limit the sintering of oxygen carrier materials, metal oxides need to be supported on various refractory materials,38e.g. CuO has been supported on Al2O3, ZrO2, or MgAl2O4, mainly by Lyngfelt's group and Adánez's group.6,26,27,33–36,39,40 One critical issue is the degree of dispersion of active phase in the materials, which significantly affects their reactivity and stability in redox cycles. Conventional methods, such as mechanical mixing or impregnation, fail to give satisfactory level of dispersion of metal oxides at high loadings and consequently poor resistance to sintering in redox cycles. Another issue is the interaction of active phase with the support. For example, when CuO is supported on various types of Al2O3, copper aluminates (CuAl2O4 and CuAlO2) are formed after redox cycles at 800–1100 °C, which critically affects the performance of the oxygen carrier.6,33–36,39–45 These issues have also been encountered for Ni, Fe, and Mn based oxygen carriers.6 Structural promoters may also be required to stabilise the active phase dispersed in the refractory support.
In this paper, we propose the novel route of preparation of oxygen carrier materials from the layered double hydroxides (LDHs) precursor. LDHs, also known as hydrotalcite-like compounds (HTLCs), are a class of two-dimensional nanostructured anionic clays.46–51 LDHs are represented by the general formula: [(M2+)n(M3+)m(OH)2(n+m)]m+[(Ax−)m/x·yH2O], where M2+ could be the metal cations, Ca2+, Mg2+, Zn2+, Co2+, Ni2+, Cu2+, Mn2+, Fe2+, etc. and M3+ could be Fe3+, Cr3+, Al3+, Ga3+, Mn3+, etc. Here, Ax− is an interlayer exchangeable anion (e.g. OH−, NO3−, CO32−, Cl−, SO42−, etc.), and m = 0.17–0.40. A naturally occurring example is hydrotalcite, Mg6Al2(OH)16CO3·4H2O, which contains carbonate anions between the brucite layers. LDHs can be synthesised by a variety of methods (coprecipitation, precipitation–deposition, hydrothermal, sol-gel, ion-exchange, rapid mixing and nucleation, delamination–restacking, etc.) from versatile combination of elements.46–51 Particularly, owing to the homogeneous dispersion of cations at the molecular level, the mixed metal oxides derived by thermal decomposition of LDHs offer high degree of dispersion of metal ion components in the support, and have shown higher reactivity, stability in many industrial applications.46,52–54 Recently, mixed metal oxides derived from LDHs have found new applications as sorbents for CO2 capture.55–57
The precursor chemistry of LDHs is versatile and sensitive to the preparation conditions. Preparation of phase-pure Cu-containing LDH precursor remains challenging because of the Jahn–Teller effect, but significant advancement has been made by Behrens et al.53 A third metal M2+ or M3+ can be added, i.e. Ni2+, Ca2+, Zn2+, Mg2+ or Fe3+, forming a ternary system (Cu2+M2+M3+).52–54 For the simple binary Cu–Al LDHs, CuAl2O4 spinel is usually formed upon calcination at high temperature.58 In an earlier work, Chuang et al.45,59 developed CuO/Al2O3 oxygen carriers by coprecipitation. However, only statistical optimisation was performed without fundamental understanding of the precursor chemistry. Another issue is that LDHs are often prepared by coprecipitation with alkali solution, which could result in residual alkali metal in the material. Generally, residual sodium was reported as damaging to the performance of catalysts, and various sodium-free methods have been developed.46,60
In this paper, using the binary Cu–Al LDH as a prototype, we studied the correlation between LDHs precursor chemistry, the structure of calcined product and their performance as an oxygen carrier in redox cycles. We synthesised a series of Cu–Al LDH precursors by co-precipitation at constant pH, with the alkali precipitating agent and molar ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al varied. We characterised the precursor and calcined mixed metal oxides with various physical and chemical techniques to understand the structure–property relationship. We then performed multiple oxygen release and storage cycles and reduction–oxidation cycles in a fluidised bed at elevated temperatures (800–1000 °C) and studied the stability of oxygen carrier materials.
:Al varied. We characterised the precursor and calcined mixed metal oxides with various physical and chemical techniques to understand the structure–property relationship. We then performed multiple oxygen release and storage cycles and reduction–oxidation cycles in a fluidised bed at elevated temperatures (800–1000 °C) and studied the stability of oxygen carrier materials.
2 Experimental
2.1 Synthesis of Cu–Al layered double hydroxides precursor
The Cu–Al LDH precursors were synthesised by co-precipitation, at room temperature and atmospheric pressure and a constant pH. A detailed description is given in the ESI.† Briefly, the precursors were prepared from an aqueous solution of Cu(NO3)2·2.5H2O and Al(NO3)3·9H2O, with the precipitate formed by mixing with a solution of NaOH and Na2CO3, at constant pH of 9.6 ± 0.1 at room temperature. The precipitate was aged for 2 h, washed with deionised water for five times, filtered, and dried at 60 °C in air for 48 h. The as-dried precursor was calcined in a muffle furnace at 950 °C for 5 h in air.One precipitate was prepared with Cu2+:Al3+ molar ratio = 1 M:1 M, with a mixture of NaOH and Na2CO3 (1 M of each). In addition, two series of LDH precursors, with fixed Cu2+:Al3+ atomic ratios of either 1:1 or 3:1 were prepared, whilst varying the ratios of [NaOH]:[Na2CO3] for each as follows: 1:0, 0.94:0.06, 0.5:0.5, and 0:1. The total concentration, i.e. [NaOH] + [Na2CO3], was fixed at 1 M throughout. Also, precipitates with different loadings of CuO (20–100 wt%) in mixture with Al2O3 were prepared by changing the molar ratio of Cu2+:Al3+ in the solution with total concentration of Cu2+ and Al3+ equal to 1 M.
2.2 Characterisation of materials
The precursors, as calcined and materials exposed to redox cycles were characterised routinely with various physical and chemical techniques. The elemental analyses were carried out using inductively coupled plasma atomic emission spectroscopy (ICP-AES, Varian Liberty) calibrated with standards. LDH precursor and calcined particles (∼100 mg) were digested in nitric acid (5 mL) for 48 h and the solution was diluted with distilled water prior to measurement. Scanning electron microscopy (SEM) and scanning transmission electron microscopy (STEM) were performed on a Hitachi S5500 electron scanning microscope. The powders were sputter-coated with a thin layer of gold prior to the SEM analysis. Energy dispersive X-ray spectroscopy (EDX) was performed using a spectrometer (Oxford Instruments INCA x-sight). Prior to the STEM analysis, the particles were crushed into powders and dispersed in isopropanol by ultra-sonication, dipped on the copper grid with carbon film and dried in air at room temperature. In some cases, high resolution TEM (HRTEM) was performed by a FEI Philips Tecnai 20 transmission electron microscope. Powder X-ray diffraction (XRD) was performed with a Philips PW1830 diffractometer, where Cu Kα radiation was generated at 40 mA and 40 kV, and the scanning rate was 0.025° per step per second. Rietveld refinements were performed using the PANalytical X'Pert HighScore Plus software. Fourier transform infrared spectroscopy (FTIR) was performed with a NICOLET analyser (Model iS10, Thermo Scientific) using the KBr pellet technique. Nitrogen adsorption and desorption isotherms of fresh and used oxygen carrier materials were performed at 77 K (Micromeritics instrument ASAP 2000).2.3 Thermal analysis
Calcination of precursor was studied in a thermogravimetric analyser (TGA), from 50 °C to 950 °C in air at a heating rate of 10 °C min−1. The oxygen storage capacity, or the reducible oxygen, was determined by temperature programmed reduction (TPR) in the TGA, with the temperature increased from 120 °C to 950 °C at a heating rate of 5 °C min−1 in 5.0 vol% H2, balance N2 (50 mL min−1, STP). The gaseous O2 release capacity of the oxygen carrier, i.e. the amount of oxygen released at a given temperature under an inert, sweep gas, was measured by temperature programmed decomposition in TGA. The sample was heated in N2 (10 mL min−1, STP) to 950 °C at a rate of 10 °C min−1, and held at 950 °C for 30 min.2.4 Cyclic oxygen release and storage in a fluidised bed
A batch of oxygen carrier materials (15.0 g) were alternately exposed to inert gas (N2, 50.0 mL s−1, STP) and oxidising gas (air, 47.4 mL s−1, STP) in a laboratory-scale fluidised bed, held at a constant temperature (800–1000 °C). The apparatus has been described elsewhere11,45 and in Fig. S1.† The stability of the oxygen carrier materials were evaluated by the apparent rate and overall amount of gaseous O2 released over multiple cycles. The detailed procedure is given in the ESI.†2.5 Cyclic reduction and oxidation in a fluidised bed
A batch of ∼0.5 g oxygen carrier materials were exposed to cyclic reduction and oxidation in the fluidised bed (typically ∼950 °C), using CO as gaseous fuel (∼2.4 vol% CO in N2, 65.8 mL s−1, STP), N2 as purging gas (50.0 mL s−1, STP), and air as oxidising gas (47.4 mL s−1, STP). The temperature was set to 800–1000 °C. The stability of the oxygen storage materials were evaluated by the apparent rate and overall production of CO2 over multiple redox cycles. For details, refer to the ESI.†3 Results
3.1 Effect of precipitant on Cu–Al LDH precursors and calcined products
We prepared a series of Cu–Al LDH precursors with fixed Cu2+:Al3+ atomic ratio of 1:1, but with the ratio of [NaOH]:[Na2CO3] in the precipitating agent varied, and found that the alkali precipitants could significantly alter the physical and chemical properties of the LDH precursors and derived Cu-based mixed metal oxides, as summarised in Table 1.
:[Al3+] = 1:1, with the concentration of NaOH and Na2CO3 varied, at constant pH of 9.6 ± 0.1
| Sample | LDHs [NaOH]:[Na2CO3] |
Precursor | Calcination | Derived metal oxides composite | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d(003) (Å) | L (LDH) (nm) | CO2 release (wt%) | L CuO (nm) | L CuAl2O4 (nm) | CuOb (wt%) | CuAl2O4b (wt%) | Al2O3b (wt%) | Na2Ob (wt%) | Nab (wt%) | R O2 (wt%) | R O (wt%) | ||
a
L
(LDH), LCuO, LCuAl2O4, crystallite size of LDH precursor d(003), CuO d(11![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and d(111), and CuAl2O4d(311), in the calcined product, respectively.
b Content quantified from elemental analyses by ICP-AES and weight ratio of crystalline CuO to CuAl2O4 from the XRD data, with uncertainty about 10%. Na could exist in the form of Na2O, NaAlO2 or other phases, here it is shown in the form of Na2O. The weight contents of Na are normalized from Na2O.
c
R
O2 is the gaseous O2 releasing capacity, as measured from the temperature programmed decomposition in TGA (Fig. S16), assumed as the weight loss of the first stage of O2 release from the crystalline CuO, with the slow oxygen release from CuAl2O4 not considered.
d
R
O is the total oxygen storage capacity of the carrier assuming complete reaction of Cu2+ → Cu0, as quantified by temperature programmed reduction in TGA (see ESI).
e Sample prepared with increasing pH method, NaOH was added dropwise to a solution of Cu–Al nitrates until the pH reached ∼4.0, and then Na2CO3 was added dropwise until the pH increased to 9.7. Details can be found in ESI (Fig. S10). ) and d(111), and CuAl2O4d(311), in the calcined product, respectively.
b Content quantified from elemental analyses by ICP-AES and weight ratio of crystalline CuO to CuAl2O4 from the XRD data, with uncertainty about 10%. Na could exist in the form of Na2O, NaAlO2 or other phases, here it is shown in the form of Na2O. The weight contents of Na are normalized from Na2O.
c
R
O2 is the gaseous O2 releasing capacity, as measured from the temperature programmed decomposition in TGA (Fig. S16), assumed as the weight loss of the first stage of O2 release from the crystalline CuO, with the slow oxygen release from CuAl2O4 not considered.
d
R
O is the total oxygen storage capacity of the carrier assuming complete reaction of Cu2+ → Cu0, as quantified by temperature programmed reduction in TGA (see ESI).
e Sample prepared with increasing pH method, NaOH was added dropwise to a solution of Cu–Al nitrates until the pH reached ∼4.0, and then Na2CO3 was added dropwise until the pH increased to 9.7. Details can be found in ESI (Fig. S10).
|
|||||||||||||
| 1 | 1 M:0 M |
7.60 | ∼28 | 1.79 | 33.2 ± 2 | 30 ± 2 | 30.17 | 68.10 | 1.73 | n.d. | n.d. | 3.0 | 12.0 |
| 2 | 0.94 M:0.06 M |
7.60 | ∼35 | 2.14 | 40.1 ± 5 | 24 ± 2 | 33.73 | 59.97 | 6.30 | 0.007 | 0.005 | 3.2 | 11.6 |
| 3 | 0.5 M:0.5 M |
7.62 | ∼40 | 3.05 | 28.2 ± 3 | 16 ± 3 | 44.49 | 33.42 | 20.64 | 1.45 | 1.08 | 4.0 | 11.2 |
| 4 | 0 M:1 M |
7.52 | ∼12 | 4.58 | 33.8 ± 3 | n.a. | 54.38 | 3.47 | 35.32 | 6.83 | 5.07 | 4.5 | 10.5 |
| 5 | 1 M:1 M |
7.60 | ∼13 | 5.38 | 37.5 ± 2 | 20.9 ± 4 | 52.83 | 5.87 | 30.66 | 10.64 | 7.90 | 5.2 | 11.9 |
| 6 | Increasing pHe | 7.55 | ∼23 | — | 48.2 ± 6 | 44.4 ± 5 | 30.8 | 66.7 | 2.50 | n.d. | n.d. | 3.2 | 12.0 |
The XRD patterns (Fig. 1A) of the precursors prepared with different precipitants correspond well to Cu–Al hydrotalcite (JCPDS 46-0099, as shown in Fig. 1) and Cu6Al2(OH)16CO3·4H2O (JCPDS 37-0630). Less crystalline material was present when the concentration of Na2CO3 in the precipitant was increased. The average crystallite size decreased to ∼13 nm when Na2CO3 was used as the precipitant (Table 1). FTIR spectra confirmed the chemistry of the LDH precursors, showing stronger absorbance associated with interlayer NO3− anions when NaOH was used as the precipitant, while stronger absorbance of interlayer CO32− anions were observed when Na2CO3 was used as the precipitant (Fig. S2†). We also confirmed that LDH could be formed with Cu:Al ratio between 1:2 to 3:1 using a mixture of NaOH and Na2CO3 (Fig. S3 and S4†).
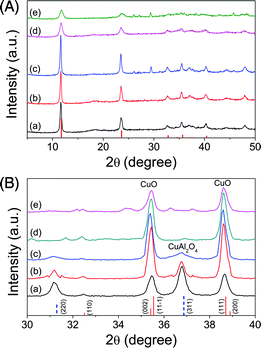 | ||
| Fig. 1 XRD patterns of (A) LDH precursor and (B) as calcined mixed metal oxides. The Cu–Al LDH precursors were prepared by coprecipitation of Cu–Al nitrates (Cu2+:Al3+ = 1:1) at constant pH of 9.6 ± 0.1. Precipitating agent for LDH precursors: (a) 1 M NaOH, (b) 0.94 M NaOH + 0.06 M Na2CO3, (c) 0.5 M NaOH + 0.5 M Na2CO3, (d) 1 M NaOH + 1 M Na2CO3, (e) 1 M Na2CO3. The crystalline pattern corresponds to Cu–Al hydrotalcite (JCPDS 46-0099, vertical lines). (B) Mixed metal oxides calcined from the precursors at 950 °C, prepared with varied precipitants. The crystalline patterns correspond to CuO (Tenorite, JCPDS 45-1548, vertical solid lines) and CuAl2O4 (JCPDS 33-0448, vertical dashed lines), respectively. | ||
The decomposition of LDH precursors was carried out in a TGA (see ESI, Fig. S4 and S5†) and gave similar patterns to those reported in the literature.58,61 The amounts of CO2 released during calcination were estimated from the TG curves. With the increase of concentration of Na2CO3, the amount of CO2 release during calcination increased as shown in Table 1, corresponding to the interlayer carbonate anions. The transformation of the precursors to mixed metal oxides was tracked with FTIR (Fig. S2†) and ex situ XRD (Fig. S6 and S7†).
For the samples prepared with Cu:Al molar ratio of 1:1, the crystalline phases of copper in the calcined material were also dependent on the concentration of Na2CO3 in the precipitant. As shown in Table 1, after calcination at 950 °C, CuAl2O4 was the dominant phase when 1 M NaOH was used as the precipitant. Increasing the concentration of Na2CO3 resulted in CuO became the dominant phase, with less crystalline CuAl2O4 formed. When 1 M Na2CO3 was used as the sole precipitant, weak peaks associated with CuAlO2 (37.08°, JCPDS 40-1037, 33.40° and 40.04°, JCPDS 21-0276) and NaAlO2 (JCPDS 33-1200) were also detected. Crystalline peaks of Al2O3 were not observed indicating that it existed as a semi-crystalline or amorphous phase.
The morphology of the LDH precursors prepared with Cu2+:Al3+ = 1:1 (total concentration 1 M) and their calcined products was also investigated with SEM and TEM (ESI, Fig. S8†). When NaOH was used as precipitant, the resulting LDH precursor consisted of densely packed crystals. After calcination, severe sintering of CuO and CuAl2O4 occurred. With the concentration of Na2CO3 increased to 1 M, nanoplatelet-like morphology was observed in the precursor and the average crystallite size of the LDH became smaller (∼12 nm). After calcination, the calcined product still had a platelet like structure with the Na2CO3 as precipitant, with a good dispersion of CuO nanocrystals in the support. This nanoplatelet structure can most clearly be seen for the material prepared with [NaOH]:[Na2CO3] = 1 M:1 M, and total cation concentration of 2 M, which is described in detail below.
3.2 Correlation of sodium with structure of LDH precursor and calcined product
An important finding is that the inhibition of formation of CuAl2O4 in the calcined oxygen carrier can be correlated with the content of residual sodium from precipitating agents. We quantified the contents of sodium in both the LDH precursors and calcined products by ICP-AES, as expressed in the form of Na2O or Na in the calcined materials in Table 1. The content of residual sodium could be correlated with the concentration of Na2CO3 in the precipitant (Table 1 and Fig. S9†). Since the oxygen carrier material with CuO loading of 60 wt% (Cu:Al = 1 M:1 M) prepared using a precipitant with [NaOH] = [Na2CO3] = 1 M had both a well defined physical structure (as discussed below) and contained little copper aluminates, this material was chosen for further investigation. We prepared many batches of 60 wt% CuO/Al2O3 but varied the degree of washing. The content of residual sodium could vary between 5 and 20 wt% and stabilised at about 5 wt% after repeated washing with distilled water for up to ∼10 times. This study indicates that the residual sodium existed as precipitate and ∼5 wt% of sodium seemed to be effective in limiting the formation of copper aluminates in the calcined material.
We further prepared Cu–Al precipitates by increasing or decreasing pH methods (see ESI, Fig. S10†), and confirmed that CuAl2O4 was the dominant phase after calcination whilst sodium was not detectable by ICP-AES, in both cases.
We investigated the crystalline phase of Na-containing species by control samples of copper-free aluminium hydroxides precursors and calcined product (see ESI, Fig. S11†). We confirmed the formation of dawsonite (NaAlCO3(OH)2, JCPDS 45-1359) in the precursors and sodium aluminates (i.e. NaAlO2) after calcination when Na2CO3 was used as the precipitant. These experiments confirmed that the presence of sodium was correlated with the inhibition of formation of copper aluminates.
We prepared Cu–Al LDH precursor with Cu:Al = 3:1, and found similar relationship between the crystalline phase and the precipitating agent (ESI, Fig. S12 and Table S1†). In the case of molar ratio of Cu:Al equal to 3, a sodium content of ∼1.4 wt% was sufficient to limit the formation of CuAl2O4 (Table S1b†). Low levels of copper aluminates were obtained when the sodium content was ∼3 wt% (obtained using Na2CO3 as the sole precipitant).
3.3 Morphology and structure of representative Cu–Al LDH precursors and derived CuO/Al2O3 nanocomposites
A representative Cu–Al LDH precursor was produced by coprecipitation of a more concentrated solution of copper and aluminium nitrates (Cu2+:Al3+ = 1:1, with total concentration of 2 M) at constant pH (9.6 ± 0.1), using concentrated mixture of NaOH and Na2CO3 (1 M of each) as precipitating agent. The XRD pattern of this sample also matched well with Cu–Al hydrotalcite (JCPDS 46-0099) as shown in Fig. 1A. SEM images (Fig. 2a) of the LDH precursors show the disordered packing of the nanoplatelets with lateral size about 500 nm and average thickness of ∼15 nm, in agreement with the average crystallite size (∼13 nm) from the XRD pattern. STEM image of the precursor with Cu:Al = 1:1 (Fig. 2b) shows the homogeneous LDH crystals although some impurities were also present.
 | ||
| Fig. 2 SEM/TEM images of Cu–Al LDH precursor and derived mixed metal oxides. (a) SEM and (b) STEM micrograph of the Cu–Al LDH precursor prepared from coprecipitation of Cu and Al nitrates, with Cu2+:Al3+ = 1:1 (total concentration of 2 M), precipitating agent of mixture of NaOH and Na2CO3 (1 M of each), (c) SEM, (d) STEM and (e) HRTEM image of as-calcined mixed metal oxides, with bottom right inset showing the SADP pattern. The lattice fringes with a separation of ∼2.5 Å, is in agreement with the (11) interplanar d spacing (2.53 Å) of monoclinic tenorite CuO (JCPDS 48-1548). | ||
The calcined mixed metal oxides at 950 °C (Fig. 1B) showed the dominant phase of crystalline CuO, with very weak peaks corresponding to CuAl2O4 (JCPDS 33-0448), CuAlO2 (JCPDS 40-1037) and NaAlO2 (JCPDS 33-1200, 2θ = 30–35°). The content of residual sodium in the calcined product was quantified as 7.9 wt% by ICP-AES. We observed large grains in SEM while the nanoplatelet-like morphology was preserved in most regions (Fig. 2c), with the thickness of each platelet being around 20–40 nm. STEM (Fig. 2d) and HRTEM (Fig. 2e) images confirmed that the CuO crystals (as confirmed by the lattice spacing) with size of 20–40 nm were reasonably well dispersed in an amorphous matrix. The selected area diffraction pattern (SADP), as shown in the inset to Fig. 2e, only showed bright electron diffraction spots, indicating the single crystalline structure of CuO. N2 adsorption isotherms showed that the specific surface area was quite low (∼5 m2 g−1) due to severe annealing at high temperature, yet the calcined materials were mesoporous.
Similar observations were made for the material prepared with Cu2+:Al3+ = 3:1, with total concentration of 1 M at constant pH (9.6 ± 0.1), using a precipitant with [NaOH] = [Na2CO3] = 0.5 M (see ESI, Fig. S13†). It is worth noting that when a precipitate was prepared with no aluminium phase present, the morphology was very different, with severe sintering observed after calcination.
3.4 Oxygen storage capacity and gaseous O2 release capacity
We carried out temperature programmed reduction in a TGA and quantified the total amount of reducible oxygen within the oxygen storage materials. For the detailed profiles refer to the ESI, Fig. S14 and S15.† According to the literature on reduction of CuO supported on Al2O3,61,62 the kinetics of reduction vary with the material, depending on the grain size, crystallite size and dispersion of the CuO, crystallinity, and degree of interaction with the support.62 Compared to the pure CuO, the CuO/Al2O3 derived from the LDH precursor showed more facile reduction, owing to the smaller crystals and more porous structure. Table 1 summarises the oxygen storage capacity of particles with nominal CuO loading of 60 wt% by different precipitant. The CuAl2O4 spinel is reducible; therefore the overall oxygen storage capacity (CuO to Cu) is approximately equal to the theoretical value of 12 wt%. When Na2CO3 was used as the precipitant, the total oxygen storage capacity (∼11 wt%) was slightly less than that expected from the precursors. A possible reason for the discrepancy is that the weight percentage of copper in the final product was different owing to the precipitation of some sodium containing species. Despite the presence of sodium, the oxygen carriers still appeared to perform well in the cycling experiments as described below.In the case of gaseous O2 release capacity, the formation of CuAl2O4 has been shown to reduce the oxygen release capacity at typical CLOU operating temperatures (800–950 °C) owing to the low equilibrium partial pressure associated with decomposition of CuAl2O4,33 and its slow kinetics.43 In the case of mixture of CuO and CuAl2O4, the rate of O2 release is mainly dominated by the crystalline CuO. Table 1 shows that a higher content of crystalline CuO gives higher gaseous O2 releasing capacity, as measured by temperature programmed decomposition in TGA (Fig. S16†).
3.5 Application of mixed metal oxides derived from LDH precursor as oxygen storage material
In the following sections, the 60 wt% CuO/Al2O3 with sodium content of 7.9 wt% prepared from a LDH precursor with Cu:Al = 1:1 and precipitating agent of NaOH and Na2CO3 (1 M of each) was extensively tested in both CLOU and CLC cycles. In the CLC cycles, the 82.5 wt% CuO/Al2O3 with sodium content of 1.4 wt% derived from Cu–Al LDH with Cu:Al = 3:1 and precipitating agent of NaOH and Na2CO3 (0.5 M of each) was also tested.
The stability of the materials is essential for economic operation in large-scale chemical-looping process. The oxygen carrier materials sampled during the multiple cycles were characterised with various physical and chemical techniques, such as SEM-EDX, STEM, XRD, N2 adsorption, ICP-AES, and TPR. Detailed characterisation results can be found in the ESI.†
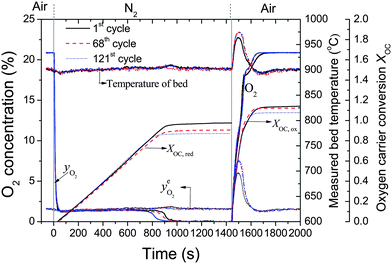 | ||
Fig. 3 Reversible oxygen release and storage of a Cu-based oxygen carrier material in a fluidised bed. The reactor was loaded with 15 g of 60 wt% CuO/Al2O3. The equilibrium O2 partial pressure, 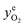 , of CuO/Cu2O couple, is calculated based on the measured bed temperature.63 Vertical lines indicate the switching of inlet gases of N2 (50.0 mL s−1, STP) during O2 release and air (47.4 mL s−1, STP) during oxidation. The oxygen carrier was Cu-based mixed metal oxides with nominal CuO loading of 60 wt% calcined from Cu–Al LDH precursor, prepared with Cu2+:Al3+ = 1:1 (total concentration of 2 M), precipitating agent of mixture of NaOH and Na2CO3 (1 M of each). The content of sodium in the fresh particles was 7.9 wt% as measured by ICP-AES. , of CuO/Cu2O couple, is calculated based on the measured bed temperature.63 Vertical lines indicate the switching of inlet gases of N2 (50.0 mL s−1, STP) during O2 release and air (47.4 mL s−1, STP) during oxidation. The oxygen carrier was Cu-based mixed metal oxides with nominal CuO loading of 60 wt% calcined from Cu–Al LDH precursor, prepared with Cu2+:Al3+ = 1:1 (total concentration of 2 M), precipitating agent of mixture of NaOH and Na2CO3 (1 M of each). The content of sodium in the fresh particles was 7.9 wt% as measured by ICP-AES. | ||
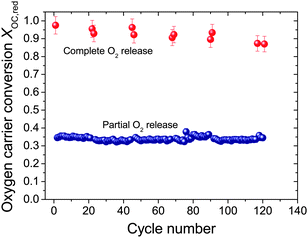 | ||
| Fig. 4 Conversion of oxygen carrier as a function of the number of oxygen uncoupling cycles. XOC,red is calculated based on the amount of O2 released by the entire batch of oxygen carriers in the reactor. Partial O2 release means the O2 release over a short period of 360 s, whilst complete O2 release was performed over extended time of 1450 s. A batch of oxygen carrier materials (15 g) were alternately exposed to inert gas (N2, 50.0 mL s−1, STP) and oxidising atmosphere (air, 47.4 mL s−1, STP) in a fluidised bed with the operation temperature set to 900 °C. The oxygen carrier was Cu-based mixed metal oxides with nominal CuO loading of 60 wt% calcined from Cu–Al LDH precursor, prepared with Cu2+:Al3+ = 1:1 (total concentration of 2 M), precipitating agent of mixture of NaOH and Na2CO3 (1 M of each). The content of sodium in the fresh particles was 7.9 wt% as measured by ICP-AES. | ||
The variation of the molar conversion of the oxygen carrier from CuO to Cu2O, XOC,red over 121 consecutive cycles at 900 °C is presented in Fig. 4. In certain cycles, after complete release of oxygen, we sampled about 200 mg of materials for characterisation analysis and added the same amount of fresh particles. The overall performance of the particles did not vary after each sampling and make-up process. Overall, the oxygen carrier particles were continuously cycled and fluidised in the reactor for nearly 40 hours. Generally, the conversion of CuO to Cu2O, during the fixed period of 360 s for partial decomposition at 900 °C, was approximately constant at 0.35, indicating that the kinetics remained fast enough for the decomposition to be limited by the equilibrium. The ultimate conversion, when CuO was fully decomposed to Cu2O, decreased from 0.99 initially to 0.88 after 121 cycles. This slight loss in the quantity of O2 released from the reactor is probably owing to a number of factors, e.g. a small amount of attrition, some slight deactivation, and accumulated experimental error over the long experiment.
Further experiments of 20 cycles at 950 °C also showed excellent stability of oxygen release and storage (Fig. S18†). We also studied the O2 release and storage over the temperature range of 800–1000 °C (Fig. S19†) and found the kinetics was fast and limited by the thermodynamic equilibrium in most cases. Ex situ temperature programmed decomposition and temperature programmed reduction in the TGA confirmed that the oxygen carrier maintained the gaseous O2 release capacity (∼5 wt%, Fig. S20†) and lattice oxygen storage capacity (∼11.5 wt% at 900 °C, Fig. S21†) as the fresh ones. Detailed characterisation of the used oxygen carrier materials can be found in the ESI,†e.g. XRD analysis (Fig. S22†) and SEM images (Fig. S23†). In addition, the particles with an initial size of 500–710 μm were robust without significant reduction of size by attrition even after continuous operation for 40 h in a fluidised bed. Agglomeration was not observed either in CLOU cycles or in redox cycles. ICP-AES confirmed that the content of Na was quite stable after multiple CLOU cycles, from 7.9 wt% in fresh materials to ∼7.6 wt% after 20 cycles at 900 and 950 °C, and slight decrease to ∼6.7 wt% in the samples after 121 cycles at 900 °C. From these cycling experiments and characterisation analyses, the Cu-based oxygen carrier with nominal CuO loading of 60 wt% has shown excellent stability for the CLOU process. Further research on the performance of oxygen carrier materials for coal combustion with the CLOU technique are underway and will be reported in the near future.
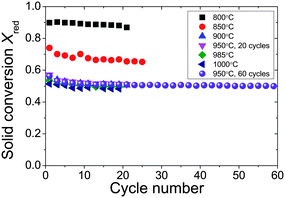 | ||
| Fig. 5 Conversion of oxygen carrier as a function of the number of reduction–oxidation cycles at representative operation temperatures. Xred is calculated based on amount of CO2 produced from the entire batch of oxygen carriers, reduced by gaseous fuel CO. A batch of ∼0.5 g oxygen carrier was exposed to cyclic reduction and oxidation in a fluidised bed, using CO as gaseous fuel (∼2.4 vol% CO in N2, 65.8 mL s−1, STP), N2 as purging gas (50.0 mL s−1, STP), and air as oxidising gas (47.4 mL s−1, STP). The operation temperature was 800–1000 °C, in isothermal mode. The oxygen carrier was Cu-based mixed metal oxides with nominal CuO loading of 60 wt% calcined from Cu–Al LDH precursor, prepared by coprecipitation of Cu and Al nitrates with Cu2+:Al3+ = 1:1 (total concentration of 2 M), precipitating agent of mixture of NaOH and Na2CO3 (1 M of each). The content of sodium in the fresh particles was 7.9 wt% as measured by ICP-AES. | ||
Previously, Chuang et al.45,59 have shown that the rate of reduction was limited by external mass transfer under similar operating conditions. Therefore, the data did not reflect the intrinsic kinetics. However, the apparent rate of reaction may still be useful to evaluate the degree of sintering, attrition and agglomeration. With a loading of CuO of 82.5 wt% (residual sodium of 1.4 wt%), the oxygen carrier materials were stable over 20 redox cycles over a temperature range 800–900 °C. However, when the sample was cycled at temperatures of 950 °C and above, the rate of production of CO2 fell, and the bed defluidised with large agglomerates recovered from the bed after 20 cycles (Fig. S28†).
The structural evolution of CuO/Al2O3 at high loading of 82.5 wt% over redox cycles at high temperature was examined. The initial particles showed nanoplatelet-like structure (Fig. 6a) and could be preserved after cycling at 800 °C. However, severe sintering and agglomeration occurred when the temperature was above 950 °C. The sintering leads to a substantial decrease of available surface area for gas solid reduction, therefore the rate of reaction fell as found in the redox cycles, albeit with the overall oxygen carrying capacity being unchanged. At temperature of 900 °C, we observed partial sintering with nanoplatelets still preserved in some regions of the particles. More detailed SEM and STEM images are given in ESI, Fig. S27–28.† Interestingly, although severe sintering occurred, the XRD patterns of all samples were quite stable, without observation of significant peaks of CuAl2O4 or crystalline Al2O3 (Fig. S29†).
 | ||
| Fig. 6 Representative SEM/STEM images of fresh and used Cu-based oxygen carrier materials. (a) Fresh 82.5 wt% CuO/Al2O3, (b) severe sintering of 82.5 wt% CuO/Al2O3 after 20 redox cycles at 950 °C, (c) partial sintering of 82.5 wt% CuO/Al2O3 after 20 cycles at 900 °C and (d) enlargement showing nanoplatelets were preserved in some regions, (e) used 60 wt% CuO/Al2O3 after 20 redox cycles at 950 °C and (f) enlargement, showing some regions have been sintered into large grains mixed with regions where the nanoplatelets were preserved. (g) SEM and (h) STEM of used 60 wt% CuO/Al2O3 after 60 redox cycles at 950 °C. The arrows indicate the zig-zag structure, which limits the nanoplatelets from face-on-face packing. The 82.5 wt% CuO/Al2O3 was calcined from Cu–Al LDH, Cu2+:Al3+ = 3:1 (total concentration of 1 M), precipitated with a mixture of NaOH and Na2CO3 (0.5 M of each). The 60 wt% CuO/Al2O3 was calcined from Cu–Al LDH, with Cu2+:Al3+ = 1:1 (total concentration of 2 M), precipitated with a mixture of NaOH and Na2CO3 (1 M of each). The content of sodium in the fresh 82.5 wt% and 60 wt% CuO/Al2O3 was 1.4 wt% and 7.9 wt%, respectively, as measured by ICP-AES. | ||
In contrast, the CuO/Al2O3 with nominal CuO loading of 60 wt% showed improved stability at high temperatures (up to 1000 °C). Agglomeration was not observed, and the solid conversion over each cycle was stable (as shown in Fig. 5) and the profiles of conversion versus time at 950 °C remain stable over 60 redox cycles. In fact, the particles appeared to be stable even at 985 and 1000 °C over 20 cycles. TPR confirmed that the reducible oxygen (Fig. S30†) remained at ∼12 wt% over 800–950 °C, with only slight loss to ∼10 wt% at 1000 °C.
We also characterised the oxygen carrier with nominal CuO loading of 60 wt% after redox cycles. Large grains were formed due to sintering at high temperature (950 °C), whilst in some regions the nanoplatelets were still preserved, as shown in Fig. 6f–g and S31–32.† One of the key concerns for Cu-based oxygen carriers supported on Al2O3 is their phase stability in redox cycles. These results show that it is possible to have reversible phase change between CuO–Cu2O–Cu, during redox cycling, as shown in Fig. 7. The crystalline phases of oxidised samples were maintained without further formation of CuAl2O4 or crystalline Al2O3 (Fig. S29†). The STEM analysis showed that the CuO nanocrystals were still well dispersed in the sintered matrix, as shown in Fig. 6h.
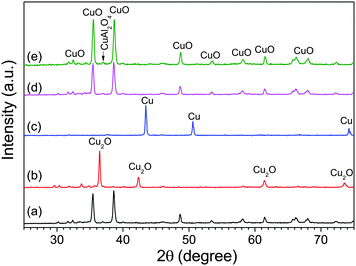 | ||
| Fig. 7 Reversible phase change of CuO–Cu2O–Cu over multiple redox cycles. XRD patterns of (a) fresh as calcined from precursor, (b) completely decomposed in N2 at 900 °C, (c) completely reduced by CO at 950 °C, (d) oxidised after 121 cycles of O2 release and storage at 900 °C, and (e) oxidised after 60 redox cycles at 950 °C. The oxygen carrier was Cu-based mixed metal oxides with nominal CuO loading of 60 wt% calcined from Cu–Al LDH precursor, prepared by coprecipitation of Cu and Al nitrates, with Cu2+:Al3+ = 1:1 (total concentration of 2 M) and precipitating agent of mixture of NaOH and Na2CO3 (1 M of each). The content of sodium in the fresh particles was 7.9 wt% as measured by ICP-AES. | ||
4 Discussion
4.1 Material chemistry
A part of this study was to establish the structure–property relationship of the LDH precursor and derived mixed metal oxides. We found that good performance of the oxygen carriers is associated with (i) well defined precursor structure, (ii) a stable, amorphous support, (iii) small, well-dispersed crystallites of CuO, and (iv) a relatively stable nanoplatelet structure.As noted in the Introduction, previous studies found that CuAl2O4 is formed when CuO is supported on various types of Al2O3 (and is in-fact the thermodynamically stable phase). A small amount of CuAl2O4 (i.e. <10 wt%) in the oxygen carrier does not appear to significantly affect the gaseous O2 release capacity.43 Our results in this paper also confirm this observation. In the case of reduction by gaseous fuel, CuAl2O4 is still reducible, therefore the oxygen storage capacity (reducible oxygen) is only slightly affected.44 The presence of a small amount of CuAl2O4 may also enhance the mechanical strength of the particles.45 However, formation of a significant amount of CuAl2O4 in the materials is not favourable for CLOU process because CuAl2O4 exerts a low equilibrium partial pressure of O2,64 and kinetically, the rate of gaseous O2 release from CuAl2O4 is much lower than that of CuO.43 Therefore the formation of the copper aluminates could lead to a lower O2 release capacity and slower reduction and reoxidation kinetics.
We found the formation of CuAl2O4 could be impeded by the presence of sodium. Our results suggest that the residual sodium could exist as dawsonite, NaAl(CO3)(OH)2, when the precipitant contains high level of sodium carbonate. These sodium containing species were mixed in with the LDH precipitate. When calcined, the decomposition of dawsonite produces NaAlO2.58,65 The NaAlO2 may further react with Al2O3 forming NaxAlyOz species (i.e. NaAl11O17) and further prevent the interaction between copper and aluminium phases.
We propose that the sodium present in the oxygen carrier prevented the crystallisation of the Al2O3 in the support, and its interaction with the copper atoms, resulting in relatively immobile copper crystallites which remained dispersed within the support, despite extensive redox cycling. Owing to the presence of sodium-containing species, the formation of spinel phase CuAl2O4 was limited. This is in stark contrast to the result expected from the phase diagram for the Cu–Al–O system,64 which indicates that CuAl2O4 is formed when they are fully oxidised in air. In an earlier study, it was found even relatively small amounts of sodium could retard the transition of Al2O3 to α-alumina.66 The same authors66 also found that the formation of copper aluminates was inhibited when sodium was present. They suggested that, if intimately mixed, NaAlO2 would concentrate at the CuO–Al2O3 grain boundaries to inhibit the diffusion of Cu2+ and Cu+ ions into the Al2O3 matrix to form CuAl2O4 and CuAlO2.65–67 This explanation may illustrate the stabiliser role of sodium in this study as well. The use of MgAl2O4 or NiAl2O4 as an alternative support has been proposed as a solution.68,69 However, we demonstrated that the oxygen carrier can be simply stabilised by controlling the precursor chemistry.
It should also be noted that the melting point of NaAlO2 is about 1650 °C, thus it is thermally stable and could also act as a promotional support along with the amorphous Al2O3. The presence of Na may also promote oxidation, and is used as a promoter in CO oxidation catalysts,70 and three-way catalysts for automobile exhausts.71 The kinetics we measured were too fast to be able to see any such affect here, and more fundamental studies are necessary to understand the effect of sodium.
Whilst LDH precursor chemistry is very flexible, precipitation methods can be difficult to scale up and are more expensive than simpler methods (e.g. mechanical mixing). Whether or not the addition of sodium (e.g. by impregnation) to CuO/Al2O3 particles produced using other methods will have the same effect requires further investigation and is beyond the scope of this paper. For other, potentially more cost effective, routes to CuO/Al2O3 particles, the impact of addition of a third metal (i.e. sodium) to tune the metal-support interaction is likely to be complicated by the initial distribution and loading of the copper (e.g. mechanical mixing results in large crystallites,45 rather than finely dispersed material in a support) and the dispersion of the sodium. If the sodium is able to diffuse to the interface between the copper phase and alumina phase, there may be some beneficial effect, though further work would be required to confirm this.
More fundamental studies are necessary for a more thorough understanding of the material chemistry and structure–property relationship. Particularly, in situ characterisation techniques can be extremely useful to understand the chemical and structural change at atomic level.72–76
4.2 Agglomeration and attrition
In chemical looping, agglomeration as a result of sintering is a major challenge for continuous operation of fluidised beds at high temperatures. In the oxygen uncoupling experiments, agglomeration was not observed regardless of the oxygen carrier used. However, when CO was used as the reducing gas, i.e. the Cu2O was further reduced to Cu, agglomeration was observed for the 82.5 wt% CuO samples when cycled at temperatures above 900 °C. By reducing the loading to 60 wt% and controlling the method of preparation, the extent of sintering was much reduced over redox cycles at 800–1000 °C due to the stable dispersion of CuO nanocrystals in the solid support. Consequently, agglomeration did not occur for this nanostructured oxygen carrier. The loading of CuO was not optimised in this study and might be further increased whilst maintaining the high degree of dispersion of CuO nanocrystals in the support, consequently a higher oxygen release and storage capacity could be obtained.Over the 40 hours of CLOU operation, about 3 wt% of the original oxygen carrier material was elutriated due to attrition. In this laboratory scale preparation, particles were obtained by crushing the calcined powder of the precipitate, whereas large-scale pelletising techniques for commercial application, e.g. spray drying of the LDH precipitate, would be capable of producing spherical particles which are more suitable for fluidisation and might give greater mechanical strength.6
4.3 LDHs-derived mixed metal oxides for energy applications”
Similar to the CLC process, many other novel processes for energy applications are based on thermochemical cycles of materials. Examples are chemical-looping reforming,6 steam-iron process for H2 production,77 calcium looping,78,79 O2 production,80 and solar hydrogen production.81,82 In all these high temperature processes, sintering of materials at high temperature is a major challenge for stable operation. According to the established chemistry of LDHs, i.e. versatile combination of cations and intercalation of anions, we anticipate that the strategy reported in this study would inspire the design of many other mixed metal oxides for application in novel high temperature redox cycles, by carefully tuning the precursor chemistry.5 Conclusions
We report the synthesis of mixed metal oxides from layered double hydroxides precursor and their applications as oxygen storage material for the chemical-looping process. Using a Cu–Al LDH as prototype, we found the physical and chemical properties of the LDH precursor could be tuned by the precursor chemistry, i.e. varying the molar ratio of the precursor solution, interlayer anions, and content of residual sodium from the precipitating agent. Calcination of the Cu–Al LDH precursor generates a nanostructured matrix, with CuO nanocrystals well dispersed in an amorphous support. The formation of CuAl2O4 was inhibited due to the presence of sodium-containing phase originated from the precursor and this inhibition is persistent after extensive redox cycles at high temperatures. Although some local sintering was observed after calcination, the nanoplatelet-like morphology was preserved. As a result of the nanoplatelet-like microstructure and high degree of dispersion of CuO in the stable support, the metal oxides nanocomposite showed a fast rate of reaction and improved cycling stability over multiple redox cycles under CLOU and CLC reaction conditions at high temperatures. The high and reversible oxygen release and storage capacity, reactivity, and excellent thermal stability make this nanostructured sodium-promoted CuO/Al2O3 composite a promising candidate oxygen carrier material for chemical looping processes. We anticipate that the powerful LDH chemistry and diverse synthetic routes can be easily used to manufacture other metal oxide composites as oxygen carriers, catalysts, and CO2 capture sorbents, for applications in high temperature reactions that are promising for clean energy production and CO2 capture.Acknowledgements
This work was financially supported by Engineering and Physical Sciences and Research Council (EPSRC, UK) and the National Basic Research Program of China (973 Program) (Grant no. 2010CB732206). Q.S. acknowledges a PhD scholarship from China Scholarship Council. The authors are grateful to Mr Andrew Moss for access to the XRD facilities and Mr Zlatko Saracevic for help with the TGA experiments and BET analysis. The authors also appreciate Dr Tamaryn Brown, Dr Shin Yong Chuang and Ms Weiyao Ma for help with the fluidised bed experiments.References
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS.
- W. K. Lewis and E. R. Gilliland, US Pat., no: 2665972, 1954.
- W. K. Lewis, E. R. Gilliland and W. A. Reed, Ind. Eng. Chem., 1949, 41, 1227–1237 CrossRef CAS.
- A. Lyngfelt, B. Leckner and T. Mattisson, Chem. Eng. Sci., 2001, 56, 3101–3113 CrossRef CAS.
- L.-S. Fan, L. Zeng, W. Wang and S. Luo, Energy Environ. Sci., 2012, 5, 7254–7280 CAS.
- J. Adánez, A. Abad, F. García-Labiano, P. Gayán and L. F. de Diego, Prog. Energy Combust. Sci., 2012, 38, 215–282 CrossRef.
- J. Adánez, L. F. de Diego, F. García-Labiano, P. Gayán, A. Abad and J. M. Palacios, Energy Fuels, 2004, 18, 371–377 CrossRef.
- J. S. Dennis, S. A. Scott and A. N. Hayhurst, J. Energy Inst., 2006, 79, 187–190 CrossRef CAS.
- S. A. Scott, J. S. Dennis, A. N. Hayhurst and T. Brown, AIChE J., 2006, 52, 3325–3328 CrossRef CAS.
- J. S. Dennis, C. R. Müller and S. A. Scott, Fuel, 2010, 89, 2353–2364 CrossRef CAS.
- J. S. Dennis and S. A. Scott, Fuel, 2010, 89, 1623–1640 CrossRef CAS.
- L. Shen, J. Wu, Z. Gao and J. Xiao, Combust. Flame, 2010, 157, 934–942 CrossRef CAS.
- R. Xiao, L. Chen, C. Saha, S. Zhang and S. Bhattacharya, Int. J. Greenhouse Gas Control, 2012, 10, 363–373 CrossRef CAS.
- L. H. Shen, J. H. Wu, J. Xiao, Q. L. Song and R. Xiao, Energy Fuels, 2009, 23, 2498–2505 CrossRef CAS.
- L. H. Shen, J. H. Wu and J. Xiao, Combust. Flame, 2009, 156, 721–728 CrossRef CAS.
- N. Berguerand and A. Lyngfelt, Fuel, 2008, 87, 2713–2726 CrossRef CAS.
- H. Leion, A. Lyngfelt, M. Johansson, E. Jerndal and T. Mattisson, Chem. Eng. Res. Des., 2008, 86, 1017–1026 CrossRef CAS.
- H. Leion, T. Mattisson and A. Lyngfelt, Int. J. Greenhouse Gas Control, 2008, 2, 180–193 CrossRef CAS.
- N. Berguerand and A. Lyngfelt, Energy Fuels, 2009, 23, 5257–5268 CrossRef CAS.
- R. Xiao, Q. L. Song, M. Song, Z. J. Lu, S. Zhang and L. H. Shen, Combust. Flame, 2010, 157, 1140–1153 CrossRef CAS.
- R. Xiao, Q. L. Song, S. Zhang, W. G. Zheng and Y. C. Yang, Energy Fuels, 2010, 24, 1449–1463 CrossRef CAS.
- Q. L. Song, R. Xiao, Z. Y. Deng, L. H. Shen, J. Xiao and M. Y. Zhang, Ind. Eng. Chem. Res., 2008, 47, 8148–8159 CrossRef CAS.
- Q. L. Song, R. Xiao, Z. Y. Deng, H. Y. Zhang, L. H. Shen, J. Xiao and M. Y. Zhang, Energy Convers. Manage., 2008, 49, 3178–3187 CrossRef CAS.
- Q. L. Song, R. Xiao, Z. Y. Deng, W. G. Zheng, L. H. Shen and J. Xiao, Energy Fuels, 2008, 22, 3661–3672 CrossRef CAS.
- R. Xiao and Q. Song, Combust. Flame, 2011, 158, 2524–2539 CrossRef CAS.
- T. Mattisson, A. Lyngfelt and H. Leion, Int. J. Greenhouse Gas Control, 2009, 3, 11–19 CrossRef CAS.
- T. Mattisson, H. Leion and A. Lyngfelt, Fuel, 2009, 88, 683–690 CrossRef CAS.
- H. Leion, Y. Larring, E. Bakken, R. Bredesen, T. Mattisson and A. Lyngfelt, Energy Fuels, 2009, 23, 5276–5283 CrossRef CAS.
- M. Rydén, A. Lyngfelt and T. Mattisson, Int. J. Greenhouse Gas Control, 2011, 5, 356–366 CrossRef.
- A. Shulman, C. Linderholm, T. Mattisson and A. Lyngfelt, Ind. Eng. Chem. Res., 2009, 48, 7400–7405 CrossRef CAS.
- A. Shulman, E. Cleverstam, T. Mattisson and A. Lyngfelt, Fuel, 2011, 90, 941–950 CrossRef CAS.
- H. Leion, T. Mattisson and A. Lyngfelt, Energy Procedia, 2009, 1, 447–453 CrossRef CAS.
- M. Arjmand, A. M. Azad, H. Leion, A. Lyngfelt and T. Mattisson, Energy Fuels, 2011, 25, 5493–5502 CrossRef CAS.
- I. Adánez-Rubio, P. Gayán, F. García-Labiano, L. F. de Diego, J. Adánez and A. Abad, Energy Procedia, 2011, 4, 417–424 CrossRef.
- P. Gayán, I. Adánez-Rubio, A. Abad, L. F. de Diego, F. García-Labiano and J. Adánez, Fuel, 2012, 96, 226–238 CrossRef.
- A. Abad, I. Adánez-Rubio, P. Gayán, F. García-Labiano, L. F. de Diego and J. Adánez, Int. J. Greenhouse Gas Control, 2012, 6, 189–200 CrossRef CAS.
- R. Naqvi and O. Bolland, Int. J. Greenhouse Gas Control, 2007, 1, 19–30 CrossRef CAS.
- H. Leion, Y. Larring, E. Bakken, R. Bredesen, T. Mattisson and A. Lyngfelt, Energy Fuels, 2009, 23, 5276–5283 CrossRef CAS.
- I. Adánez-Rubio, A. Abad, P. Gayán, L. F. de Diego, F. García-Labiano and J. Adánez, Fuel, 2012, 102, 634–645 CrossRef.
- I. Adánez-Rubio, P. Gayán, A. Abad, L. F. de Diego, F. García-Labiano and J. Adánez, Energy Fuels, 2012, 26, 3069–3081 CrossRef.
- T. Tsuchida, R. Furuichi, T. Sukegawa, M. Furudate and T. Ishii, Thermochim. Acta, 1984, 78, 71–80 CrossRef CAS.
- T. James, M. Padmanabhan, K. G. K. Warrier and S. Sugunan, Mater. Chem. Phys., 2007, 103, 248–254 CrossRef CAS.
- M. Arjmand, A. M. Azad, H. Leion, A. Lyngfelt and T. Mattisson, Energy Fuels, 2011, 25, 5493–5502 CrossRef CAS.
- L. F. de Diego, F. García-Labiano, P. Gayán, J. Celaya, J. M. Palacios and J. Adánez, Fuel, 2007, 86, 1036–1045 CrossRef CAS.
- S. Y. Chuang, J. S. Dennis, A. N. Hayhurst and S. A. Scott, Combust. Flame, 2008, 154, 109–121 CrossRef CAS.
- F. Li, X. Duan, X. Duan and D. Evans, Applications of Layered Double Hydroxides, Springer, Berlin/Heidelberg, 119th edn, 2006, pp. 193–223 Search PubMed.
- V. R. L. Constantino and T. J. Pinnavaia, Inorg. Chem., 1995, 34, 883–892 CrossRef CAS.
- V. Rives and M. a. Angeles Ulibarri, Coord. Chem. Rev., 1999, 181, 61–120 CrossRef CAS.
- S. P. Newman and W. Jones, New J. Chem., 1998, 22, 105–115 RSC.
- A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191–3198 RSC.
- G. R. Williams and D. O'Hare, J. Mater. Chem., 2006, 16, 3065–3074 RSC.
- C. Baltes, S. Vukojevic and F. Schüth, J. Catal., 2008, 258, 334–344 CrossRef CAS.
- M. Behrens, I. Kasatkin, S. Kühl and G. Weinberg, Chem. Mater., 2009, 22, 386–397 CrossRef.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS.
- S. Wang, S. Yan, X. Ma and J. Gong, Energy Environ. Sci., 2011, 4, 3805–3819 CAS.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS.
- P. H. Chang, Y. P. Chang, S. Y. Chen, C. T. Yu and Y. P. Chyou, ChemSusChem, 2011, 4, 1844–1851 CrossRef CAS.
- A. Alejandre, F. Medina, X. Rodriguez, P. Salagre and J. E. Sueiras, J. Catal., 1999, 188, 311–324 CrossRef CAS.
- S. Y. Chuang, J. S. Dennis, A. N. Hayhurst and S. A. Scott, Proc. Combust. Inst., 2009, 32, 2633–2640 CrossRef CAS.
- A. Naghash, T. H. Etsell and B. Lu, J. Mater. Chem., 2008, 18, 2562–2568 RSC.
- B. Bridier, N. López and J. Pérez-Ramírez, J. Catal., 2010, 269, 80–92 CrossRef CAS.
- M. F. Luo, P. Fang, M. He and Y. L. Xie, J. Mol. Catal. A: Chem., 2005, 239, 243–248 CrossRef CAS.
- I. Barin, F. Sauert and G. Patzki, Thermochemical Data of Pure Substances, Wiley-VCH, Weinheim, New York, 1995 Search PubMed.
- K. T. Jacob and C. B. Alcock, J. Am. Ceram. Soc., 1975, 58, 192–195 CrossRef CAS.
- M. M. Selim and N. A. Youssef, Thermochim. Acta, 1987, 118, 57–63 CrossRef CAS.
- G. A. El-Shobaky, T. El-Nabarawy, G. A. Fagal and N. H. Amin, Thermochim. Acta, 1989, 141, 195–203 CrossRef CAS.
- G. A. El-Shobaky, T. El-Nabarawy and G. A. Fagal, Appl. Catal., 1989, 52, 33–40 CrossRef CAS.
- C. R. Forero, P. Gayán, F. García-Labiano, L. F. de Diego, A. Abad and J. Adánez, Int. J. Greenhouse Gas Control, 2011, 5, 659–667 CrossRef CAS.
- P. Gayán, C. R. Forero, A. Abad, L. F. De Diego, F. García-Labiano and J. Adánez, Energy Fuels, 2011, 25, 1316–1326 CrossRef.
- N. Macleod, J. Isaac and R. M. Lambert, J. Catal., 2000, 193, 115–122 CrossRef CAS.
- V. Matsouka, M. Konsolakis, R. M. Lambert and I. V. Yentekakis, Appl. Catal., B, 2008, 84, 715–722 CrossRef CAS.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS.
- M. A. Newton, C. Belver-Coldeira, A. Martinez-Arias and M. Fernandez-Garcia, Nat. Mater., 2007, 6, 528–532 CrossRef CAS.
- P. Nolte, A. Stierle, N. Y. Jin-Phillipp, N. Kasper, T. U. Schulli and H. Dosch, Science, 2008, 321, 1654–1658 CrossRef CAS.
- F. Tao and M. Salmeron, Science, 2011, 331, 171–174 CrossRef CAS.
- C. D. Bohn, C. R. Muller, J. P. Cleeton, A. N. Hayhurst, J. F. Davidson, S. A. Scott and J. S. Dennis, Ind. Eng. Chem. Res., 2008, 47, 7623–7630 CrossRef CAS.
- J. S. Dennis and R. Pacciani, Chem. Eng. Sci., 2009, 64, 2147–2157 CrossRef CAS.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- Z. S. Li, T. Zhang and N. S. Cai, Ind. Eng. Chem. Res., 2008, 47, 7147–7153 CrossRef CAS.
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048–4077 CrossRef CAS.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, detailed characterisation of Cu–Al LDH precursors and derived mixed metal oxides, including SEM/TEM, XRD, FTIR, thermal analyses, TPR profiles, detailed profiles of O2 release and storage cycles, detailed profiles of cyclic reduction and oxidation cycles, characterisation of used oxygen carrier materials with various techniques, including XRD, SEM, STEM, and TPR. See DOI: 10.1039/c2ee22801g |
| This journal is © The Royal Society of Chemistry 2013 |