 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-lived photoinduced charge separation for solar cell applications in supramolecular complexes of multi-metalloporphyrins and fullerenes
Shunichi Fukuzumi*ab and Kei Ohkuboa
aDepartment of Material and Life Science, Division of Advanced Science and Biotechnology, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370; Tel: +81-6-6879-7368
bDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
First published on 3rd October 2013
Abstract
Monomers, dimers, trimers, dendrimers and oligomers of metalloporphyrins form supramolecular complexes with fullerene derivatives via electrostatic interactions, π–π interactions and coordination bonds. Photoexcitation of the supramolecular complexes resulted in photoinduced electron transfer from the porphyrin moiety to the fullerene moiety to produce the charge-separated states as revealed by laser flash photolysis measurements. The rate constants of photoinduced charge separation and charge recombination in supramolecular complexes of multi-metalloporphyrins and fullerenes were also determined by laser flash photolysis measurements and the results depending on the number of porphyrins in the supramolecular complexes are discussed in terms of efficiency of photoinduced energy transfer and charge separation as well as the lifetimes of charge-separated states. The photoelectrochemical performances of solar cells composed of supramolecular complexes of monomers, dimers, dendrimers and oligomers of metalloporphyrins with fullerenes are compared in relation to the rate constants of photoinduced charge separation and charge recombination.
 Shunichi Fukuzumi | Shunichi Fukuzumi earned a Ph.D. degree in applied chemistry at the Tokyo Institute of Technology in 1978. After working as a postdoctoral fellow (1978–1981) at Indiana University in USA, he joined the Department of Applied Chemistry, Osaka University, as an Assistant Professor in 1981 and was promoted to a Full Professor in 1994. He is now a special distinguished professor at Osaka University and the director of an ALCA (Advanced Carbon Technology Research and Development) project that started in 2011. |
 Kei Ohkubo | Kei Ohkubo earned his Ph.D. degree from the Graduate School of Engineering, Osaka University in 2001. He was working as a JSPS fellow and a JST research fellow at Osaka University from 2001 to 2005. He has been a designated associate professor at Osaka University since 2005. |
1. Introduction
Photosynthesis is one of the most fundamental and indispensable processes in nature, because it converts light energy into chemical energy required to maintain life.1,2 Photosynthesis is initiated by the multistep electron-transfer reactions in the photosynthetic reaction centres following light energy harvesting by antenna chlorophylls, funnelled to a bacteriochlorophyll dimer, the so-called special pair, to attain the long-lived charge-separated (CS) state.1,2 The redox-active components such as chlorophyll, pheophytin and quinones are appropriately located in the protein matrix by non-covalent interactions.1,2 Extensive efforts have so far been devoted to the design of electron donor–acceptor composites using covalently and non-covalently linked systems to form the long-lived CS state upon photoexcitation for artificial photosynthesis.3–29 Porphyrins, which are involved in a number of important biological electron-transfer systems including the primary photochemical reactions of chlorophylls (porphyrin derivatives) in the photosynthetic reaction centres, are particularly attractive building blocks as electron acceptors as well as light-harvesting compounds for the construction of supramolecular electron donor–acceptor composites due to their excellent photophysical and electron-transfer properties.8–29 With regard to electron acceptors, fullerenes, which are extensively conjugated three-dimensional π systems, are ideal electron acceptors because of the minimal changes of structure and solvation associated with the electron-transfer reduction.30–38 Thus, combination of porphyrins and fullerenes is regarded as ideal donor–acceptor ensembles, because the combination results in a small reorganization energy, which allows to accelerate photoinduced electron transfer and to slow down charge recombination, leading to the generation of long-lived CS states with high quantum yields.39–63 However, non-covalent binding between monomer porphyrins and fullerenes is usually not strong enough in polar solvents which are generally used for studies on photoinduced electron-transfer reactions.64–68 Among non-covalent interactions, an electrostatic interaction is relatively strong in polar solvents.69–73 Muti-point binding sites can be introduced by using multi-metalloporphyrins (dimers, trimers, dendrimers and oligomers), allowing strong binding between multi-metalloporphyrins and fullerenes in polar solvents.29,30In this perspective, we review our recent development on photoinduced charge separation in supramolecular complexes of porphyrin anions and fullerene cations with electrostatic interactions and those composed of multi-metalloporphyrins and fullerenes, which are strongly bound in polar solvents, towards construction of supramolecular solar cells based on the long-lived photoinduced charge separation.
2. Supramolecular complexes of monomer porphyrin sulfonates and Li+@C60
Zinc tetraphenylporphyrin tetrasulfonate anion [(Bu4N+)4ZnTPPS4−] forms a strong supramolecular binding with a cationic lithium ion encapsulated fullerene (Li+@C60)74–77 in benzonitrile (PhCN) by electrostatic and π–π interactions (Scheme 1).78 The Job's plots of the absorbance change confirmed the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between ZnTPPS4− and Li+@C60.78 Free base tetraphenylporphyrin tetrasulfonate anion [(Bu4N+)4H2TPPS4−] also forms a 1:1 complex with Li+@C60. The formation constants (K) of the ZnTPPS4−/Li+@C60 and H2TPPS4−/Li+@C60 complexes were determined from the absorption change to be 1.6 × 105 and 3.0 × 105 M−1, respectively.78 The same formation constants were obtained from the fluorescence quenching of ZnTPPS4− and H2TPPS4− and by Li+@C60 in PhCN.78
:1 stoichiometry between ZnTPPS4− and Li+@C60.78 Free base tetraphenylporphyrin tetrasulfonate anion [(Bu4N+)4H2TPPS4−] also forms a 1:1 complex with Li+@C60. The formation constants (K) of the ZnTPPS4−/Li+@C60 and H2TPPS4−/Li+@C60 complexes were determined from the absorption change to be 1.6 × 105 and 3.0 × 105 M−1, respectively.78 The same formation constants were obtained from the fluorescence quenching of ZnTPPS4− and H2TPPS4− and by Li+@C60 in PhCN.78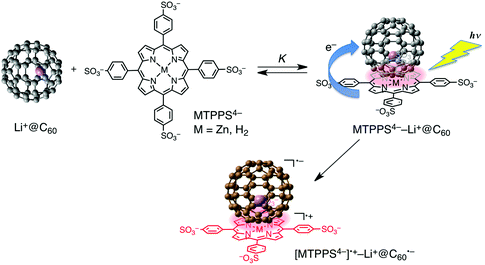 | ||
| Scheme 1 Supramolecular complex formation and photoinduced charge separation of MTPPS4− (M = Zn and H2) with Li+@C60 in PhCN. | ||
The occurrence of the photoinduced energy transfer in the supramolecular complex was confirmed by the transient absorption spectra of the ZnTPPS4−–Li+@C60 complex measured in PhCN using femtosecond laser flash photolysis (Fig. 1a).78 The transient absorption bands taken at 2 ps observed at 620 and 737 nm are assigned to the singlet excited state of ZnTPPS4−. This band decays with the rate constant (kEN) of 9.7 × 1010 s−1 (Fig. 1b) to form the singlet excited state of Li+@C60 at 100 ps (Fig. 1a). The decay rate constant of 1[Li+@C60]* was determined to be 8.9 × 108 s−1, which agrees with the rate constant of the intersystem crossing of Li+@C60.78 Thus, efficient energy transfer occurred from 1[ZnTPPS4−]* to Li+@C60 rather than electron transfer.
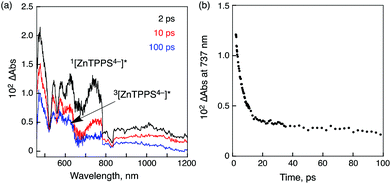 | ||
| Fig. 1 (a) Transient absorption spectra of ZnTPPS4− (2.5 × 10−5 M) in the presence of Li+@C60 (5.0 × 10−5 M) in deaerated PhCN at 298 K taken at 2, 10 and 100 ps after femtosecond laser excitation at 388 nm. (b) Time profile at 737 nm. | ||
The transient absorption spectra taken by nanosecond laser flash photolysis shown in Fig. 2a demonstrate the formation of [ZnTPPS4−]˙+ (λmax = 670 nm) and that of Li+@C60 radical anion (λmax = 1035 nm).78 Thus, the electron transfer from ZnTPPS4− to 3[Li+@C60]* or from 3[ZnTPPS4−]* to Li+@C60 occurs in the supramolecular complex to produce the triplet charge-separated (CS) state. The lifetime of the triplet CS state of the supramolecular complex was determined to be 300 μs for ZnTPPS4− from the first-order decay of the CS state (Fig. 2b).78 It was confirmed that back electron transfer occurred in the supramolecular complex, because the first-order decay rate constant remains the same irrespective of the difference in the laser intensity (inset of Fig. 2b).78 Similarly the CS lifetime of 310 μs was determined for [(H2TPPS4−)˙+–Li+@C60˙−].78 This is the longest lifetime of the CS state ever reported for monomer porphyrin/fullerene systems linked non-covalently in solution. The quantum yield of the CS state is determined to be 0.39 using the absorption of the CS state (Li+@C60˙−: ε1035 = 7300 M−1 cm−1).78
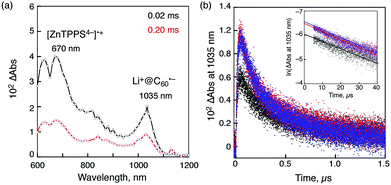 | ||
| Fig. 2 (a) Transient absorption spectra of ZnTPPS4− (2.5 × 10−5 M) in the presence of Li+@C60 (5.0 × 10−5 M) in deaerated PhCN at 298 K taken at 20 and 200 μs after nanosecond laser excitation at 550 nm; (b) decay time profiles at 1035 nm with different laser intensities (1, 3, 6 mJ per pulse). Inset: first-order plots. | ||
The activation enthalpies of the charge-recombination (CR) processes were determined to be 3.0 kcal mol−1 for ZnTPPS4−–Li+@C60 and 5.4 kcal mol−1 for H2TPPS4−–Li+@C60.78 This indicates that there is a significant energy difference between the singlet and triplet CS states and that the CR processes may occur through the thermally activated singlet CS state. The lifetime of the CS state at 77 K is estimated as long as 60 h for H2TPPS4−–Li+@C60.78 Such a long-lived triplet CS state was detected by the EPR measurements by photoirradiation of the H2TPPS4−–Li+@C60 complex in frozen PhCN as shown in Fig. 3. The spin–spin interaction in the triplet radical ion pair of the supramolecular complex is clearly shown at 77 K, where the fine structure due to the triplet CS state is clearly observed at g = 2. From the zero-field splitting values (D = 52 G for ZnTPPS4− and 56 G for H2TPPS4−) the distances (r) between two electron spins were estimated using the relation, D = 27800/r3,79,80 to be 8.1 and 7.9 Å, respectively.78 These r values agree with the centre-to-centre distance of a reported crystal structure of porphyrin/C60.
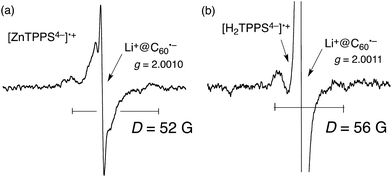 | ||
| Fig. 3 EPR spectra of (a) (ZnTPPS4−)˙+–Li+@C60˙− and (b) (H2TPPS4−)˙+–Li+@C60˙− in PhCN generated by photoirradiation with a high-pressure Hg lamp (1000 W) at 77 K. | ||
By mixing PhCN solutions of the supramolecular complexes of MTPPS4− and Li+@C60 with acetonitrile (MeCN), nanoclusters were produced and they were deposited on an optically transparent electrode (OTE) of nanostructured SnO2 (OTE/SnO2) by application of a dc electric field (∼100 V cm−1) to construct photovoltaic cells.81 The (MTPPS4−/Li+@C60)n films are composed of closely packed Li+@C60 clusters of about 80 nm size, which render a nanoporous morphology to the film as shown in the TEM images in Fig. 4.81
 | ||
| Fig. 4 TEM images of (a) Li+@C60/ZnTPPS4− and (b) Li+@C60/H2TPPS4− nanoclusters. | ||
The photoelectrochemical measurements of a robust thin film of OTE/SnO2/(MTPPS4−/Li+@C60)n were performed using a standard two-electrode system consisting of a working electrode and a Pt wire gauze electrode in air-saturated MeCN containing 0.5 M LiI and 0.01 M I2 (Fig. 5).77 The IPCE (incident photon-to-photocurrent efficiency) values were determined by normalizing the photocurrent values for incident light energy and intensity and using eqn (1).82–85
| IPCE (%) = 100 × 1240 × isc/(Iinc × λ) | (1) |
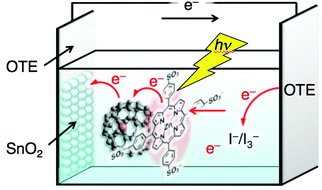 | ||
| Fig. 5 Schematic image of photoelectrochemical cell of OTE/SnO2/MTPPS4−/Li+@C60 and electron-transfer pathways to generate photocurrent. | ||
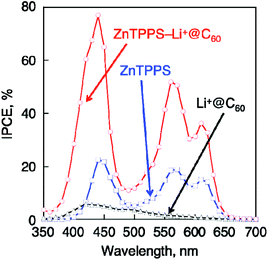 | ||
| Fig. 6 Photocurrent action spectra of OTE/SnO2/(ZnTPPS4−/Li+@C60)n, (red) OTE/SnO2/(ZnTPPS4−)n (blue) and OTE/SnO2/(Li+@C60)n (black). Electrolyte: 0.5 M LiI and 0.01 M I2 in MeCN–PhCN (3:1 v/v). | ||
The power conversion efficiency (η) of the OTE/SnO2/(ZnTPPS4−/Li+@C60)n electrode was calculated by using eqn (2):82–85
| η = FF × Isc × Voc/Win | (2) |
3. Supramolecular complexes of cyclic porphyrin dimers with C60 and Li+@C60
As compared to porphyrin monomers, porphyrin dimers with appropriate linkage can accommodate electron acceptor guest molecules by π–π interactions to form sandwich complexes.86–96 For example, a cyclic Ni porphyrin dimer (Ni-CPDPy) linked by butadiyne moieties bearing 4-pyridyl groups (Fig. 7) forms a sandwich complex with C60 (C60 ⊂ Ni2-CPDPy) as shown in the X-ray crystal structure (Fig. 8), where the dimer bites a C60 molecule by tilting the porphyrin rings with respect to each other and there are strong π–π interactions between the porphyrin rings and C60.97 The adjacent dimers are linked by hydrogen bonds and π–π interactions.97 The C60 molecules are linearly arranged in the inner channel to give a supramolecular peapod.97–102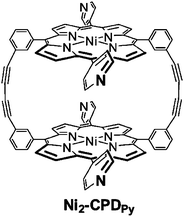 | ||
| Fig. 7 Chemical structure of Ni2-CPDPy. | ||
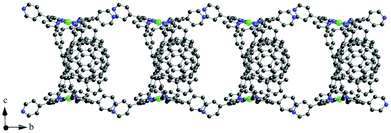 | ||
| Fig. 8 Crystal structures of tubular assemblies of C60 ⊂ Ni2-CPDPy. Hydrogen atoms are omitted for clarity. | ||
The linear arrangement of C60 in C60 ⊂ Ni2-CPDPy high electron mobilities of ∑μ = 0.72 and 0.20 cm2 V−1 s−1 along the b and c axes, respectively, which were determined by flash-photolysis time-resolved microwave conductivity (FP-TRMC) measurements.97 The TRMC technique can evaluate the intrinsic mobility without being affected by the chemical or physical defects in the material and/or the organic/metal–electrode interfaces.103–105 The observed value along the b axis of the single crystal of C60 ⊂ Ni2-CPDPy is comparable to that of the single crystal of C60 (∑μ = 0.50 cm2 V−1 s−1 measured by TOF).106 The observed high electron mobility along the b axis is due to the well-ordered linear arrangement of C60 in the porphyrin nanotube. However, the expected charge-separated state could not be observed in the time-resolved transient absorption spectra of C60 ⊂ Ni2-CPDPy because the singlet excited state of the nickel porphyrin immediately changes to the triplet excited state by intersystem crossing and the low energy triplet excited state of C60 (3C60*) is formed by energy transfer.97 The estimated energy level of the charge-separated state (1.98 eV) is higher than that of 3C60* (1.60 eV).97 When Ni2-CPDPy was replaced by a free base porphyrin dimer (H4-CPDPy), a complete charge-separated state {H4-CPDPy˙+ + C60˙−)} was observed by femtosecond laser flash photolysis of C60 ⊂ H4-CPDPy in the solid state with a lifetime of 470 ps.107 The photovoltaic activity of C60 ⊂ Ni2-CPDPy and C60 ⊂ H4-CPDPy was evaluated by using solar cells composed of modified electrodes and I−/I3− solution.107 The C60 ⊂ H4-CPDPy-modified electrode exhibited IPCE of 17% and a power conversion efficiency (η) of 0.33%, which was more than 16 times larger than that of OTE/SnO2/(C60 ⊂ Ni2-CPDPy)n (0.02%).107 Such a significant enhancement of the η value demonstrates that the formation of highly ordered clusters and the efficient charge separation of (C60 ⊂ H4-CPDPy)n contributes to the improvement of the light energy conversion properties.107
When C60 is replaced by Li+@C60, porphyrin dimers with four long alkoxy substituents on the meso-phenyl groups (MCPDPy(OC6) in Fig. 9) form strong supramolecular complexes in even a polar solvent such as PhCN.108 The association constants (Kassoc) of Li+@C60 ⊂ MCPDPy(OC6) in PhCN at 298 K were determined to be 2.6 × 105 M−1 for Li+@C60 ⊂ H4-CPDPy(OC6) and 3.5 × 105 M−1 for Li+@C60 ⊂ Ni2-CPDPy(OC6).108
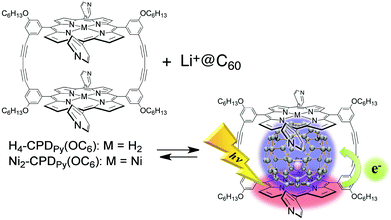 | ||
| Fig. 9 Supramolecular formation and photoinduced charge separation between MCPDPy(OC6) and Li+@C60. | ||
Upon laser excitation of Li+@C60 ⊂ Ni2-CPDPy(OC6), transient absorption bands due to Ni2-CPDPy(OC6)˙+ and Li+@C60˙− were observed as shown in Fig. 10a.108 In this case, electron transfer occurs from Ni2-CPDPy(OC6) to the triplet excited state of Li+@C60 (3Li+@C60*) rather than from 3[Ni2-CPDPy(OC6)]* to Li+@C60 as indicated by the disappearance of the absorption band at 750 nm due to 3Li+@C60*, accompanied by the appearance of the absorption band at 1035 nm due to Li+@C60˙− (Fig. 10b).108 The rate constant of electron transfer from Ni2-CPDPy(OC6) to 3Li+@C60* to produce the CS state was determined from the rise in the absorbance at 1035 nm due to Li+@C60˙− to be 5.7 × 107 s−1.108 The absorbance at 1035 nm due to Li+@C60˙− in the CS state decayed obeying first-order kinetics with the same slope irrespective of the difference in the laser intensity (Fig. 10c).108 This clearly indicates that the decay of the CS state occurs via intrasupramolecular back electron transfer rather than a bimolecular back electron-transfer reaction between the CS states. The CS lifetime was determined from the slope of the first-order plots in Fig. 10c to be 0.67 ms, which is the longest value ever reported for non-covalent monomer dimer porphyrin-fullerene supramolecules in solution.108 The CS state was also observed for Li+@C60 ⊂ H4-CPDPy(OC6). The quantum yields of the CS states were estimated to be 0.13 for Li+@C60 ⊂ Ni2-CPDPy(OC6) and 0.32 for Li+@C60 ⊂ H4-CPDPy(OC6) and by means of the comparative method with the absorption intensities of the CS states (Li+@C60˙−: ε(1035 nm) = 7300 M−1 cm−1).108 When Li+@C60 was replaced by pristine C60, no CS states were produced as predicted by their higher energy levels than those of the triplet excited states of CPDPy(OC6) and C60.108
![(a) Transient absorption spectra of Ni2-CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 4 and 28 μs after nanosecond laser excitation at 520 nm. [Ni2-CPDPy(OC6)] = 2.5 × 10−5 M, [Li+@C60] = 5.0 × 10−5 M. (b) Rise and (c) decay time profiles at 1035 nm with different laser intensities (1, 3, 5 mJ per pluse). Inset: first-order plots.](/image/article/2013/DT/c3dt51883c/c3dt51883c-f10.gif) | ||
| Fig. 10 (a) Transient absorption spectra of Ni2-CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 4 and 28 μs after nanosecond laser excitation at 520 nm. [Ni2-CPDPy(OC6)] = 2.5 × 10−5 M, [Li+@C60] = 5.0 × 10−5 M. (b) Rise and (c) decay time profiles at 1035 nm with different laser intensities (1, 3, 5 mJ per pluse). Inset: first-order plots. | ||
The mechanisms of intrasupramolecular photoinduced charge separation in Li+@C60 ⊂ Ni2-CPDPy(OC6) are shown in Scheme 2.108 The singlet excited state of Ni2-CPDPy(OC6) (1[Ni2-CPDPy(OC6)]*) is generated upon photoexcitation of Li+@C60 ⊂ Ni2-CPDPy(OC6) at 420 nm, where the porphyrin moiety is exclusively excited. Even if the Li+@C60 moiety is excited, energy transfer from 1[Li+@C60]* (Es = 1.94 eV)77 to Ni2-CPDPy(OC6) (Es = 1.97 eV) occurs to produce 1[Ni2-CPDPy(OC6)]*. Although electron transfer from 1[Ni2-CPDPy(OC6)]* to Li+@C60 is energetically possible (Scheme 2), the fast intersystem crossing occurs to generate 3[Ni2-CPDPy(OC6)]* (kISC > 1012 s−1).108 Then, electron transfer occurs from 3[Ni2-CPDPy(OC6)]* to Li+@C60 with the driving force of 0.30 eV to produce the CS state. The CS state decays slowly via intrasupramolecular BET with the lifetime of 0.67 ms (Scheme 2).108
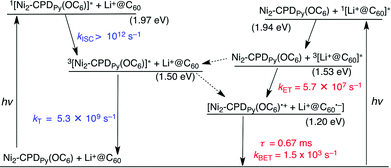 | ||
| Scheme 2 Energy diagram for Li+@C60 ⊂ Ni2-CPDPy(OC6); broken arrow: minor pathway. | ||
4. Supramolecular complex of a porphyrin tripod with C60
The tripod conformation of a porphyrin trimer (TPZn3) in Fig. 11 makes it possible to capture a fullerene derivative containing a pyridine moiety (PyC60)109 inside the cavity strongly by π–π interactions together with the coordination bond between Zn2+ and the pyridine moiety (Scheme 3).110–113 The association constant of TPZn3 with PyC60 (1.1 × 105 M−1 in toluene) determined from the UV-vis absorption spectral titration (Fig. 12a) is much larger as compared with those of the corresponding monomer (MPZn) and dimer porphyrin (DPZn2).109 The 1H NMR signals of TPZn3 exhibit downfield shifts upon complexation with PyC60, whereas the pyridyl protons of PyC60 exhibit large upfield shifts by the complexation, which is ascribed to the influence of the large porphyrin aromatic ring current.113 This result clearly shows that the pyridyl group of PyC60 coordinates to the central zinc ions of TPZn3. The encapsulation of PyC60 into the cavity of TPZn3 was supported by the DFT-optimized structure (B3LYP/3-21G(*) basis set) in Fig. 12b.113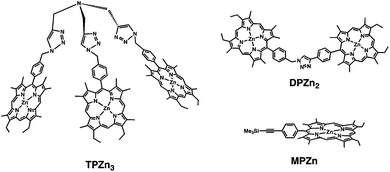 | ||
| Fig. 11 A porphyrin tripod and the reference dimer and monomer. | ||
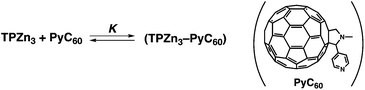 | ||
| Scheme 3 Formation of a supramolecular complex between TPZn3 and PyC60. | ||
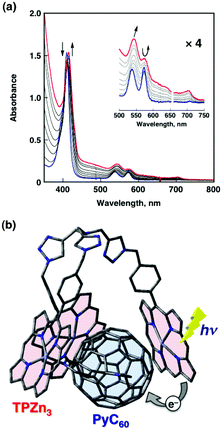 | ||
| Fig. 12 (a) UV-Vis spectral changes upon addition of PyC60 (0 to 48 μM) to an o-DCB solution of TPZn3 (3 μM) at 298 K. (b) Schematic view of photoinduced electron transfer in the TPZn3–PyC60 complex. The structure of the TPZn3–PyC60 complex was optimized by DFT at the B3LYP/3-21G(*) level. | ||
The occurrence of photoinduced electron transfer from 1TPZn3* to PyC60 was confirmed by femtosecond laser flash photolysis measurements in Fig. 13a, where the transient absorption spectrum due to 1TPZn3* changes as time elapses to afford the absorption bands at λmax = 1000 nm due to the monofunctionalized fullerene radical anion114,115 and at 670 nm due to the one-electron oxidized species of TPZn3 (TPZn3˙+).113,116,117
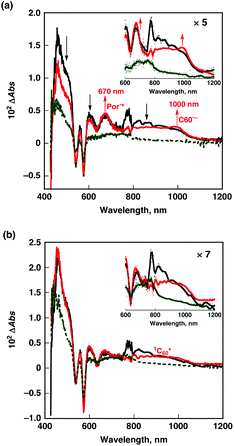 | ||
| Fig. 13 Transient absorption spectra of (a) TPZn3 (7.0 × 10−6 M) and (b) MPZn (1.1 × 10−5 M) in the presence of PyC60 (2.3 × 10−5 M) obtained at 2 ps (black), 62 ps (red), and 2800 ps (green) after femtosecond laser pulse irradiation at 410 nm in deaerated toluene at 298 K. | ||
In sharp contrast to the TPZn3–PyC60 complex, the transient absorption spectrum of the monomer porphyrin (MPZn) in the presence of PyC60 (Fig. 13b) exhibits the absorbance change due to the energy transfer from 1MPZn* to PyC60 to give the singlet excited state 1PyC60* (1.76 eV), followed by the conversion to the triplet excited states 3MPZn* and 3PyC60* at 2800 ps (green line in Fig. 13b), accompanied by the recovery of the ground state.113
The energy diagrams of photodynamics for TPZn3 and MPZn in the presence of PyC60 in toluene are shown in Scheme 4a and 4b, respectively.113 The energy level (1.49 eV) of the CS state (TPZn3˙+−PyC60˙−) is lower than the energy level of the triplet excited state of PyC60 moieties (1.56 eV). The rate constant (kET) of photoinduce electron transfer from 1TPZn3* to PyC60 is larger than the rate constant of intersystem crossing. From the rate constant of back electron transfer (kBET = 1.9 × 109 s−1), the lifetime of the CS state is determined to be τCS = 0.53 ns. In contrast, only energy transfer from 1MPZn* to PyC60 occurs to produce 1PyC60*, in competition with intersystem crossing to 3MPZn*.113
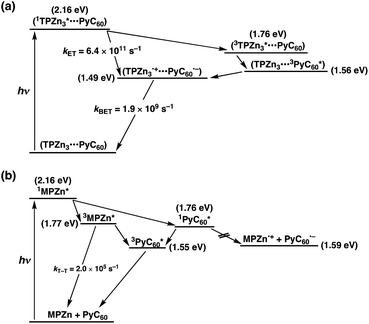 | ||
| Scheme 4 Energy diagrams for photodynamics of (a) TPZn3 and (b) MPZn in the presence of PyC60 in toluene. | ||
TPZn3 also forms a stable 1:1 complex with gold(III) tetra(4-pyridyl)porphyrin (AuTPyP+) in nonpolar solvents.118 The strong binding of TPZn3 with AuTPyP+ results from the encapsulation of AuTPyP+ inside the cavity of TPZn3 through multiple coordination bonds. The efficient quenching of the singlet excited state of TPZn3 occurs via a photoinduced electron-transfer pathway in the TPZn3–AuTPyP+ complex as the case of TPZn3–PyC60 complex.118
5. Supramolecular complexes of porphyrin oligopeptides and C60
Multiple photosynthetic reaction centres composed of light-harvesting multiporphyrin units and charge-separation units were obtained by using both the coordination bond and π–π interaction. Zinc porphyrinic oligopeptides with various numbers of porphyrin units [P(ZnP)n; n = 2, 4, 8]119,120 were used as light-harvesting multiporphyrin units (Fig. 14), which are bound to electron acceptors of fulleropyrrolidine bearing a pyridine (PyC60)113 or imidazole coordinating ligand (ImC60)82 as shown in Fig. 15.121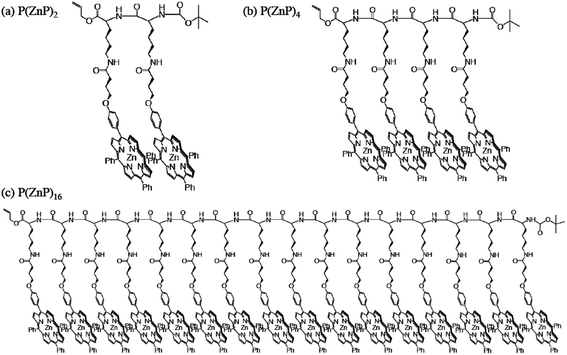 | ||
| Fig. 14 Chemical structures of P(ZnP)2, P(ZnP)4 and P(ZnP)16. | ||
![Illustration of supramolecular complex composed of porphyrin-peptide octamer [P(ZnP)8, Ar = 3,5-(t-Bu)2C6H3] and PyC60 or ImC60.](/image/article/2013/DT/c3dt51883c/c3dt51883c-f15.gif) | ||
| Fig. 15 Illustration of supramolecular complex composed of porphyrin-peptide octamer [P(ZnP)8, Ar = 3,5-(t-Bu)2C6H3] and PyC60 or ImC60. | ||
The binding constant (K) of PyC60 to P(ZnP)n increased with increasing number of zinc porphyrins in an oligopeptide unit.121 No supramolecular complex formation was observed in the case of zinc tetraphenylporphyrin in PhCN.121 The strong binding between P(ZnP)8 and PyC60 results from the strong π–π interactions between two zinc porphyrins and PyC60 in addition to the axial coordination of PyC60 to zinc ions of the porphyrins. In the case of ImC60, however, the highest K value was obtained in the P(ZnP)4–ImC60 complex. This indicates that ImC60 is much more strongly bound by the oligopeptide, P(ZnP)4, than PyC60.121 The apparent binding constants (K) determined from the fluorescence quenching of P(ZnP)n were significantly larger than those determined from the UV-vis spectral change, and the difference in the values increased with increasing the generation of porphyrinic oligopeptides (with increasing the number of the porphyrin units).121 This indicates that the excited energy migration between the porphyrin units occurs efficiently prior to the electron transfer to the bound C60 moiety. An extremely efficient energy transfer also occurs in P(ZnP)8–ImC60 judging from the large difference in the K values determined by the absorption change and by the fluorescence quenching (1.5 × 104vs. 3.3 × 105 M−1).
The occurrence of photoinduced electron transfer in the supramolecular complex in PhCN was confirmed by the transient absorption spectra of the supramolecular complex of P(ZnP)8 with PyC60 using nanosecond laser flash photolysis.121 The laser photoexcitation at 561 nm of the supramolecular complex of P(ZnP)8 with PyC60 results in formation of the CS state as indicated by the transient absorption spectra in Fig. 16a, where the absorption band due to PyC60˙− is clearly observed at 1000 nm together with that due to ZnP˙+ at 630 nm.121 The CS state detected decays obeying first-order kinetics (Fig. 16b) and the first-order plots at different initial CS concentrations afford linear correlations with the same slope (inset of Fig. 16b).121 If there is any contribution of intermolecular back electron transfer from unbound PyC60˙− to ZnP˙+, the second-order kinetics would be involved for the decay time profile. In fact, the corresponding second-order plots (Fig. 16c) are clearly non-linear and the initial slope varies depending on the CS concentration. Thus, the decay process is ascribed to back electron transfer in the supramolecular complex rather than intermolecular back electron transfer between ZnP˙+ and PyC60˙−.121 The CS lifetimes of the supramolecular complexes of other porphyrins [P(ZnP)n] and the fullerene derivative (ImC60) become longer with increasing generation of porphyrinic oligopeptides (with increasing the number of the porphyrin units).121 Such elongation of the CS lifetimes results from efficient hole migration between the porphyrin units following the photoinduced electron transfer in the supramolecular complexes.
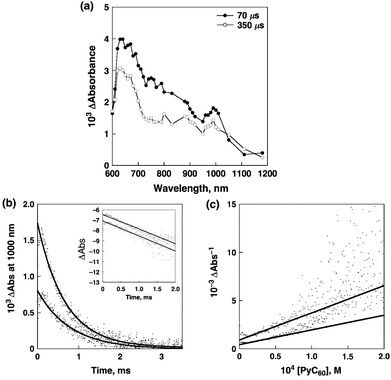 | ||
| Fig. 16 (a) Transient absorption spectra of P(ZnP)8 (2.9 × 10−6 M) in the presence of PyC60 (4.9 × 10−3 M) in deaerated PhCN at 298 K taken at 70 (solid line with black circles) and 350 μs (solid line with white circles) after laser excitation at 561 nm (4 mJ per pulse), respectively. (b) Time profiles of the absorption at 1000 nm due to PyC60˙− with different laser powers (4 and 1 mJ per pulse) at 298 K. Inset: first-order plots. (c) Second-order plots. | ||
Multiple photosynthetic reaction centres have also been constructed using supramolecular complexes of zinc porphyrin dendrimers [D(ZnP)n: n = 4, 8, 16] with PyC60.122 Efficient energy migration occurs more efficiently between the ZnP units of dendrimers prior to the photoinduced electron transfer with increasing the generation of dendrimers to attain an extremely long CS lifetime e.g., 0.25 ms for the D(ZnP)16–PyC60 complex in PhCN at 298 K.122
Multiple photosynthetic reaction centres composed of supramolecular complexes of harvesting multiporphyrin units and charge-separation units have enabled us to construct supramolecular organic solar cells by the electrodeposition of mixed porphyrin-peptide oligomers [P(H2P)n or P(ZnP)n] and C60 clusters [(P(H2P)n + C60)m or (P(ZnP)n + C60)m] onto an optically transparent electrode (OTE) of a nanostructured SnO2 electrode (OTE/SnO2), to obtain modified electrodes [denoted as (P(H2P)n + C60)m or (P(ZnP)n + C60)m (n = 1, 2, 4, 8, 16)].123 The IPCE value increased with increasing the number of porphyrins in a polypeptide unit in both (P(H2P)n + C60)m and (P(ZnP)n + C60)m (n = 1, 2, 4, 8, 16) systems as shown in Fig. 17. Such a good photoelectrochemical performance results from efficient photoinduced electron-transfer from the excited state of the porphyrin unit to C60 in the supramolecular complex with longer CS lifetimes as the number of porphyrins in a polypeptide unit increases (vide supra). The maximum IPCE value of (P(ZnP)16 + C60)m (56%) is larger than that of (P(H2P)16 + C60)m (48%) probably because of the larger driving force of the photoinduced electron transfer.
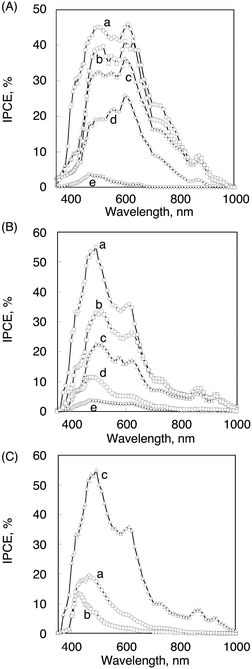 | ||
| Fig. 17 (A) The photocurrent action spectra (IPCE vs. wavelength) of (a) (P(H2P)16 + C60)m, (b) (P(H2P)8 + C60)m, (c) (P(H2P)4 + C60)m, (d) (P(H2P)2 + C60)m and (e) (P(H2P)1 + C60)m modified OTE/SnO2 electrodes. (B) The photocurrent action spectra of (a) (P(ZnP)16 + C60)m, (b) (P(ZnP)8 + C60)m, (c) (P(ZnP)4 + C60)m, (d) (P(ZnP)2 + C60)m and (e) (P(ZnP)1 + C60)m modified electrodes. (C) The photocurrent action spectra of (a) (P(ZnP)16 + ImC60)m, (b) (P(ZnP)16 + PyC60)m and (c) (P(ZnP)16 + C60)m modified OTE/SnO2 electrodes. See text for the employed concentration of the individual species. | ||
The maximum IPCE values of (P(ZnP)16 + PyC60)m (20%) and (P(ZnP)16 + ImC60)m (15%) are much smaller than that of (P(ZnP)16 + C60)m (56%), whereas the binding constant of P(ZnP)16–C60 is smaller than those of P(ZnP)16–ImC60 and P(ZnP)16–PyC60.123 The lower IPCE values of P(ZnP)16–ImC60 and P(ZnP)16–PyC60 systems as compared with that of P(ZnP)16–C60 system may result from the poor electron-transport properties of C60 derivatives due to the steric hindrance of the ligand moiety.123 Thus, a key element for efficient photocurrent generation is mainly the hole and electron transport in the thin film rather than the charge separation between porphyrins and C60.123
I/V characteristics of (a) (P(H2P)16 + C60)m, (b) (P(H2P)8 + C60)m and (c) (P(H2P)1 + C60)m modified electrodes under visible light irradiation (λ > 400 nm) are shown in Fig. 18. The (P(H2P)16 + C60)m system has a larger fill factor (FF) of 0.47, an open circuit voltage (Voc) of 320 mV, a short circuit current density (Isc) of 0.36 mA cm−2, and the overall power conversion efficiency (η) of 1.6% at input power (Win) of 3.4 mW cm−2.123
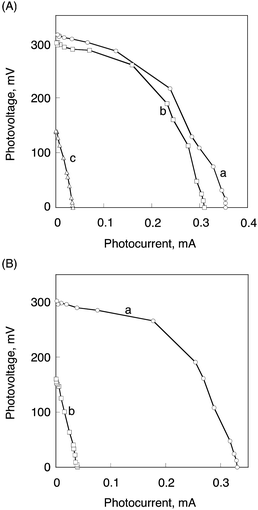 | ||
| Fig. 18 (A) Current–voltage characteristics of (a) (P(H2P)16 + C60)m, (b) (P(H2P)8 + C60)m, and (c) (P(H2P)1 + C60)m modified electrodes. (B) Current–voltage characteristics of (a) (P(ZnP)16 + C60)m and (b) (P(ZnP)1 + C60)m. Electrolyte: 0.5 M NaI and 0.01 M I2 in acetonitrile. Input power: 3.4 mW cm−2, λ > 400 nm. | ||
The η values of the (P(H2P)16 + C60)m system was remarkably enhanced (around 40 times) in comparison with the (P(H2P)1 + C60)m modified electrode (η = 0.043%) under the same experimental conditions. The η value of (P(ZnP)16 + C60)m is also determined as 1.4% and this value is much larger than that of (P(ZnP)1 + C60)m (0.047%) as shown in Fig. 18B.123
6. Conclusions
As described above, porphyrin monomers, dimers, trimers and oligomers form supramolecular complexes with fullerene derivatives via electrostatic interactions, π–π interactions and coordination bonds. Photoexcitation of the supramolecular complexes resulted in efficient photoinduced electron transfer from the porphyrin moiety to the fullerene moiety to produce the long-lived CS states as revealed by laser flash photolysis measurements. In particular, a supramolecular complex of a cyclic Ni porphyrin dimer with Li+@C60 [Li+@C60 ⊂ Ni2-CPDPy(OC6)] affords a long-lived triplet CS state with 0.63 ms lifetime. A high IPCE value (77% at 450 nm) was achieved for a supramolecular solar cell using the OTE/SnO2/(ZnTPPS4−/Li+@C60)n electrode. The use of porphyrin oligomer peptides has also enabled to construct multiple photosynthetic reaction centres composed of light-harvesting multiporphyrin units and charge-separation units linked by both the coordination bond and π–π interactions, which afforded long-lived CS states. Supramolecular organic solar cells composed of porphyrinic oligopeptides and C60 exhibited higher overall power conversion efficiency with increasing the number of porphyrin units. Supramolecular complexes formed between porphyrins and fullerenes in particular Li+@C60 provide promising materials for more efficient solar energy conversion.Acknowledgements
The authors gratefully acknowledge the contributions of their collaborators and co-workers mentioned in the cited references, in particular Prof. Fumito Tani (Kyushu University) and Prof. Taku Hasobe (Keio University). Financial supports from the Grants-in-Aid (no. 20108010 to S.F. and 23750014 to K.O.) from MEXT of Japan and KOSEF/MEST of Korea through WCU project (R31-2008-000-10010-0) are gratefully acknowledged.References
- Anoxygenic Photosynthetic Bacteria, ed. R. E. Blankenship, M. T. Madigan and C. E. Bauer, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1995 Search PubMed.
- W. Leibl and P. Mathis, in Electron Transfer in Photosynthesis. Series on Photoconversion of Solar Energy, 2004, vol. 2, p. 117 Search PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695 Search PubMed.
- S. Fukuzumi, Y. Yamada, T. Suenobu, K. Ohkubo and H. Kotani, Energy Environ. Sci., 2011, 4, 2754 CAS.
- A. Harriman and J.-P. Sauvage, Chem. Soc. Rev., 1996, 25, 41 RSC.
- M. N. Paddon-Row, Adv. Phys. Org. Chem., 2003, 38, 1 CrossRef CAS.
- M. N. Paddon-Row, Acc. Chem. Res., 1994, 27, 18 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Faraday Discuss., 2012, 155, 9 RSC.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS PubMed.
- C. Thilgen and F. Diederich, Chem. Rev., 2006, 106, 5049 CrossRef CAS PubMed.
- F. Giacalone and N. Martín, Chem. Rev., 2006, 106, 5136 CrossRef CAS PubMed.
- W.-S. Li and T. Aida, Chem. Rev., 2009, 109, 6047 CrossRef CAS PubMed.
- C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630 CrossRef CAS PubMed.
- (a) F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410 CrossRef CAS PubMed; (b) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (c) F. D'Souza, A. S. D. Sandanayaka and O. Ito, J. Phys. Chem. Lett., 2010, 1, 2586 CrossRef CAS; (d) F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86 RSC.
- (a) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177 CrossRef CAS; (b) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283 RSC; (c) S. Fukuzumi, Pure Appl. Chem., 2007, 79, 981 CrossRef CAS; (d) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609 RSC.
- H. Imahori and S. Fukuzumi, Adv. Funct. Mater., 2004, 14, 525 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, F. Zerbetto and M. Prato, Acc. Chem. Res., 2005, 38, 871 CrossRef CAS PubMed.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 676 CrossRef PubMed.
- S. Fukuzumi, T. Honda, K. Ohkubo and T. Kojima, Dalton Trans., 2009, 3880 RSC.
- S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427 RSC.
- D. M. Guldi and V. Sgobba, Chem. Commun., 2011, 47, 606 RSC.
- D. M. Guldi, Phys. Chem. Chem. Phys., 2007, 9, 1400 RSC.
- S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575 RSC.
- (a) K. Ohkubo and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2008, 12, 993 CrossRef CAS; (b) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303 CrossRef CAS.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS PubMed.
- M. V. Martinez-Diaz, N. S. Fender, M. S. Rodriguez-Morgade, M. Gomez-Lopez, F. Diederich, L. Echegoyen, J. F. Stoddart and T. Torres, J. Mater. Chem., 2002, 12, 2095 RSC.
- (a) C. G. Claessens and T. Torres, Chem. Commun., 2004, 1298 RSC; (b) C. G. Claessens and T. Torres, J. Am. Chem. Soc., 2002, 124, 14522 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, F. D'Souza and J. L. Sessler, Chem. Commun., 2012, 48, 9801 RSC.
- J.-F. Nierengarten, U. Hahn, T. M. F. Duarte, F. Cardinali, N. Solladié, M. E. Walther, A. Van Dorsselaer, H. Herschbach, E. Leize, A.-M. Albrecht-Gary, A. Trabolsi and M. Elhabiri, C. R. Chim., 2006, 9, 1022 CrossRef CAS PubMed.
- L. Echegoyen, F. Diederich and L. E. Echegoyen, in Fullerenes: Chemistry, Physics and Technology, ed. K. M. Kadish and R. S. Ruoff, Wiley-Interscience, New York, 2000, pp. 1–51 Search PubMed.
- T. M. Figueira-Duarte, A. Gegout and J.-F. Nierengarten, Chem. Commun., 2007, 109 RSC.
- S. Fukuzumi and D. M. Guldi, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 2, pp. 270–337 Search PubMed.
- S. Fukuzumi, in Functional Organic Materials, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, 2007, pp. 465–510 Search PubMed.
- J.-F. Nierengarten, New J. Chem., 2004, 28, 1177 RSC.
- S. Fukuzumi, I. Nakanishi, T. Suenobu and K. M. Kadish, J. Am. Chem. Soc., 1999, 121, 3468 CrossRef CAS.
- S. Fukuzumi, K. Ohkubo, H. Imahori and D. M. Guldi, Chem.–Eur. J., 2003, 9, 1585 CrossRef CAS PubMed.
- Y. Kawashima, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2013, 117, 6737 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, H. Imahori, J. Shao, Z. Ou, G. Zheng, Y. Chen, R. K. Pandey, M. Fujitsuka, O. Ito and K. M. Kadish, J. Am. Chem. Soc., 2001, 123, 10676 CrossRef CAS PubMed.
- D. Wróbel and A. Graja, Coord. Chem. Rev., 2011, 255, 2555 CrossRef PubMed.
- H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607 CrossRef CAS PubMed.
- H. Imahori, D. M. Guldi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 6617 CrossRef CAS PubMed.
- K. Ohkubo, H. Imahori, J. Shao, Z. Ou, K. M. Kadish, Y. Chen, G. Zheng, R. K. Pandey, M. Fujitsuka, O. Ito and S. Fukuzumi, J. Phys. Chem. A, 2002, 106, 10991 CrossRef CAS.
- H. Imahori, K. Tamaki, Y. Araki, Y. Sekiguchi, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2002, 124, 5165 CrossRef CAS PubMed.
- K. Ohkubo, H. Kotani, J. Shao, Z. Ou, K. M. Kadish, G. Li, R. K. Pandey, M. Fujitsuka, O. Ito, H. Imahori and S. Fukuzumi, Angew. Chem., Int. Ed., 2004, 43, 853 CrossRef CAS PubMed.
- F. D'Souza, P. M. Smith, M. E. Zandler, A. L. McCarty, M. Itou, Y. Araki and O. Ito, J. Am. Chem. Soc., 2004, 126, 7898 CrossRef CAS PubMed.
- H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem.–Eur. J., 2004, 10, 3184 CrossRef CAS PubMed.
- F. D'Souza, R. Chitta, S. Gadde, S. D.-M. Islam, A. L. Schumacher, M. E. Zandler, Y. Araki and O. Ito, J. Phys. Chem. B, 2006, 110, 25240 CrossRef CAS PubMed.
- F. D'Souza, R. Chitta, S. Gadde, L. M. Rogers, P. A. Karr, M. E. Zandler, A. S. D. Sandanayaka, Y. Araki and O. Ito, Chem.–Eur. J., 2007, 13, 916 CrossRef CAS PubMed.
- R. Chitta, K. Ohkubo, M. Tasior, N. K. Subbaiyan, M. E. Zandler, D. T. Gryko, S. Fukuzumi and F. D'Souza, J. Am. Chem. Soc., 2008, 130, 14263 CrossRef PubMed.
- F. D'Souza, E. Maligaspe, P. A. Karr, A. L. Schumacher, M. El Ojaimi, C. P. Gros, J.-M. Barbe, K. Ohkubo and S. Fukuzumi, Chem.–Eur. J., 2008, 14, 674 CrossRef CAS PubMed.
- F. D'Souza, E. Maligaspe, K. Ohkubo, M. E. Zandler, N. K. Subbaiyan and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 8787 CrossRef CAS PubMed.
- F. D'Souza, G. M. Venukadasula, K. Yamanaka, N. K. Subbaiyan, M. E. Zandler and O. Ito, Org. Biomol. Chem., 2009, 7, 1076 CAS.
- F. D'Souza, N. K. Subbaiyan, Y. Xie, J. P. Hill, K. Ariga, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 16138 CrossRef CAS PubMed.
- E. Maligaspe, T. Kumpulainen, N. K. Subbaiyan, M. E. Zandler, H. Lemmetyinen, N. V. Tkachenko and F. D'Souza, Phys. Chem. Chem. Phys., 2010, 12, 7434 RSC.
- M. E. El-Khouly, D. K. Ju, K.-Y. Kay, F. D'Souza and S. Fukuzumi, Chem.–Eur. J., 2010, 16, 6193 CrossRef CAS PubMed.
- F. D'Souza, C. A. Wijesinghe, M. E. El-Khouly, J. Hudson, M. Niemi, H. Lemmetyinen, N. V. Tkachenko, M. E. Zandler and S. Fukuzumi, Phys. Chem. Chem. Phys., 2011, 13, 18168 RSC.
- F. D'Souza, A. N. Amin, M. E. El-Khouly, N. K. Subbaiyan, M. E. Zandler and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 654 CrossRef CAS PubMed.
- S.-H. Lee, A. G. Larsen, K. Ohkubo, J. R. Reimers, S. Fukuzumi and M. J. Crossley, Chem. Sci., 2012, 3, 257 RSC.
- V. Garg, G. Kodis, M. Chachisvilis, M. Hambourger, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2011, 133, 2944 CrossRef CAS PubMed.
- J. Frey, G. Kodis, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, J. Phys. Chem. A, 2013, 117, 607 CrossRef CAS PubMed.
- Y. Kashiwagi, K. Ohkubo, J. A. McDonald, I. M. Blake, M. J. Crossley, Y. Araki, O. Ito, H. Imahori and S. Fukuzumi, Org. Lett., 2003, 5, 2719 CrossRef CAS PubMed.
- D. Curiel, K. Ohkubo, J. R. Reimers, S. Fukuzumi and M. J. Crossley, Phys. Chem. Chem. Phys., 2007, 9, 5260 RSC.
- J. Iehl, M. Vartanian, M. Holler, J.-F. Nierengarten, B. Delavaux-Nicot, J.-M. Strub, A. Van Dorsselaer, Y. Wu, J. Mohanraj, K. Yoosaf and N. Armaroli, J. Mater. Chem., 2011, 21, 1562 RSC.
- F. D'Souza, G. R. Deviprasad, M. S. Rahman and J.-P. Choi, Inorg. Chem., 1999, 38, 2157 CrossRef CAS PubMed.
- F. D'Souza, G. R. Deviprasad, M. E. Zandler, V. T. Hoang, A. Klykov, M. VanStipdonk, A. Perera, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2002, 106, 3243 CrossRef CAS.
- T. Da Ros, M. Prato, D. M. Guldi, M. Ruzzi and L. Pasimeni, Chem.–Eur. J., 2001, 7, 816 CrossRef CAS.
- F. D'Souza, G. R. Deviprasad, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 5277 CrossRef CAS PubMed.
- T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 10155 CrossRef CAS PubMed.
- T. Honda, T. Kojima, N. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2011, 50, 2725 CrossRef CAS PubMed.
- S. Fukuzumi, T. Honda and T. Kojima, Coord. Chem. Rev., 2012, 256, 2488 CrossRef CAS PubMed.
- J. L. Sessler, E. Karnas, S. K. Kim, Z. Ou, M. Zhang, K. M. Kadish, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 15256 CrossRef CAS PubMed.
- E. Karnas, S. K. Kima, K. A. Johnson, J. L. Sessler, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 16617 CrossRef CAS PubMed.
- S. Aoyagi, E. Nishibori, H. Sawa, K. Sugimoto, M. Takata, Y. Miyata, R. Kitaura, H. Shinohara, H. Okada, T. Sakai, Y. Ono, K. Kawachi, K. Yokoo, S. Ono, K. Omote, Y. Kasama, S. Ishikawa, T. Komuro and H. Tobita, Nat. Chem., 2010, 4, 678 CrossRef PubMed.
- S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938 CrossRef CAS PubMed.
- N. L. Bill, M. Ishida, S. Bähring, J. M. Lim, S. Lee, C. M. Davis, V. M. Lynch, K. A. Nielsen, J. O. Jeppesen, K. Ohkubo, S. Fukuzumi, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2013, 135, 10852 CrossRef CAS PubMed.
- Y. Kawashima, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2012, 116, 8942 CrossRef CAS PubMed.
- K. Ohkubo, Y. Kawashima and S. Fukuzumi, Chem. Commun., 2012, 48, 4314 RSC.
- J. S. Park, E. Karnas, K. Ohkubo, P. Chen, K. M. Kadish, S. Fukuzumi, C. W. Bielawski, T. W. Hudnall, V. M. Lynch and J. L. Sessler, Science, 2010, 329, 1324 CrossRef CAS PubMed.
- M. Murakami, K. Ohkubo, T. Nanjo, K. Souma, N. Suzuki and S. Fukuzumi, ChemPhysChem, 2010, 11, 2594 CrossRef CAS PubMed.
- K. Ohkubo, Y. Kawashima, H. Sakai, T. Hasobe and S. Fukuzumi, Chem. Commun., 2013, 49, 4474 RSC.
- T. Hasobe, Y. Kashiwagi, M. A. Absalom, J. Sly, K. Hosomizu, M. J. Crossley, H. Imahori, P. V. Kamat and S. Fukuzumi, Adv. Mater., 2004, 16, 975 CrossRef CAS.
- T. Hasobe, H. Imahori, S. Fukuzumi and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 12105 CrossRef CAS.
- T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, D. Kim, T. Hanada, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216 CrossRef CAS PubMed.
- T. Hasobe, P. V. Kamat, V. Troiani, N. Solladié, T. K. Ahn, S. K. Kim, D. Kim, A. Kongkanand, S. Kuwabata and S. Fukuzumi, J. Phys. Chem. B, 2005, 109, 19 CrossRef CAS PubMed.
- D. Sun, F. S. Tham, C. A. Reed, L. Chaker, M. Burgess and P. D. W. Boyd, J. Am. Chem. Soc., 2000, 122, 10704 CrossRef CAS.
- D. Sun, F. S. Tham, C. A. Reed, L. Chaker and P. D. W. Boyd, J. Am. Chem. Soc., 2002, 124, 6604 CrossRef CAS PubMed.
- M. Tanaka, K. Ohkubo, C. P. Gros, R. Guilard and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 14625 CrossRef CAS PubMed.
- L. P. Hernández-Eguía, E. C. Escudero-Adán, I. C. Pintre, B. Ventura, L. Flamigni and P. Ballester, Chem.–Eur. J., 2011, 17, 14564 CrossRef PubMed.
- A. L. Kieran, S. I. Pascu, T. Jarrosson and J. K. M. Sanders, Chem. Commun., 2005, 1276 RSC.
- B. Grimm, J. Schornbaum, C. M. Cardona, J. D. van Paauwe, P. D. W. Boyd and D. M. Guldi, Chem. Sci., 2011, 2, 1530 RSC.
- L. P. Hernández-Eguía, E. C. Escudero-Adán, J. R. Pinzón, L. Echegoyen and P. Ballester, J. Org. Chem., 2011, 76, 3258 CrossRef PubMed.
- S. K. Samanta and M. Schmittel, Org. Biomol. Chem., 2013, 11, 3108 CAS.
- K. Börjesson, J. G. Woller, E. Parsa, J. Mårtensson and B. Albinsson, Chem. Commun., 2012, 48, 1793 RSC.
- B. Kang, R. K. Totten, M. H. Weston, J. T. Hupp and S. T. Nguyen, Dalton Trans., 2012, 41, 12156 RSC.
- S. Fukuzumi, I. Amasaki, K. Ohkubo, C. P. Gros, R. Guilard and J.-M. Barbe, RSC Adv., 2012, 2, 3741 RSC.
- H. Nobukuni, F. Tani, Y. Shimazaki, Y. Naruta, K. Ohkubo, T. Nakanishi, T. Kojima, S. Fukuzumi and S. Seki, J. Phys. Chem. C, 2009, 113, 19694 CAS.
- T. Yamaguchi, N. Ishii, K. Tashiro and T. Aida, J. Am. Chem. Soc., 2003, 125, 13934 CrossRef CAS PubMed.
- R. Harada, Y. Matsuda, H. Okawa and T. Kojima, Angew. Chem., Int. Ed., 2004, 43, 1825 CrossRef CAS PubMed.
- T. Kojima, T. Nakanishi, R. Harada, K. Ohkubo, S. Yamauchi and S. Fukuzumi, Chem.–Eur. J., 2007, 13, 8714 CrossRef CAS PubMed.
- T. Nakanishi, T. Kojima, K. Ohkubo, T. Hasobe, K. Nakayama and S. Fukuzumi, Chem. Mater., 2008, 20, 7492 CrossRef CAS.
- T. Kojima, R. Harada, T. Nakanishi, K. Kaneko and S. Fukuzumi, Chem. Mater., 2007, 19, 51 CrossRef CAS.
- A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193 CrossRef CAS PubMed.
- R. J. O. M. Hoofman, M. P. de Hass, L. D. A. Siebbeles and J. M. Warman, Nature, 1998, 392, 54 CrossRef CAS PubMed.
- J. H. Choi, T. Honda, S. Seki and S. Fukuzumi, Chem. Commun., 2011, 47, 11213 RSC.
- E. Frankevich, Y. Maruyama and H. Ogata, Chem. Phys. Lett., 1993, 214, 39 CrossRef CAS.
- H. Nobukuni, Y. Shimazaki, H. Uno, Y. Naruta, K. Ohkubo, T. Kojima, S. Fukuzumi, S. Seki, H. Sakai, T. Hasobe and F. Tani, Chem.–Eur. J., 2010, 16, 11611 CrossRef CAS PubMed.
- T. Kamimura, K. Ohkubo, Y. Kawashima, H. Nobukuni, Y. Naruta, F. Tani and S. Fukuzumi, Chem. Sci., 2013, 4, 1451 RSC.
- F. D'Souza, P. M. Smith, S. Gadde, A. L. McCarty, M. J. Kullman, M. E. Zandler, M. Itou, Y. Araki and O. Ito, J. Phys. Chem. B, 2004, 108, 11333 CrossRef CAS.
- N. Armaroli, F. Diederich, L. Echegoyen, T. Habicher, L. Flamigni, G. Marconi and J.-F. Nierengarten, New J. Chem., 1999, 77 RSC.
- T. Da Ros, M. Prato, D. M. Guldi, E. Alessio, M. Ruzzi and L. Pasimeni, Chem. Commun., 1999, 635 RSC.
- F. Hauke, A. Swartz, D. M. Guldi and A. Hirsch, J. Mater. Chem., 2002, 12, 2088 RSC.
- A. Takai, M. Chkounda, A. Eggenspiller, C. P. Gros, M. Lachkar, J.-M. Barbe and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 4477 CrossRef CAS PubMed.
- F. D'Souza, G. R. Deviprasad, M. E. Zandler, V. T. Hoang, A. Klykov, M. VanStipdonk, A. Perera, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2002, 106, 3243 CrossRef CAS.
- D. I. Schuster, K. Loa, D. M. Guldi, A. Palkar, L. Echegoyen, C. Stanisky, R. J. Cross, M. Niemi, M. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2007, 129, 15973 CrossRef CAS PubMed.
- A. Takai, C. P. Gros, R. Guilard and S. Fukuzumi, Chem.–Eur. J., 2009, 15, 3110 CrossRef CAS PubMed.
- A. Takai, C. P. Gros, J.-M. Barbe and S. Fukuzumi, Chem.–Eur. J., 2011, 17, 3420 CrossRef CAS PubMed.
- A. Takai, C. P. Gros, J.-M. Barbe and S. Fukuzumi, Phys. Chem. Chem. Phys., 2010, 12, 12160 RSC.
- N. Solladié, A. Hamel and M. Gross, Tetrahedron Lett., 2000, 41, 6075 CrossRef.
- F. Aziat, R. Rein, J. Peon, E. Rivera and N. Solladié, J. Porphyrins Phthalocyanines, 2008, 12, 1232 CrossRef CAS.
- S. Fukuzumi, K. Saito, K. Ohkubo, V. Troiani, H. Qiu, S. Gadde, F. D'Souza and N. Solladié, Phys. Chem. Chem. Phys., 2011, 13, 17019 RSC.
- S. Fukuzumi, K. Saito, K. Ohkubo, T. Khoury, Y. Kashiwagi, M. A. Absalom, S. Gadde, F. D'Souza, Y. Araki, O. Ito and M. J. Crossley, Chem. Commun., 2011, 47, 7980 RSC.
- T. Hasobe, K. Saito, P. V. Kamat, V. Troiani, H. Qiu, N. Solladié, T. K. Ahn, K. S. Kim, S. K. Kim, D. Kim, F. D'Souza and S. Fukuzumi, J. Mater. Chem., 2007, 17, 4160 RSC.
| This journal is © The Royal Society of Chemistry 2013 |