 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure, EPR/ENDOR and DFT characterisation of a [CuII(en)2](OTf)2 complex†
Emma Carter*a,
E. Louise Hazelandab,
Damien M. Murphya and
Benjamin D. Ward*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: CarterE4@Cardiff.ac.uk; WardBD@Cardiff.ac.uk; Fax: +44 (0)29 208 74030; Tel: +44 (0)29 208 70302
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK
First published on 23rd August 2013
Abstract
The Jahn–Teller distorted Cu(II) complex [Cu(en)2](OTf)2 1 (en = 1,2-diaminoethane) has been reported and characterised using X-ray crystallography, EPR and ENDOR spectroscopy, and DFT calculations. The solid state structure shows an intra- and inter-molecular hydrogen-bonded network via the N–H groups and the coordinated triflate anions. CW and pulsed EPR/ENDOR were used to determine the spin Hamiltonian parameters of the Cu(II) complex, which were in excellent agreement with the DFT. The structure of the complex, as determined by angular selective ENDOR, is also in good agreement with the crystal structure, confirming the axial coordination of the counter-ion(s) in the frozen solution. The small 14N superhyperfine couplings are also consistent with the sp3 hybridised nature of the coordinating nitrogens. These results show that the correlation between the 14N hyperfine coupling and hybridisation of donor nitrogens can be useful to determine not only the coordination around the Cu(II) metal centre but also the nature of the donor in unknown Cu(II) systems.
Introduction
The coordination chemistry of copper(II), with its significant Jahn–Teller distortion along the z-axis, is somewhat more complex than for the majority of transition metals, since a variety of coordination geometries are commonplace.1 However, copper complexes have found widespread application in functional metal complexes, such as catalysts. Being relatively inexpensive, they have been highlighted as potential candidates for environmentally benign catalysts based upon non-toxic and inexpensive metals.2 The paramagnetic nature of copper(II), with its single unpaired electron, means that analysis using Electron Paramagnetic Resonance (EPR) techniques can divulge a significant amount of information that can be helpful in further understanding the intricacies of its complex coordination chemistry,3 and it is this aspect that we report here.As a means to embark on these investigations, we have employed the ubiquitous 1,2-diaminoethane (ethylene diamine, en) as a supporting ligand for copper(II). Ethylene diamine is well established as an excellent supporting ligand for Cu(II), since there are many complexes found with two en ligands occupying the equatorial plane of a Jahn–Teller distorted octahedral complex. It was with some surprise therefore to discover that the crystal structure of the anhydrous complex [Cu(en)2](OTf)2 1 has not been reported and we sought to use this relatively simple complex as a means to establish the synergic analysis of such complexes using a combination of synthetic, spectroscopic and theoretical methods.
Whilst EPR studies of coordinated Cu(II) complexes are numerous, considerably fewer studies involving Electron Nuclear Double Resonance (ENDOR) spectroscopy of these complexes have been reported.4 Owing to the higher resolving power of ENDOR, a more detailed analysis of the ligand hyperfine couplings can be extracted from the EPR spectra, which in turn provides more information on the metal–ligand orientation and unpaired spin densities, even in orientationally disordered systems,4,5 as illustrated by the many excellent ENDOR reviews of copper proteins.6 Furthermore, it is well known that the correlation between the Cu g∥ and A∥ values can provide useful insights into the local coordination environment surrounding the Cu(II) centre.7 However, an equally important correlation is also found between the hyperfine coupling constants of the donor nitrogens and the structure of the first coordination sphere in the Cu(II) complex, as revealed by the 14N ENDOR spectra.8 As a result, both single crystal and powder (frozen solution) ENDOR studies of Cu(II) complexes bearing an N4, N2S2, cis-N2O2 and trans-N2O2, have all been investigated in the past9 in order to study the relationship between the structure of the coordinated Cu(II) ion and the spin Hamiltonian parameters. Surprisingly, no detailed ENDOR studies have ever been reported for perhaps one of the simplest Cu(II) complexes bearing an N4 coordination sphere (i.e., [CuII(en)2]2+). Historically, only a limited number of EPR reports have appeared on the hydrated mono- and bis-ethylenediamine Cu(II) complex,10–13 and only the g values were reported in these works.
Herein, we provide the first detailed EPR and ENDOR analysis of the [CuII(en)2](OTf)2 complex bearing the weakly coordinated triflate counterions (1), in frozen acetonitrile–THF solution, and compare the experimental spin Hamiltonian parameters to the theoretical values extracted by DFT. The structural parameters derived from the angular selective ENDOR methodology are then compared to the crystal structure.
Results and discussion
Crystal structure
The ethylene diamine copper(II) complex [Cu(en)2](OTf)2 1 was prepared using anhydrous copper(II) triflate in dry acetonitrile. X-ray quality single crystals were grown directly from the reaction mixture; the molecular structure is displayed in Fig. 1, with principal metric parameters listed in Table 1. The structure contains two independent copper centres, each displaying a distorted octahedral geometry, as evidenced by the angles subtended at copper, which deviate from the ideal 90°. As expected for Cu(II), the complex displays a significant Jahn–Teller distortion along the O–Cu–O axis, the bond lengths being significantly greater than the Cu–N bond distances by ca. 0.5 Å. The triflate anions, whilst loosely coordinated to the copper centres, are also held in place by a series of intra- and inter-molecular hydrogen bonds, which were found between non-coordinating oxygen atoms and N–H groups of the ethylene diamine ligands. The metric parameters lie within the expected ranges found within the Cambridge Structural Database.142. H atoms omitted for clarity, except those bonded to N, which are shown as spheres of arbitrary radius.](/image/article/2013/DT/c3dt51694f/c3dt51694f-f1.gif) | ||
| Fig. 1 Displacement ellipsoid plot (35%) of [Cu(en)2](OTf)2. H atoms omitted for clarity, except those bonded to N, which are shown as spheres of arbitrary radius. | ||
| Cu(1)–N(1) | 2.0202(15) | Cu(2)–N(5) | 2.0003(14) |
| Cu(1)–N(2) | 2.0190(14) | Cu(2)–N(6) | 2.0214(15) |
| Cu(1)–N(3) | 2.0154(15) | Cu(2)–N(7) | 1.9964(15) |
| Cu(1)–N(4) | 2.0136(15) | Cu(2)–N(8) | 2.0169(15) |
| Cu(1)–O(1) | 2.5104(12) | Cu(2)–O(7) | 2.5824(12) |
| Cu(1)–O(4) | 2.4787(12) | Cu(2)–O(11) | 2.6903(12) |
| N(3)–O(3) | 2.978(2) | N(6)–O(9) | 3.196(2) |
| N(4)–O(12) | 3.1107(19) | N(8)–O(10) | 3.1486(19) |
| N(1)–Cu(1)–N(3) | 175.91(6) | N(5)–Cu(2)–N(7) | 179.11(6) |
| N(2)–Cu(1)–N(4) | 179.24(6) | N(6)–Cu(2)–N(8) | 179.03(7) |
| N(1)–Cu(1)–O(1) | 91.77(5) | N(5)–Cu(2)–O(7) | 84.11(5) |
| N(2)–Cu(1)–O(1) | 86.25(5) | N(6)–Cu(2)–O(7) | 91.04(5) |
| N(3)–Cu(1)–O(1) | 92.31(5) | N(7)–Cu(2)–O(7) | 96.39(5) |
| N(4)–Cu(1)–O(1) | 93.13(5) | N(8)–Cu(2)–O(7) | 89.90(5) |
| N(3)–H(3D)–O(3) | 137(2) | N(6)–H(6D)–O(9) | 148.4(18) |
| N(4)–H(4C)–O(12) | 146.0(19) | N(8)–H(8D)–O(10) | 152.9(19) |
Whilst the structure of 1 has not been reported, the structural motif containing two ethylene diamine units occupying the equatorial plane of a Cu(II) complex is by no means uncommon. There are many such complexes reported, the most common being those in which water ligands occupy the axial positions,1 although examples have been reported containing coordinated counterions, such as nitrate,1d,15 oxalate,16 sulphonate,17 and carboxylate.18
The mass spectrometry data are supportive that the solid state structure is largely representative of the structure in solution: as expected from a weakly coordinated triflate, the parent ion was not observed, and the largest identifiable peak was that attributed to the cation formed via loss of one triflate. Nevertheless, the fact that one triflate remains coordinated under such conditions shows a significant degree of triflate coordination that is likely to exist in solution, and this has been confirmed using ENDOR measurements (vide infra).
DFT calculations
The structure of [Cu(en)2](OTf)2 1 was calculated by density functional theory calculations, using the unrestricted B3LYP hybrid functional and the 6-31+G(d,p) basis set incorporated into the Gaussian 09 software package.19 The optimised structure was found to be in excellent agreement with the X-ray structure; TD-DFT calculations were performed and indicated an excited state absorption at 536 nm, corresponding largely to a formally forbidden transition between MOs containing d character (Fig. 2 and Fig. S1†). This matches extremely well with the experimentally observed UV-vis absorption at 545 nm (ε = 75.6 mol−1 dm3 cm−1).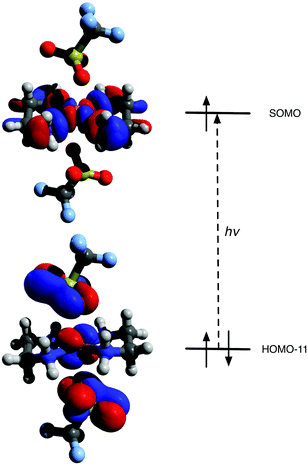 | ||
| Fig. 2 Principal β MOs involved in TD-DFT computed absorption (HOMO − 11 → SOMO). | ||
The spin Hamiltonian parameters for Cu(II) and the corresponding ligand nuclei, 1H, 14N and 19F, were calculated for [Cu(en)2](OTf)2 1 in order to compare to the experimental EPR and ENDOR data. The calculations were performed using the ORCA package20 employing the atomic coordinates of the DFT-optimised structure. The relevant EPR parameters are listed in Tables 2–4 and discussed further below.
| g1a | g2a | g3a | A1b | A2b | A3c | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| The Cu hyperfine values (A) values are given in MHz.a ±0.005.b ±5.c ±3 MHz.d No hyperfine values reported. | ||||||||
| [Cu(en)2](OTf)2 | Expt | 2.040 | 2.046 | 2.202 | −78.0 | −82.0 | −602.0 | |
| DFT | 2.046 | 2.049 | 2.152 | −83.3 | −98.7 | −858.0 | ||
| [Cu(en)](Cl)2 | 2.049 | 2.049 | 2.239 | — | — | — | 10d | |
| [Cu(en)2](Cl)2 | 2.047 | 2.047 | 2.205 | — | — | — | 11d | |
| [Cu(en)2](BF4)2 | 2.048 | 2.048 | 2.198 | — | — | — | 11d | |
| [Cu(en)2](NO3)2 | 2.059 | 2.059 | 2.189 | — | — | — | 12d | |
| [Cu(en)2](SO4) | 2.054 | 2.054 | 2.166 | — | — | — | 13d | |
| [Cu(gly)2] | 2.0434 | 2.0715 | 2.2644 | 156.4 | 39.7 | 468.7 | 9h | |
| [Cu(Box)](OTf)2 | 2.064 | 2.073 | 2.313 | 15.0 | 14.5 | 506.7 | 25 | |
| [Cu(Box)]2 | 2.054 | 2.063 | 2.254 | 25.9 | 28.9 | 461.3 | 25 | |
| [CuPc] | 2.0405 | 2.0405 | 2.1625 | −86.0 | −86.0 | −643.0 | 9b | |
| Coordination | Complexe | A1a | A2 | A3 | P1b | P2 | P3 | e2qQ/hc | ηd | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| All values are given in MHz.a ±0.2 MHz.b ±0.1 MHz.c ±0.2 MHz.d ±0.1.e The structures of these complexes are given in Fig. S5; T.W. = this work. | ||||||||||
| cis-N2O2 (imino) | [Cu(salen)] | 50.5 | 37.4 | 38.5 | −1.15 | 0.70 | 0.45 | −2.3 | 0.2 | 9e |
| [Cu(acacen)] | 48.7 | 39.1 | 39.1 | — | — | — | — | — | 9g | |
| trans-N2O2 | [Cu(sal)2] | 51.9 | 42.1 | 43.6 | −1.71 | 1.91 | −0.20 | 9d | ||
| [Cu(gly)2] | 32.8 | 20.6 | 17.40 | 1.26 | −1.81 | 0.55 | 9h | |||
| N2 | [Cu(Box)](OTf)2 | 45.6 | 35.9 | 36.7 | −0.87 | 0.97 | −0.10 | −2.3 | 0.2 | 25 |
| [Cu(Box)]Cl2 | 41.9 | 32.5 | 32.8 | −0.87 | 0.97 | −0.10 | −2.5 | 0.15 | 25 | |
| N4 (aza) | [Cu(TPP)] | 54.2 | 42.7 | 44.0 | −0.62 | 0.93 | −0.31 | 9a | ||
| [CuPc] | 56.4 | 44.8 | 45.7 | −0.79 | 0.82 | 0.03 | 9b | |||
| [Cu(Box)2] | 39.8 | 33.1 | 32.9 | −0.57 | 0.52 | 0.05 | 25 | |||
| N4 (amine) | [Cu(en)2](OTf)2 | 39.35 | 26.0 | 26.4 | −1.25 | 0.86 | 0.39 | T.W. | ||
| A1(x)a | A2(y) | A3(z) | aiso | αb | βc | γ | Adip | ||
|---|---|---|---|---|---|---|---|---|---|
| All A values are given in MHz, with relative signs.a ±0.2 MHz.b ±10°.c ±5°.d Note this angle of rotation affects the ordering of the A tensor. | |||||||||
| C–Hax | Expt | 5.70 | 10.0 | 4.76 | 6.77 | 0 | 10 | 0 | −2.17 |
| DFT | 5.33 | 9.52 | 4.16 | 6.34 | 16.4 | 9.7 | 94.5 | −2.18 | |
| C–Heq | Expt | −2.63 | −1.70 | 4.30 | −0.01 | 0 | 60 | 0 | 4.31 |
| DFT | −2.63 | −1.77 | 4.00 | −0.13 | −15.2 | 63.1 | 43.5 | 4.13 | |
| N–H | Expt | −8.00 | 5.52 | −13.65 | −5.38 | 35 | 50 | 0 | −8.27 |
| DFT | −9.66 | 5.52 | −17.09 | −7.08 | 41.1 | 47.7 | −13.7 | −10.01 | |
| 19F | Expt | −0.35 | −0.35 | 0.80 | 0.1 | 0 | 0 | 0 | 0.7 |
| DFT | 0.79 | −0.40 | −0.38 | 0.01 | −4.0 | 86.6d | 6.3 | 0.79 | |
EPR spectroscopy
The X-band continuous wave (CW) EPR spectrum of 1 was recorded in a frozen solution of acetonitrile–tetrahydrofuran (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 10 K (Fig. 3a). This MeCN–THF solution was found to give the best quality spectrum. The isotropic room temperature EPR spectrum and the X-band pulsed (FSED) EPR spectrum are also shown in the ESI for completeness (Fig. S2 and S3†). No extra resolution was observed in the pulsed EPR spectrum. The low temperature CW spectrum (Fig. 3a) was found to be axially symmetric (Table 2). Although the copper hyperfine splitting could be readily extracted from the spectrum, the contributions to the spin Hamiltonian from the two 63,65Cu isotopes and the 14N ligand nuclei could not be determined. In general, large 14N superhyperfine couplings can be directly observed in the X-band EPR spectra of Cu(II) macrocycles (e.g., in Cu(II) porphyrins, phthalocyanines) or salen complexes. The absence of such resolved features and resolution in Fig. 3a arises from the considerably smaller magnitude of the 14N coupling in 1 (vide infra).
:1) at 10 K (Fig. 3a). This MeCN–THF solution was found to give the best quality spectrum. The isotropic room temperature EPR spectrum and the X-band pulsed (FSED) EPR spectrum are also shown in the ESI for completeness (Fig. S2 and S3†). No extra resolution was observed in the pulsed EPR spectrum. The low temperature CW spectrum (Fig. 3a) was found to be axially symmetric (Table 2). Although the copper hyperfine splitting could be readily extracted from the spectrum, the contributions to the spin Hamiltonian from the two 63,65Cu isotopes and the 14N ligand nuclei could not be determined. In general, large 14N superhyperfine couplings can be directly observed in the X-band EPR spectra of Cu(II) macrocycles (e.g., in Cu(II) porphyrins, phthalocyanines) or salen complexes. The absence of such resolved features and resolution in Fig. 3a arises from the considerably smaller magnitude of the 14N coupling in 1 (vide infra).
2 1 recorded in a frozen acetonitrile–THF (1 : 1) solution (a) X-band, (b) Q-band, (c) road-map showing the angular dependency profile. The corresponding simulations are given in a′ and b′.](/image/article/2013/DT/c3dt51694f/c3dt51694f-f3.gif) | ||
| Fig. 3 CW EPR spectra (10 K) of [Cu(en)2](OTf)2 1 recorded in a frozen acetonitrile–THF (1:1) solution (a) X-band, (b) Q-band, (c) road-map showing the angular dependency profile. The corresponding simulations are given in a′ and b′. | ||
To obtain more accurate g values, the Q-band EPR spectrum was also recorded (Fig. 3b). Owing to the magnitude of the g3 and A3 components, a considerable overshoot appears in the X-band spectrum, common for such Cu(II) complexes,21 and this complicates the extraction of the perpendicular g values from X-band data alone. Owing to the large g strain experienced at the higher field, a reliable estimate of the unresolved perpendicular A values is also difficult; nevertheless, analysis of the aiso coupling from the room temperature spectrum (Fig. S2†) enables an accurate value to be found. The resulting X- and Q-band simulations are shown in Fig. 3a′ and b′, and the associated spin Hamiltonian parameters are listed in Table 2.
The g values for 1 are similar to those previously reported for [Cu(en)2] and typical of those expected of Cu(II) bearing a dx2−y2 ground state. The literature references in Table 2 were all studied at X-band frequency, hence only axially symmetric g values were reported. In this work, a very small rhombic distortion can be observed in g for [Cu(en)2](OTf)2 afforded by the increased resolution at Q-band frequency (Table 2). The variation in g3 is clearly influenced by the choice of the counterion, although the chloride, sulphate and nitrate bearing complexes also reportedly contain water (i.e., [Cu(en)2](X)·2H2O) compared to the anhydrous 1. The ACu values for the bis-ethylenediamine complex have not previously been reported, due to the spectra being recorded on either undiluted powders or single crystals.10–13 The experimental values found in this work were found to be in good agreement with the DFT derived values (Table 2), not withstanding the known limitations of DFT in calculating metal hyperfine splitting, where CuA values are often overestimated.22 These g/A parameters confirm the tetragonally octahedral environment of copper in 1.
ENDOR of ligand nuclei
ENDOR spectroscopy provides more information on the extent of spin delocalisation onto the surrounding ligand nuclei. In ideal cases (single crystals), analysis of the spectra yields the complete hyperfine tensor for interacting nuclei, including the orientation of the respective tensors. In frozen solutions, the analysis is considerably more complicated. Nevertheless, by recording the ENDOR data at selected magnetic field positions (Fig. 3c), one can observe spectra from the copper complexes having a specific orientation with respect to the external magnetic field.The theory of angular selective ENDOR for disordered systems was initiated by Rist and Hyde,5a and completed for general cases by Hoffman and co-workers5b,c (with later adaptations by Kreilick),5d,e and has been explained in several reviews.23 An important aspect to note, is that the two unique ‘single crystal-like’ turning points in the powder EPR pattern, namely corresponding to a g = g⊥ and g = g∥ position (shown by arrows in Fig. 3c) can be more readily resolved at Q-band, since considerable overlap of multiple Cu mI transitions occurs close to g = g⊥ at X-band, rendering the data more difficult to simulate. Hence, the angular selective Q-band 14N, 1H and 19F ENDOR spectra recorded at multiple field positions are shown in Fig. 4 and 5.
2 1 recorded in a frozen d3-acetonitrile–d8-THF (1 : 1) solution. The ENDOR spectra were recorded at the field (a) 1198, (b) 1193.5, (c) 1188.9, (d) 1135.2, (e) 1095.6 and (f) 1077.5 mT. Corresponding simulations are shown with a dotted line. Peaks appearing at lower frequencies arise from solvent 2H.](/image/article/2013/DT/c3dt51694f/c3dt51694f-f4.gif) | ||
| Fig. 4 CW Q-band 14N ENDOR spectra (10 K) of [Cu(en)2](OTf)2 1 recorded in a frozen d3-acetonitrile–d8-THF (1:1) solution. The ENDOR spectra were recorded at the field (a) 1198, (b) 1193.5, (c) 1188.9, (d) 1135.2, (e) 1095.6 and (f) 1077.5 mT. Corresponding simulations are shown with a dotted line. Peaks appearing at lower frequencies arise from solvent 2H. | ||
2 1 recorded at the field positions (a) 1198, (b) 1193.5, (c) 1188.9, (d) 1183.6, (e) 1175.9, (f) 1164.7, (g) 1151.6, (h) 1135.2, (i) 1116.2, (j) 1095.6 and (k) 1077.5 mT. Corresponding simulations are shown with a dotted line.](/image/article/2013/DT/c3dt51694f/c3dt51694f-f5.gif) | ||
| Fig. 5 CW Q-band 1H ENDOR spectra (10 K) of [Cu(en)2](OTf)2 1 recorded at the field positions (a) 1198, (b) 1193.5, (c) 1188.9, (d) 1183.6, (e) 1175.9, (f) 1164.7, (g) 1151.6, (h) 1135.2, (i) 1116.2, (j) 1095.6 and (k) 1077.5 mT. Corresponding simulations are shown with a dotted line. | ||
The experimental and simulated angular selective 14N ENDOR spectra are shown in Fig. 4. The resulting hyperfine and quadrupolar values are listed in Table 3. The hyperfine tensor is very nearly axially symmetric, with the largest principal axis directed approximately along the Cu–N bond. There is no evidence of any inequivalency among the four 14N nuclei in the ENDOR spectra, as suggested by the slight rhombicity in the g tensor. Any minor in-plane distortion leading to in-equivalencies in the nitrogen couplings, would likely be unresolved in the intrinsically broad 14N spectra. The hyperfine coupling is noticeably smaller in 1 compared to a range of other Cu(II) complexes bearing coordinated nitrogen ligands in an N2, N4 or N2O2 environment (see Table 3). This indicates that the delocalisation of the unpaired spin onto the ligand nitrogen nuclei is significantly smaller in this complex (aiso = 30.5 MHz for 1, compared to 42.13 MHz for [Cu(salen)],9e for example). Interactions of Cu(II) complexes with electron-donor molecules causes a decrease of the nitrogen coupling constants in the square-planar array.8 In the present case of [Cu(en)2](OTf)2, electron donation from the oxygen atoms of the counterions in the two axial positions may be responsible for the much smaller hyperfine couplings detected.
The magnitude of the 14N hyperfine coupling in any Cu(II) complex will depend on the extent of distortions to the in-plane arrangement of the ligands9f (which in turn affects the unpaired electron distribution) but also from hybridisation of the nitrogen coordinating orbitals.8 Complexes bearing coordinated nitrogens with planar conformation, such as imino nitrogens and aromatic aza nitrogens (salen and porphyrin complexes, Table 3), are expected to produce large hyperfine couplings, compared to those with nitrogens possessing a tetrahedral conformation, such as amine nitrogens. As a result the sp3 hybridised nature of the nitrogens in the en ligand accounts for the lower observed 14N hyperfine couplings in 1. Although in-plane distortions may partly contribute to the lower NA values in 1, the hybridised 14N orbitals of en which overlap with the |x2 − y2〉 metal orbitals, would appear to be the dominant factor.
The results presented herein demonstrate that the N-donor hybridisation might be predicted based on the magnitude of the 14N hyperfine coupling determined through ENDOR spectroscopy. This diagnostic tool may prove particularly useful in investigations of samples with unknown or competing donor molecules.
The magnitude of the 14N couplings (Table 3) are such that at X-band frequencies, the 1H and 14N peaks are completely overlapped, owing to the large nuclear Larmor frequency of the proton. This can be seen in the pulsed X-band Davies ENDOR spectrum (Fig. S4†) of 1, where the 14N couplings are completely buried and unresolved under the 1H peaks. For this reason, we recorded the ligand 1H couplings at Q-band frequencies (Fig. 5), since νn = 51 MHz for 1H at 1200 mT, avoiding any overlap and distortion with the 14N peaks which now appear in the 5–30 MHz region (Fig. 4).
The 1H ENDOR spectra were extremely well resolved, facilitating the simulations and extraction of the hyperfine couplings. The resulting principal components of the hyperfine values are listed in Table 4. Three distinct sets of proton couplings were identified. By comparison to the values calculated by DFT, these couplings were assigned to the –NH amine proton and the axial and equatorially positioned methine protons of the carbon backbone (labelled CHax/eq). The agreement between the experimental and calculated hyperfine couplings is excellent. The –NH proton possesses a relatively large aiso and Adip value, reflecting both the unpaired spin density in the sp3 hybridised 14N orbitals, and the close proximity of the amine proton to the copper centre. Whilst the axial –CH proton also has a large aiso component, the dipolar contribution to the hyperfine tensor is considerably less owing to the Cu⋯H distance. An almost opposite trend is manifested for the equatorial –CH proton, and once again this is expected owing to the orientation of these protons in positions parallel and orthogonal to the primary Cu–N4 plane.
Although the crystal structure clearly evidences the presence of an internal H-bond between the –NH proton and the oxygen of the triflate counter-ion, we were unable to confirm this via our ENDOR measurements. In principle, H-bonding to a proton will result in a broadening of the ENDOR peaks, and in some favourable cases, a shift in the hyperfine coupling.24 Recording the ENDOR spectra of [Cu(en)2]2+ in the absence of a potential H-bonding counter-ion, and in a solvent of different dielectric properties, would be required to extract the experimental –NH coupling in the absence of H-bonding and to examine how solvent affects the strength of the H-bond.
The 19F hyperfine coupling, arising from the coordinated triflate counter-ion, was also clearly resolved in the ENDOR spectra. Since νn = 48 MHz for 19F at 1200 mT, the fluorine couplings are partly overlaid on the 1H spectra (Fig. 5). Since the theoretical aiso for 19F is very large (52808 MHz), even a small spin density at the nucleus is sufficient to produce a resolvable coupling by ENDOR (Table 4) and this confirms that the counter-ion remains coordinated to the copper in solution. According to the crystal structure, the Cu⋯F distances vary from 4.51–5.96 Å, with coordination to copper along the axial (z) axis. The 19F ENDOR data also support this, since the largest component of the coupling occurs along the z-axis (at g = g∥). The ENDOR data clearly shows we have [Cu(en)2](OTf)x where x = 1 or 2. Whilst it is not possible to determine the number of coordinating (OTf)− ions from the ENDOR data directly, the relatively narrow linewidths in the 19F ENDOR spectrum indicates a well-defined Cu–F interaction, and the absence of a 19F matrix line would suggest that there are no (OTf)− ions at a more remote distance from the copper centre (i.e. non-coordinating). Therefore, in combination with the XRD evidence we suggest that x = 2, i.e. [Cu(en)2](OTf)2. This situation is in fact analogous to a recent example reported by us for a [Cu(bis-oxazoline)](OTf)2 complex,25 illustrating the advantages of ENDOR to study the coordination mode of counter-ions in metal complexes.
Experimental
[Cu(en)2](OTf)2 was prepared and handled under an atmosphere of argon or dinitrogen using standard Schlenk and glove box techniques. Solvents were dried by passing through an alumina drying column incorporated into a MBraun SPS800 solvent purification system. All solvents were degassed, saturated with argon, and stored under argon in Teflon valve ampoules prior to use. All other reagents were purchased from commercial suppliers and used as received unless otherwise stated.Infrared spectra were prepared as KBr pellets and were recorded on a Jasco 660-Plus FTIR spectrometer. Infrared data are quoted in wavenumbers (cm−1). UV/vis data were measured on a Perkin Elmer Lambda 20 UV/vis spectrometer. Elemental analyses were recorded by Mr Stephen Boyer at London Metropolitan University, and mass spectra were recorded by the EPSRC National Mass Spectrometry Service.
[Cu(en)2](OTf)2
A solution of copper(II) triflate (0.5 g, 1.38 mmol) in acetonitrile was added to a solution of ethylene diamine (2 eq., 2.76 mmol, 0.18 ml) in acetonitrile. The reaction was stirred overnight before concentrating and cooling to 5 °C. The title complex formed as blue crystals on standing overnight. Yield: 0.44 g, 65%. Anal. Calc. for C6H16CuF6N4O6S2: C 14.96, H 3.35, N 11.63. Found: C 15.06, H 3.29, N 11.53. UV/vis: λmax = 545 nm, ε = 75.6 mol−1 dm3 cm−1. IR (KBr) [ν/cm−1]: 3337 (N–H), 3284 (N–H), 2967 (C–H), 2905 (C–H), 1160 (C–F), 1028 (C–N). Accurate mass ES-MS for [Cu{C2H4(NH2)2}2(OTf)]+: m/z = 332.0186 (calc. for C5H16CuF3N4O3S: 332.0191).EPR/ENDOR measurements
For CW and pulsed EPR/ENDOR measurements, the [Cu(en)2](OTf)2 1 was dissolved in d3-acetonitrile–d8-tetrahydrofuran (1:1, 21 mM). All X-band EPR spectra were recorded on a Bruker EMX spectrometer operating at 100 kHz field modulation and equipped with a high sensitivity X-band cavity (ER 4119HS). The spectra were recorded at a microwave power of 10 mW at 140 or 298 K. The CW Q-band ENDOR spectra were recorded at 10 K on a CW Bruker ESP 300E series spectrometer equipped with an ESP360 DICE ENDOR unit, operating at 12.5 kHz field modulation in a Q-band ENDOR cavity (Bruker ER 5106 QT-E). The ENDOR spectra were obtained using 8 dB RF power from an ENI A-300 RF amplifier and 50 or 200 kHz RF modulation depth and 1 mW microwave power. The pulsed X-band EPR/ENDOR spectra were recorded on a Bruker Elexsys E580 spectrometer equipped with a liquid Helium cryostat from Oxford Inc. Simulations were performed using the Easyspin toolbox in Matlab.26
DFT calculations
Geometry optimisation and TD-DFT calculations were performed using the Gaussian 09 program.19 The structure of [Cu(en)2](OTf)2 1 was optimised without geometry restraints using the unrestricted B3LYP hybrid functional, employing the 6-31+G(d,p) basis set on all atoms. The geometry optimisation was followed by a frequency calculation to ascertain the nature of the stationary point (minimum vs. saddle point). TD-DFT calculations were performed on the optimised geometry. The 12 lowest excitation energies were calculated.The EPR parameters were calculated via spin-unrestricted density functional computations using the ORCA package20 using the DFT-optimised coordinates for 1. The computations were performed with the B3LYP functional. Basis sets with significant flexibility in the core region were used (ORCA basis sets ‘CoreProp’ (CP(III))27 for copper, and a Barone basis set ‘EPRII’28 for the remaining atoms).
Crystallography
Single crystals of [Cu(en)2](OTf)2 suitable for X-ray analysis were grown from a saturated solution in acetonitrile.‡ X-ray data were collected by the EPSRC National Crystallographic Service29 on a Rigaku Saturn 724+ CCD diffractometer. Intensity data were measured at 100 K using Mo-Kα radiation (λ = 0.71073 Å). The structure was solved using direct methods with absorption corrections being applied as part of the data reduction scaling procedure. After refinement of the heavy atoms, difference Fourier maps revealed the maxima of residual electron density close to the positions expected for the hydrogen atoms; they were introduced as fixed contributors in the structure factor calculations and treated with a riding model, with isotropic temperature factors (Uiso(H) = 1.3Ueq(C)) but not refined, except for the hydrogens attached to the nitrogen atoms, which were taken from the Fourier difference synthesis and refined. The N–H bond distances were restrained to 0.91(2) Å. Full least-square refinement was carried out on F2. A final difference map revealed no significant maxima of residual electron density. Structure solution and refinement were performed using the SHELX software suite.30 CCDC reference number: 946821.Conclusions
The crystal structure of the anhydrous [Cu(en)2](OTf)2 1 complex, bearing coordinated triflate counter-ions, has been reported for the first time. A series of intra- and inter-molecular hydrogen-bonded networks involving the ligand N–H groups and the weakly coordinated triflate counter-ion were identified in the solid state structure. The structure and electronic properties of the frozen solution complex 1 were further characterised by advanced EPR/ENDOR techniques. No previous ENDOR studies have ever been reported for any [Cu(en)2] complex. The 14N hyperfine and quadrupole parameters were found to be relatively small and shown to be consistent with the sp3 hybridised nature of the coordinated ligand nitrogens. The principal hyperfine values for the ligand 1H's were also determined, including the axial and equatorial C–H protons and the N–H protons. The magnitude of the anisotropic and isotropic components to the hyperfine (Adip and aiso) were in good agreement with those expected from the crystal structure and confirmed via DFT calculations. Evidence for the retention of the triflate coordination in frozen solution was also confirmed via the 19F ENDOR data. The orientation and magnitude of the 19F tensor was entirely consistent with the crystallographic data and the DFT calculations.This work confirms the correlation between the hyperfine coupling and hybridisation of donor nitrogens8,9g and demonstrates that the 14N hyperfine constants may be useful for estimation of copper binding sites. This relationship may be of particular significance in determining the identity of unknown Cu(II) sites, particularly in samples of biological interest in which nitrogen-containing heterocycles are abundant and whose crystal structures are not readily available. There are several examples in the literature of how the full range of paramagnetic techniques have been employed to study nitrogen interactions in metalloproteins and enzymes.31 Indeed, this result may prove useful to an investigation currently underway in our group focussed towards identifying the coordination modes of a series of N-heterocycles to Cu(II) for which multiple binding conformations are possible.32
Notes and references
- For selected examples see: (a) M.-T. Li, C.-G. Wang, Y. Wu and X.-C. Fu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1660 CAS; (b) J.-C. Lee, H. Takahashi and Y. Matsui, Z. Kristallogr. - New Cryst. Struct., 2005, 220, 491 CAS; (c) R. P. Sharma, A. Singh, A. Saini, P. Venugopalan, A. Molinari and V. Ferretti, J. Mol. Struct., 2009, 923, 78 CrossRef CAS; (d) Y. Komiyama and E. C. Lingafelter, Acta Crystallogr., 1964, 17, 1145 CrossRef CAS.
- For selected reviews concerning the catalytic applications of copper see: (a) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS PubMed; (b) T. Liu and H. Fu, Synthesis, 2012, 2805 CAS; (c) C. Fehr, Synlett, 2012, 990 CrossRef CAS; (d) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (e) M. Zhang, Appl. Organomet. Chem., 2010, 24, 269 CAS.
- (a) A. Schweiger and G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance, Oxford University Press, New York, 2001 Search PubMed; (b) J. R. Pilbrow, Transition Ion Electron Paramagnetic Resonance, Oxford Science Publications, Oxford University Press, 1st edn, 1990 Search PubMed; (c) B. R. McGarvey, Electron Paramagnetic Resonance of Transition-Metal Complexes, Transition Metal Chemistry, New York, 1966, vol. 3, p. 89 Search PubMed.
- A. Schweiger, Struct. Bonding, 1982, 51, 1 CrossRef.
- (a) G. H. Rist and J. S. Hyde, J. Chem. Phys., 1970, 52, 4633 CrossRef CAS; (b) B. M. Hoffman, J. Martinsen and R. A. Venters, J. Magn. Reson., 1984, 59, 110 CAS; (c) B. M. Hoffman, R. A. Venters and J. Martinsen, J. Magn. Reson., 1985, 62, 537 CAS; (d) G. C. Hurst, T. A. Henderson and R. W. Kreilick, J. Am. Chem. Soc., 1985, 107, 7294 CrossRef CAS; (e) S. P. Greiner, R. W. Kreilick and K. A. Kraft, J. Am. Chem. Soc., 1992, 114, 391 CrossRef CAS.
- (a) B. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3575 CrossRef CAS PubMed; (b) B. M. Hoffman, Acc. Chem. Res., 2003, 36, 522 CrossRef CAS PubMed; (c) S. van Doorslaer and E. Vinck, Phys. Chem. Chem. Phys., 2007, 9, 4620 RSC; (d) C. Calle, A. Sreekanth, M. V. Fedin, J. Forrer, I. Garcia-Rubio, I. A. Gromov, D. Hinderberger, B. Kasumaj, P. Leger, B. Mancosu, G. Mitrikas, M. G. Santangelo, S. Stoll, A. Schweiger, R. Tschaggelar and J. Harmer, Helv. Chim. Acta, 2006, 89, 2495 CrossRef CAS; (e) D. Goldfarb and D. Arieli, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 441 CrossRef CAS PubMed; (f) J. Huttermann and R. Kappl, Met. Ions Biol. Syst., 1987, 22, 1 Search PubMed; (g) P. Doan, Paramagnetic Resonance of Metallobiomolecules, ACS Symp. Ser., 2003, 858, 55–81 CrossRef CAS.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691 CrossRef CAS PubMed.
- M. Iwaizumi, T. Kudo and S. Kita, Inorg. Chem., 1986, 25, 1546 CrossRef CAS.
- (a) T. G. Brown and B. M. Hoffman, Mol. Phys., 1980, 39, 1073 CrossRef CAS; (b) C. Finazzo, C. Calle, S. Stoll, S. van Doorslaer and A. Schweiger, Phys. Chem. Chem. Phys., 2006, 8, 1942 RSC; (c) C. Calle, A. Schweiger and G. Mitrikas, Inorg. Chem., 2007, 46, 1847 CrossRef CAS PubMed; (d) A. Schweiger and H. Gunthard, Chem. Phys., 1978, 32, 35 CrossRef CAS; (e) S. Kita, M. Hashimoto and M. Iwaizumi, Inorg. Chem., 1979, 18, 3432 CrossRef CAS; (f) R. Jirmse, J. Stach and R. Bottcher, Chem. Phys. Lett., 1980, 75, 565 CrossRef; (g) R. Miyamoto, Y. Ohba and M. Iwaizumi, Inorg. Chem., 1992, 31, 3138 CrossRef CAS; (h) M. Fujimoto, C. A. McDowell and T. Takui, J. Chem. Phys., 1979, 70, 3694 CrossRef CAS; (i) A. Schweiger, M. Rudin and J. Forrer, Chem. Phys. Lett., 1981, 80, 376 CrossRef CAS; (j) D. M. Murphy, I. Caretti, E. Carter, I. A. Fallis, M. C. Göbel, J. Landon, S. van Doorslaer and D. J. Willock, Inorg. Chem., 2011, 50, 6944 CrossRef CAS PubMed.
- R. Rajan, Physica, 1963, 29, 1191 CrossRef CAS.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143 CrossRef CAS.
- R. Rajan, J. Chem. Phys., 1962, 37, 1901 CrossRef CAS.
- R. Rajan and J. R. Reddy, J. Chem. Phys., 1963, 39, 1140 CrossRef CAS.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746 CrossRef CAS (The UK Chemical Database Service: CSD version 5.34 updated November 2012).
- For selected examples see: (a) V. Vrabel and J. Garaj, Collect. Czech. Chem. Commun., 1982, 47, 409 CrossRef CAS; (b) V. Manriquez, M. Campos-Vallette, N. Lara, N. Gonzalez-Tejeda, O. Wittke, G. Diaz, R. Munoz and L. Kriskovic, J. Chem. Crystallogr., 1996, 26, 15 CrossRef CAS.
- M. Hernandez-Molina, P. A. Lorenzo-Luis and C. Ruiz-Perez, CrystEngComm, 2001, 3, 60 RSC.
- P. J. M. W. Birker, P. T. Crisp and C. J. Moore, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 3194 CrossRef.
- E. V. Karpova, M. A. Zakharov, S. I. Gutnikov and A. I. Boltalin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m658 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- (a) F. Neese, J. Chem. Phys., 2001, 115, 11080 CrossRef CAS; (b) F. Neese, J. Phys. Chem. A, 2001, 105, 4290 CrossRef CAS; (c) F. Neese, J. Chem. Phys., 2003, 118, 3939 CrossRef CAS; (d) F. Neese, J. Chem. Phys., 2005, 122, 34107 CrossRef PubMed.
- (a) F. E. Mabbs and D. Collison, Electron Paramagnetic Resonance of d Transition Metal Compounds, Elsevier, Netherlands, 1992 Search PubMed; (b) P. H. Rieger, Electron Spin Resonance, Analysis and Interpretation, RSC Publishing, Cambridge, 2007 Search PubMed.
- (a) M. P. Waller, A. Robertazzi, J. A. Platts, D. E. Hibbs and P. A. Williams, J. Comput. Chem., 2006, 27, 491–504 CrossRef CAS PubMed; (b) F. Neese, J. Biol. Inorg. Chem., 2006, 11, 702–711 CrossRef CAS PubMed; (c) F. Neese and M. L. Munzarova, in Calculation of NMR and EPR parameters, ed. M. Kaupp, M. Buhl and V. G. Malkin, Wiley-VCH, Weinheim, 2004, p. 21 Search PubMed; (d) M. Stein, E. van Lenthe, E. J. Baerends and W. Lubitz, J. Am. Chem. Soc., 2001, 123, 5839 CrossRef CAS PubMed; (e) F. Neese, Coord. Chem. Rev., 2009, 256, 526 CrossRef.
- (a) D. M. Murphy and R. D. Farley, Chem. Soc. Rev., 2006, 35, 249 RSC; (b) D. Attanasio, J. Phys. Chem., 1986, 90, 4952 CrossRef CAS (and refs therein).
- D. M. Murphy, I. A. Fallis, D. J. Willock, J. Landon, E. Carter and E. Vincks, Angew. Chem., Int. Ed., 2008, 47, 1414 CrossRef CAS PubMed.
- M. E. Owen, E. Carter, G. J. Hutchings, B. D. Ward and D. M. Murphy, Dalton Trans., 2012, 41, 11085 RSC.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 45 CrossRef PubMed.
- This basis is based on the TurboMole DZ basis developed by Ahlrichs and co-workers and obtained from the basis set library under http://ftp.chemie.uni-karlsruhe.de/pub/basen; R. Ahlrichs and co-workers (unpublished).
- V. Barone, Recent Advances in Density Functional Methods, World Scientific Publ. Co., Singapore, 1996 Search PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) H. L. van Camp, R. H. Sands and J. A. Fee, Biochim. Biophys. Acta, 1982, 704, 75 CrossRef CAS PubMed; (b) C. P. Scholes, A. Lapidot, R. Mascarenhas, T. Inubushi, R. A. Isaacson and G. Feher, J. Am. Chem. Soc., 1982, 104, 2724 CrossRef CAS; (c) H.-J. Scholl and J. Hüttermann, J. Phys. Chem., 1992, 96, 9684 CrossRef CAS; (d) H. Reinhard, R. Kappl, J. Hüttermann and M.-S. Viezzoli, J. Phys. Chem., 1994, 98, 8806 CrossRef CAS; (e) T. Prisner, M. Rohrer and F. MacMillan, Annu. Rev. Phys. Chem., 2001, 52, 279 CrossRef CAS PubMed; (f) M. Ubbink, J. A. R. Worrall, G. W. Canters, E. J. J. Groenen and M. Huber, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 393 CrossRef CAS PubMed; (g) M. Vogt, S. Lahiri, C. G. Hoogstraten, R. D. Britt and V. J. DeRose, J. Am. Chem. Soc., 2006, 128, 16764 CrossRef CAS PubMed; (h) M. J. Colaneri, J. Vitali and J. Peisach, J. Phys. Chem. A, 2009, 113, 5700 CrossRef CAS PubMed.
- K. M. Sharples, E. Carter, D. M. Murphy and J. A. Platts, 2013, in prep.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray data in CIF format, coordinates of the calculated structure of 1, computational data, and additional EPR and ENDOR data. CCDC 946821. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51694f |
| ‡ X-ray data for 1: C6H16CuF6N4O6S2, M = 481.89; crystal system monoclinic, space group P21/n, a = 16.7332(6) Å, b = 10.3150(4) Å, c = 20.3164(14) Å, α = 90°, β = 101.664(7)°, γ = 90°, V = 3434.3(3) Å3, T = 100 K, Z = 8, 7846 unique reflections, R(int) = 0.0242, R1 [I > 2σ(I)] = 0.0251, wR2 (all data) = 0.0703. |
| This journal is © The Royal Society of Chemistry 2013 |