 Open Access Article
Open Access ArticleDecomposition kinetics of peroxynitrite: influence of pH and buffer
Christian
Molina
,
Reinhard
Kissner
and
Willem H.
Koppenol
*
Institute of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland. E-mail: koppenol@inorg.chem.ethz.ch
First published on 23rd May 2013
Abstract
The decay of ONOOH near neutral pH has been examined as a function of isomerization to H+ and NO3−, and decomposition to NO2− and O2via O2NOO−. We find that in phosphate buffer kisomerization = 1.11 ± 0.01 s−1 and kdisproportionation = (1.3 ± 0.1) × 103 M−1 s−1 at 25 °C and I = 0.2 M. In the presence of 0.1 M tris(hydroxymethyl)aminomethane (Tris), the decay proceeds more rapidly: kdisproportionation = 9 × 103 M−1 s−1. The measured first half-life of the absorbance of peroxynitrite correlates with [Tris]0·([ONOO−]0 + [ONOOH]0)2, where the subscript 0 indicates initial concentrations; if this function exceeds 6.3 × 10−12 M3, then Tris significantly accelerates the decomposition of peroxynitrite.
Introduction
Peroxynitrite† is formed from the reaction of NO˙ with O˙−2,3 which proceeds at a diffusion-controlled rate, k = 1.6 × 1010 M−1 s−1.4,5 Because both NO˙ and O˙−2 are released by activated macrophages and neutrophils,6–11 peroxynitrite can be formed in vivo.12 Due to its oxidizing power and nitrating activity, peroxynitrite is cytotoxic and is associated with inflammatory diseases.13ONOO− is stable in a cold alkaline solution in the absence of carbonyl compounds and metals. The pKa of ONOOH increases from 6.5 to 7.3 with increasing phosphate concentration.14 Isomerization of ONOOH to HNO3,15,16 reaction 1,‡ is first-order in ONOOH. Rate constants for the isomerization, obtained over a period of ca. 40 years, have slightly decreased, from 1.5 to 1.1 s−1, most likely because of purer preparations of peroxynitrite (Table 1). ONOOH is a powerful one-electron and two-electron oxidant.17,18 A review of the literature shows that evidence for the much-cited homolysis of ONOOH to HO˙ and NO˙2 is weak: at most 5% of ONOOH may undergo this reaction.19 In any case, the reaction of otherwise stable ONOO− with CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 is far more relevant to biology (k = (3–5.8) × 104 M−1 s−1)21,22 as it yields, in part, very oxidising species, NO˙2 and CO˙−3 with E°(NO˙2/NO2−) = 1.04 V23 and E°(CO˙−3/CO32−) = 1.59 V.24 These species are responsible for the higher yield of nitration of phenols compared to nitration by peroxynitrite alone (4–9% without and 14–19% with 20 mM HCO3−).22,25–27
20 is far more relevant to biology (k = (3–5.8) × 104 M−1 s−1)21,22 as it yields, in part, very oxidising species, NO˙2 and CO˙−3 with E°(NO˙2/NO2−) = 1.04 V23 and E°(CO˙−3/CO32−) = 1.59 V.24 These species are responsible for the higher yield of nitration of phenols compared to nitration by peroxynitrite alone (4–9% without and 14–19% with 20 mM HCO3−).22,25–27
Deviations from first-order behaviour33 and formation of O234 and NO2− in a 1:2 stoichiometric ratio35 have been observed. Both phenomena can be explained with an additional reaction, namely between ONOOH and ONOO−, a disproportionation that, given a pKa of ONOOH of nearly 7, is only relevant near and above neutral pH (Scheme 1). This disproportionation thus fits the observation that NO2− yields increase with pH and with peroxynitrite concentration.36,37 Gupta et al.38 showed that the disproportionation reaction yields initially O2NOO− and NO2−. O2NOOH has a pKa of 5.939,40 and is relatively stable. When one compares the wavelengths of maximal absorptivity and the extinction coefficients of ONOO− and O2NOO−, one sees that these parameters are quite similar: O2NOO− absorbs with εmax = 1650 M−1 cm−1 at 285 nm,39 and ONOO− with εmax = 1705 M−1 cm−1 at 302 nm.41 Furthermore, the rates of decay of both ONOOH and O2NOO− are close to 1 s−1 (see below). For these reasons O2NOO− went unnoticed as an intermediate during the decay of peroxynitrite. The decomposition of O2NOO− yields NO2− and O2,39 of which ca. 1–2% is in an excited (1ΔgO2) state.42 Estimates for decomposition reaction 3 at 25 °C are k3 = 1.0 ± 0.2 s−1,39 1.35 ± 0.03 s−1,43 and 0.58 s−1.38k4 and k−4 are estimated to be 1.05 ± 0.23 s−143 and (4.5 ± 1.0) × 109 M−1 s−1,39 respectively. Redox reaction 5 is thermodynamically favoured, Δrxn5G°′ = −118 kJ mol−1. ΔfG0 (kJ mol−1) values in aqueous solution are 62.5 for NO˙2,44 33.8 for O˙−2,45 −38 for NO2−23,44 and 16.4 for O2;46 the reaction proceeds with k5 = 1 × 108 M−1 s−1.47 We emphasise that the formation of O2NOO−, a reaction that is second-order in peroxynitrite, is biologically insignificant, because the concentration of peroxynitrite that may be generated by activated macrophages is very low. However, upon bolus additions of peroxynitrite during in vitro experiments that result in initial concentrations exceeding 0.1 mM, the formation of peroxynitrate should be taken into account.
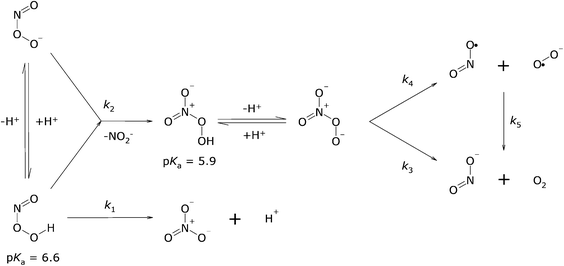 | ||
| Scheme 1 | ||
In neutral 0.1 M tris(hydroxymethyl)aminomethane (Tris) buffer, decomposition of peroxynitrite yields up to 40% peroxynitrate with k2 = 3 × 104 M−1 s−1.38 Decomposition of peroxynitrite in various neutral amine buffers, e.g. 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) or 1,4-piperazinediethanesulfonic acid (PIPES), yields an oxidant that was suggested to be H2O2.48 However, it could well have been O2NOOH. The pH of peroxynitrite containing samples is usually controlled by phosphate buffer, and rarely by Tris or HEPES buffer (e.g.ref. 49 and 50). At equal pH, similar initial peroxynitrite concentrations yield more NO2− in Tris buffer50 than in phosphate buffer.36 This indicates that a Tris-dependent conversion of peroxynitrite to NO2− takes place. The possibility that the buffer influences the course or result of a reaction has rarely been considered.48,51 We therefore also compared decomposition kinetics of peroxynitrite at pH = 6.8 in buffers that contain variable concentrations of phosphate and Tris and refined the decomposition model by taking into account reactions (1)–(3).
Materials and methods
Materials
(Me4N)OONO was prepared according to the work by Bohle et al.,52 dissolved in 0.01 M NaOH and frozen as 25 mM solutions. NaH2PO4·H2O (s), Na2HPO4·12H2O (s), H2NC(CH2OH)3 (s), NaOH (s), 85% H3PO4 (aq), and 70% HClO4 (aq), all of analytical grade, were purchased from Sigma-Aldrich/Fluka (Buchs, CH), Brenntag Schweizerhall AG (Basel, CH), Merck (Darmstadt, DE), Riedel-de Haën (Seelze, DE) and used as received. Ar and N2, both of 99.995% purity, were taken from the in-house gas supply. All solutions were prepared in deionized water that was further purified by using a Milli-Q unit from Millipore AG (Zug, CH).Preparation of solutions
All solutions were prepared, stored, transferred and mixed at room temperature in glass vessels under Ar or N2 and used within one day. Because ONOO− is sensitive to acid, heat and light, samples (1.4 g) of deeply frozen 25 mM (Me4N)OONO (aq) were dissolved in ice-cold 20 mM aqueous NaOH to produce alkaline ONOO− solutions that were stored and transferred at 0 °C and kept in the dark. The fresh alkaline ONOO− solutions contained ONOO−, NO2− and NO3− in a molar ratio of 10:1:1 (see the section ‘Determination of peroxynitrite, nitrite and nitrate’). The impurity by NO2− and NO3− in the alkaline ONOO− solutions is most probably formed during the thawing of frozen (Me4N)OONO (aq).53 Because the pKa of ONOOH depends on the ionic strength of the medium,14 the ionic strength of the Tris buffers and the pH-varied 0.07 M phosphate buffers was adjusted to 0.2 M by the addition of NaClO4, which we assume to be inert. Considering the ion-pairing of Na+ with HPO42−,54 we calculated the ionic strength of our samples at pH 6–8 to be between 0.197 M and 0.206 M. The pKa of ONOOH is 6.7 under these conditions.
Determination of peroxynitrite, nitrite and nitrate
The content of peroxynitrite and of its typical ionic decomposition products – NO2− and NO3− – were quantified for the lot of deeply frozen (Me4N)OONO (aq) used for kinetics experiments as described before.36 Briefly, a sample (1.4 g) of deeply frozen (Me4N)OONO (aq) was subsequently weighted, dissolved in ice-cold 2 mM NaOH and analysed by spectrophotometry at 302 nm (Specord 250 from Analytik Jena AG, Jena, DE). To determine the content of NO2− and NO3−, a sample (1.4 g) of deeply frozen (Me4N)OONO (aq) was dissolved in 30 ml ice-cold 2 mM NaOH. 1 ml of this alkaline solution was added to 5 ml ice-cold 2 mM phosphoric acid to yield a mixture with pH = 3. In an acidic solution, peroxynitrite is mainly present as ONOOH, which isomerises to NO3−. This acidic solution was neutralised after 5 min by the addition of 4 ml 2 mM NaOH and then analysed by ion chromatography (Super Sep IC Anion Column from Metrohm AG, Herisau, CH) with a phthalate eluent of pH = 4.7 and by conductometric detection (732 IC Detektor from Metrohm AG, Herisau, CH). To determine the content of NO3− in the (Me4N)OONO (aq) sample, the peroxynitrite content was subtracted from the content of NO3−, which resulted after isomerization of ONOOH. The amount of peroxynitrate was not determined because it decays very fast in an alkaline medium.Stopped-flow experiments
The decay of peroxynitrite was initiated by a rapid 1:1 mixing of an alkaline ONOO− solution with an acidic buffer solution in a thermostated (25 °C) stopped-flow unit (SX17MN) from Applied Photophysics (Leatherhead, UK) and observed for 10 s by spectrophotometry. Monochromatic light at the absorption maximum of ONOO−, 302 nm, was used.
For each sample the decay of the absorption was measured at least four times, and the decay traces were averaged before analysis. The initial absorption of each mixture was used to calculate [ONOO−]0: the optical path length is 1 cm, and ε302 nm(ONOO−) = 1705 M−1 cm−1.41
Prior to each decay experiment, the total initial peroxynitrite concentration in the sample, [ONOO−]0, total, was derived from the absorbance of a 1:1 mixture of an alkaline ONOO− solution with 20 mM NaOH. Then the experiments were carried out at the desired pH in a specific buffer. Prior to switching to another pH and buffer, the standardization with NaOH was repeated. For each mixture, the final pH value was measured by an Orion Microprocessor Ionalyzer 901 from Thermo Scientific after mixing, outside the stopped-flow apparatus. For the decay simulations, the pKa of ONOOH was calculated from the measured pH, [ONOO−]0 and [ONOO−]0, total at pH = 6.8.
Computations
We assume that at pH < 6 ONOOH isomerises to NO3− and H+ exclusively; the decay of ONOOH was fitted to a single exponential function that resulted in kobs. The rate constant k1 was derived from kobs, which is a function of pH, pKa(ONOOH) and k1: | (1) |
Reactions (1)–(3)§ lead to rate laws 2 and 3 for peroxynitrite and peroxynitrate.
 | (2) |
 | (3) |
Given the pKa values of ONOOH and O2NOOOH, we calculated the time-dependent concentrations of the absorbing species ONOO− and O2NOO−.
 | (4) |
 | (5) |
With the extinction coefficients [ε302 nm(ONOO−) = 1705 M−1 cm−1; ε302 nm(O2NOO−) = 1370 M−1 cm−1 from the spectrum in ref. 38], we reproduced the absorption trace of peroxynitrite solutions at 302 nm. The calculated absorbance trace was fitted to the measured absorbance trace by the variation of the rate constants according to the method of the least squares. All data analysis and model calculations were carried out in Microsoft Excel by macro programmes (which are available from the authors upon request). The kinetics of the mixed-order decomposition of peroxynitrite was quantitatively compared by the use of the first half-life of the absorbance.
Results
Computation of the decomposition rate constants of peroxynitrite in phosphate buffer
From the measurement of the initial absorbance at various pH values, we derive a pKa(ONOOH) of 6.7 at I = 0.2 M which fits with the dependence of the pKa on ionic strength.A rate constant k1 = 1.11 ± 0.01 s−1 (n = 7, 95% confidence level) was determined by fitting absorbance traces at pH = 5.3 and I = 0.2 M in 70 mM phosphate buffer (see Fig. 1). At this pH, [ONOO−] is too low to contribute to significant disproportionation, and [HNO2], from the nitrite present in the (Me4N)OONO preparation, is too low to cause undesired reactions.
 | ||
| Fig. 1 Decomposition of 0.25 mM peroxynitrite at 25 °C and pH = 5.3 in 0.07 M phosphate buffer (I = 0.2 M) and simulation according to isomerization only with k1 = 1.11 s−1. (a) Actual decay curve measured (black) and simulated (yellow); (b) residuals (green) of the model. | ||
It became clear that the results obtained at higher pH (Fig. 2) could not be fitted with k1 = 1.11 s−1 (Fig. 3). We therefore take into account an additional process, disproportionation of peroxynitrite to O2NOO− and NO2−.38 For the decay of O2NOO− to NO2− and O2, we take a value of 1.35 s−1.43 The combination of these three processes allows us to fit the obtained results with significantly smaller errors (Fig. 4).
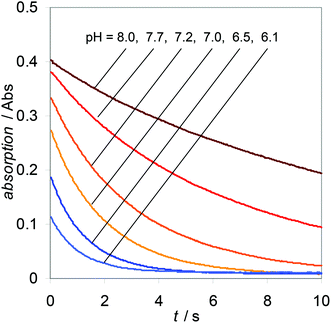 | ||
| Fig. 2 Four to six decay traces per pH value (0.25 mM peroxynitrite in 70 mM phosphate buffer at 25 °C and I = 0.2 M, see Table 2) were measured at 302 nm and averaged. | ||
 | ||
| Fig. 3 Four to six decay traces per pH value (0.25 mM peroxynitrite in 70 mM phosphate buffer at 25 °C and I = 0.2 M, see Table 2) were averaged and fitted with k1 = 1.11 s−1. The residuals are much larger than in Fig. 4, the range of which is indicated by the two dashed lines. | ||
 | ||
| Fig. 4 The same as Fig. 3, but fitted with k1 = 1.11 s−1 and k3 = 1.35 s−1.43k2 was varied to minimise the residuals shown here. | ||
Fig. 5 shows an actual decay curve of 0.25 mM peroxynitrite at pH = 6.5 which was fitted according to rate laws 2 and 3 with k1 = 1.11 s−1 and k3 = 1.35 s−1.43 In addition, the fit required that k2 be 1163 M−1 s−1: the residuals are very small and do not show any systematic error. This procedure was repeated at various other pH values, and the results for k2 are presented in Table 2.
![Decomposition of 0.25 mM peroxynitrite at 25 °C and pH = 6.5 in 0.07 M phosphate buffer (I = 0.2 M) and simulation according to rate laws 2 and 3 with k1 = 1.11 s−1 and k3 = 1.35 s−1,43 resulting in k2 = 1163 M−1 s−1. (a) Actual decay curve measured (black) and simulated (yellow); (b) residuals (green) of the model; (c) concentrations calculated for ONOO− (blue) and O2NOO− (red), [ONOOH] is not shown.](/image/article/2013/DT/c3dt50945a/c3dt50945a-f5.gif) | ||
| Fig. 5 Decomposition of 0.25 mM peroxynitrite at 25 °C and pH = 6.5 in 0.07 M phosphate buffer (I = 0.2 M) and simulation according to rate laws 2 and 3 with k1 = 1.11 s−1 and k3 = 1.35 s−1,43 resulting in k2 = 1163 M−1 s−1. (a) Actual decay curve measured (black) and simulated (yellow); (b) residuals (green) of the model; (c) concentrations calculated for ONOO− (blue) and O2NOO− (red), [ONOOH] is not shown. | ||
| pH | First t1/2/s | k 2/(103 M−1 s−1) | n |
|---|---|---|---|
| 5.3 | 0.69 ± 0.01 | — | 7 |
| 6.1 | 0.76 ± 0.04 | 0.9 ± 1.0 | 4 |
| 6.5 | 0.92 ± 0.03 | 1.3 ± 0.3 | 4 |
| 7.0 | 1.41 ± 0.02 | 1.7 ± 0.1 | 4 |
| 7.2 | 2.21 ± 0.06 | 1.1 ± 0.1 | 6 |
| 7.7 | 4.6 ± 0.1 | 1.4 ± 0.1 | 5 |
| 8.0 | 9.4 ± 0.1 | 1.7 ± 0.1 | 4 |
| Over all pH values | 1.3 ± 0.1 | 27 |
The first half-life of the absorbance increases with pH (Fig. 6) because ONOO− is stable and the isomerization of ONOOH is the predominant decomposition pathway of peroxynitrite. The first half-life of the absorbance at pH 6–8 can be calculated according to rate laws 2 and 3 with k1 = 1.11 s−1, k2 = 1.3 × 103 M−1 s−1 and k3 = 1.35 s−1 much more accurately than by a model based on a first-order decay only.
 | ||
| Fig. 6 Decomposition of 0.25 mM peroxynitrite at pH = 5.3, 6.1, 6.5, 7.0, 7.2, 7.7 and 8.05 in 70 mM phosphate buffer (I = 0.2 M). The first half-life of the absorbance measured (+) and calculated (blue lines) based on the best fit of two decomposition models: (1) (dashed line) according to isomerization only with k1 = 1.11 s−1; (2) (solid line) according to rate laws 2 and 3 with k1 = 1.11 s−1, k2 = 1.3 × 103 M−1 s−1 and k3 = 1.35 s−1. | ||
Decomposition of peroxynitrite in phosphate buffer and Tris buffer
The decomposition of peroxynitrite is decelerated by phosphate and accelerated by Tris: t1/2 = 0.75–0.88 s in 0.1 M phosphate and 0.1 M Tris, t1/2 = 1.33–1.42 s in 0.1 M phosphate only and t1/2 = 0.63 s in 0.1 M Tris only (Tables 3 and 4, and Fig. 7).![Decomposition of 0.047 mM (blue), 0.25 mM (green) and 0.34 mM (black) peroxynitrite at pH = 6.8 in 0.1 M phosphate buffers with varied Tris concentrations. The solid horizontal lines indicate the first half-life of the absorbance of peroxynitrite in the absence of Tris. When [Tris]0·[ONOO−]0, total2 exceeds 6.3 × 10−12 M3, Tris significantly catalyses the disproportionation.](/image/article/2013/DT/c3dt50945a/c3dt50945a-f7.gif) | ||
| Fig. 7 Decomposition of 0.047 mM (blue), 0.25 mM (green) and 0.34 mM (black) peroxynitrite at pH = 6.8 in 0.1 M phosphate buffers with varied Tris concentrations. The solid horizontal lines indicate the first half-life of the absorbance of peroxynitrite in the absence of Tris. When [Tris]0·[ONOO−]0, total2 exceeds 6.3 × 10−12 M3, Tris significantly catalyses the disproportionation. | ||
| [Phosphate]/M | First t1/2/s | k 2/(103 M−1 s−1) | n |
|---|---|---|---|
| 0 | 0.65 ± 0.00 | 14 ± 2 | 2 |
| 0.001 | 0.68 ± 0.02 | 13 ± 1 | 5 |
| 0.005 | 0.69 ± 0.09 | 13 ± 4 | 3 |
| 0.025 | 0.73 ± 0.04 | 12 ± 3 | 3 |
| 0.1 | 0.91 ± 0.02 | 9.3 ± 0.4 | 6 |
Interestingly, up to a certain concentration, Tris shows no significant influence on the decay of peroxynitrite. However, this concentration decreases with increasing initial peroxynitrite concentration. We find that the measured first half-life of the absorbance of peroxynitrite correlates with [Tris]0·[ONOO−]0, total2: if [Tris]0·[ONOO−]0, total2 exceeds 6.3 × 10−12 M3 (1 × 10−11.2 M3), then Tris significantly accelerates the decomposition of peroxynitrite (Fig. 7).
Discussion
According to rate laws 2 and 3, absorption traces are fitted (e.g.Fig. 5) and values of the first half-life of the absorbance are simulated (Fig. 6) without a systematic error for the pH range 6–8. A model restricted to homolysis37 does not simulate the decay of peroxynitrite in this pH range very well.The first-order rate constant of the isomerization agrees with earlier results.
The disproportionation can be regarded as a two-step reaction: a reversible association and a subsequent irreversible conversion to the products:

 | (8) |
Earlier estimates of the parameters of this sequence are listed in Table 5. The first two are in good agreement with the disproportionation rate constant determined here; however, the third estimate is higher.
According to rate laws 2 and 3, the concentration of peroxynitrate reaches its concentration maximum ca. 1 s after the decay of peroxynitrite is initiated (Fig. 5c). After that peak concentration, the second-order disproportionation becomes negligible in comparison with first-order decay processes 1 and 3. Therefore, decay traces of peroxynitrite deviate from first-order kinetics in the first 1–2 s.33
The decomposition model according to reactions (1)–(3)§ and (9) fails to reproduce the Tris-dependent first half-life of the absorbance. It fits the absorption curves with a residual that indicates a systematic error that increases with the Tris concentration (not shown). Moreover, it results in k2 values that increase non-linearly with the Tris concentration if the latter is higher than 10−4 M (Table 4). Saturation may cause a non-linear dependence. The accelerating effect of Tris will be addressed in a future publication.
We find that k2 is one order of magnitude lower in phosphate buffer (this work: 1.3 × 103 M−1 s−1) than in Tris buffer (this work: 9 × 103 M−1 s−1, Gupta et al.:38 3 × 104 M−1 s−1, both in 0.1 M Tris) and that 0.25 mM peroxynitrite decomposes in 0.1 M Tris buffer twice as fast as it does in 0.1 M phosphate buffer. Therefore, Tris accelerates the disproportionation of peroxynitrite and thereby increases k2. Indeed, the peroxynitrate yield is, being dependent on the square of the initial peroxynitrite concentration, 40 times higher in Tris buffer than it is in phosphate buffer.38 The observed rate of decay of peroxynitrite accelerates with increasing Tris concentration (Fig. 7). Furthermore, the first half-life of the absorbance correlates with the concentration product [Tris]0·[ONOO−]0, total2. The concentration product [Tris]·[ONOO−]total2 is likely part of the rate law of the additional Tris-dependent decomposition pathway. If the Tris concentration is high enough, this additional decomposition pathway contributes significantly to the overall decomposition. If Tris participates in the disproportionation according to reaction (9) and if k2, in Tris = k2 + k9·[Tris], then we obtain an estimate of k9 = (8–29) × 103 M−2 s−1:
 | (9) |
It is likely that a more accurate decomposition model can be achieved by considering a chain of association equilibria that involve peroxynitrite and Tris.
Peroxyacids, in general, decompose in reactions that are first-order in both the acid and its conjugate base: typically with rate constants of 1 × 10−3–1 × 10−1 M−1 s−1.56–59 These rate constants are much smaller than those found for peroxynitrite (1.3 × 103–3 × 104 M−1 s−1). However, in contrast to other peroxyacids, the atom attached to the OOH group of peroxynitrite has a lone pair that allows additional reactions – electrophilic attack and oxidative addition – that may allow faster decomposition (Scheme 2).
 | ||
| Scheme 2 | ||
At peroxynitrite concentrations >10−4 M, but at lower concentration in the presence of Tris, disproportionation of peroxynitrite in samples competes with reactions of peroxynitrite with target molecules and produces peroxynitrate that is an artificial reactive intermediate.
Acknowledgements
We thank the SNF and the ETH Zurich for financial support.References
- N. G. Connelly, T. Damhus, R. M. Hartshorn and A. T. Hutton, Nomenclature of Inorganic Chemistry. IUPAC Recommendations 2005, 2005 Search PubMed.
- W. H. Koppenol, Pure Appl. Chem., 2000, 72, 437–446 CrossRef CAS.
- N. V. Blough and O. C. Zafiriou, Inorg. Chem., 1985, 24, 3502–3504 CrossRef CAS.
- T. Nauser and W. H. Koppenol, J. Phys. Chem. A, 2002, 106, 4084–4086 CrossRef CAS.
- H. Botti, M. Möller, D. Steinmann, T. Nauser, W. H. Koppenol, A. Denicola and R. Radi, J. Phys. Chem. B, 2010, 114, 16584–16593 CrossRef CAS.
- S. Moncada, M. W. Radomski and R. M. J. Palmer, Biochem. Pharmacol., 1988, 37, 2495–2501 CrossRef CAS.
- J. B. Hibbs, R. R. Taintor, Z. Vavrin and E. M. Rachlin, Biochem. Biophys. Res. Commun., 1988, 157, 87–94 CrossRef CAS.
- M. R. Green, H. A. O. Hill, M. J. Okolow-Zubkowska and A. W. Segal, FEBS Lett., 1979, 100, 23–26 CrossRef CAS.
- R. J. Gryglewski, R. M. J. Palmer and S. Moncada, Nature, 1986, 320, 454–456 CrossRef CAS.
- T. B. McCall, N. K. Boughton-Smith, R. M. J. Palmer, B. J. R. Whittle and S. Moncada, Biochem. J., 1989, 261, 293–296 CAS.
- Y. Xia, V. L. Dawson, T. M. Dawson, S. H. Snyder and J. L. Zweier, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6770–6774 CrossRef CAS.
- J. S. Beckman, T. W. Beckman, J. Chen, P. A. Marshall and B. A. Freeman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1620–1624 CrossRef CAS.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS.
- R. Kissner, T. Nauser, P. Bugnon, P. G. Lye and W. H. Koppenol, Chem. Res. Toxicol., 1997, 10, 1285–1292 CrossRef CAS.
- M. Anbar and H. Taube, J. Am. Chem. Soc., 1954, 76, 6243–6247 CrossRef CAS.
- J. D. Ray, J. Inorg. Nucl. Chem., 1962, 24, 1159–1162 CrossRef CAS.
- W. H. Koppenol, J. J. Moreno, W. A. Pryor, H. Ischiropoulos and J. S. Beckman, Chem. Res. Toxicol., 1992, 5, 834–842 CrossRef CAS.
- C. Kurz, X. Zeng, S. Hannemann, R. Kissner and W. H. Koppenol, J. Phys. Chem. A, 2005, 109, 965–969 CrossRef CAS.
- W. H. Koppenol, P. L. Bounds, T. Nauser, R. Kissner and H. Ruegger, Dalton Trans., 2012, 41, 13779–13787 RSC.
- W. G. Keith and R. E. Powell, J. Chem. Soc. A, 1969, 90–90 RSC.
- S. V. Lymar and J. K. Hurst, J. Am. Chem. Soc., 1995, 117, 8867–8868 CrossRef CAS.
- A. Denicola, B. A. Freeman, M. Trujillo and R. Radi, Arch. Biochem. Biophys., 1996, 333, 49–58 CrossRef CAS.
- D. M. Stanbury, Adv. Inorg. Chem., 1989, 33, 69–138 CrossRef CAS.
- R. E. Huie, C. L. Clifton and P. Neta, Radiat. Phys. Chem., 1991, 38, 477–481 CrossRef CAS.
- S. V. Lymar, Q. Jiang and J. K. Hurst, Biochemistry, 1996, 35, 7855–7861 CrossRef CAS.
- M. S. Ramezanian, S. Padmaja and W. H. Koppenol, Chem. Res. Toxicol., 1996, 9, 232–240 CrossRef CAS.
- J. N. Lemercier, S. Padmaja, R. Cueto, G. L. Squadrito, R. M. Uppu and W. A. Pryor, Arch. Biochem. Biophys., 1997, 345, 160–170 CrossRef CAS.
- F. Barat, L. Gilles, B. Hickel and J. Sutton, J. Chem. Soc. A, 1970, 1982–1986 RSC.
- T. Løgager and K. Sehested, J. Phys. Chem., 1993, 97, 6664–6669 CrossRef.
- B. Alvarez and R. Radi, First Int. Conf. Chemistry and Biology of Peroxynitrite, Ascona, 1997, 2–2.
- W. H. Koppenol and R. Kissner, Chem. Res. Toxicol., 1998, 11, 87–90 CrossRef CAS.
- P. Maurer, C. F. Thomas, R. Kissner, H. Rüegger, O. Greter, U. Röthlisberger and W. H. Koppenol, J. Phys. Chem. A, 2003, 107, 1763–1769 CrossRef CAS.
- J. S. Beckman, H. Ischiropoulos, L. Zhu, M. van der Woerd, C. D. Smith, J. Chen, J. Harrison, J. C. Martin and M. Tsai, Arch. Biochem. Biophys., 1992, 298, 438–445 CrossRef CAS.
- R. Radi, J. S. Beckman, K. M. Bush and B. A. Freeman, Arch. Biochem. Biophys., 1991, 288, 481–487 CrossRef CAS.
- S. Pfeiffer, A. C. F. Gorren, K. Schmidt, E. R. Werner, B. Hansert, D. S. Bohle and B. Mayer, J. Biol. Chem., 1997, 272, 3465–3470 CrossRef CAS.
- R. Kissner and W. H. Koppenol, J. Am. Chem. Soc., 2002, 124, 234–239 CrossRef CAS.
- M. Kirsch, H.-G. Korth, A. Wensing, R. Sustmann and H. de Groot, Arch. Biochem. Biophys., 2003, 418, 133–150 CrossRef CAS.
- D. Gupta, B. Harish, R. Kissner and W. H. Koppenol, Dalton Trans., 2009, 5730–5736 RSC.
- T. Løgager and K. Sehested, J. Phys. Chem., 1993, 97, 10047–10052 CrossRef.
- S. Goldstein and G. Czapski, Inorg. Chem., 1997, 36, 4156–4162 CrossRef CAS.
- D. S. Bohle, B. Hansert, S. C. Paulson and B. D. Smith, J. Am. Chem. Soc., 1994, 116, 7423–7424 CrossRef CAS.
- S. Miyamoto, G. E. Ronsein, T. C. Corrêa, G. R. Martinez, M. H. G. Medeiros and P. Di Mascio, Dalton Trans., 2009, 5720–5729 RSC.
- S. Goldstein, G. Czapski, J. Lind and G. Merényi, Inorg. Chem., 1998, 37, 3943–3947 CrossRef CAS.
- W. H. Koppenol, Inorg. Chem., 2012, 51, 5637–5641 CrossRef CAS.
- W. H. Koppenol, D. M. Stanbury and P. L. Bounds, Free Radical Biol. Med., 2010, 49, 317–322 CrossRef CAS.
- D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney and R. L. Nuttal, J. Phys. Chem. Ref. Data, 1982, 11(Suppl. 2), 37–38 Search PubMed.
- P. Warneck and C. Wurzinger, J. Phys. Chem., 1988, 92, 6278–6283 CrossRef CAS.
- M. Kirsch, E. E. Lomonosova, H.-G. Korth, R. Sustmann and H. de Groot, J. Biol. Chem., 1998, 273, 12716–12724 CrossRef CAS.
- S. Kapiotis, M. Hermann, I. Held, A. Mühl and B. Gmeiner, FEBS Lett., 1997, 409, 223–226 CrossRef CAS.
- D. Tsikas, K. Denker and J. C. Frölich, J. Chromatogr., A, 2001, 915, 107–116 CrossRef CAS.
- E. Pollet, J. A. Martínez, B. Metha, B. P. Watts Jr. and J. F. Turrens, Arch. Biochem. Biophys., 1998, 349, 74–80 CrossRef CAS.
- D. S. Bohle, P. A. Glassbrenner and B. Hansert, Methods Enzymol., 1996, 269, 302–311 CAS.
- P. Latal, R. Kissner, D. S. Bohle and W. H. Koppenol, Inorg. Chem., 2004, 43, 6519–6521 CrossRef CAS.
- R. M. Smith and A. E. Martell, Critical Stability Constants: Volume 4: Inorganic Complexes, 1976 Search PubMed.
- R. Kissner, T. Nauser, C. Kurz and W. H. Koppenol, IUBMB Life, 2003, 55, 567–572 CrossRef CAS.
- D. F. Evans and M. W. Upton, J. Chem. Soc., Dalton Trans., 1985, 1151–1153 RSC.
- J. F. Goodman, P. Robson and E. R. Wilson, Trans. Faraday Soc., 1962, 58, 1846–1851 RSC.
- E. Koubek, M. L. Haggett, C. J. Bataglia, K. M. Ibne-Rasa, H. Y. Pyun and J. O. Edwards, J. Am. Chem. Soc., 1963, 85, 2263–2268 CrossRef CAS.
- Z. Yuan, Y. Ni and A. R. P. van Heiningen, Can. J. Chem. Eng., 1997, 75, 37–41 CrossRef CAS.
Footnotes |
| † “Peroxynitrite” refers to a mixture of ONOO− and ONOOH, depending on the pH; “peroxynitrate” refers to a mixture of O2NOO− and O2NOOH, depending on the pH. Buffers: Tris, tris(hydroxymethyl)aminomethane; Hepes, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid; Pipes, 1,4-piperazinediethanesulfonic acid. Formulae, trivial names and systematic names: ONOO−, peroxynitrite, (dioxido)oxidonitrate(1−); ONOOH, peroxynitrous acid, (hydridodioxido)oxidonitrogen; O2NOO−, peroxynitrate, (dioxido)dioxidonitrate(1−); O2NOOH, peroxynitric acid, (hydridodioxido)dioxidonitrogen; NO˙, nitric oxide, oxidonitrogen(˙) or nitrogen monoxide; O˙−2, superoxide, dioxide(˙1−) or dioxidanidyl; HNO3, nitric acid, hydroxidodioxidonitrogen; HO˙, hydroxyl, hydridooxygen(˙) or oxidanyl; NO˙2, pernitric oxide, nitrogen dioxide or dioxidonitrogen(˙); CO2, carbon dioxide, dioxidocarbon; CO˙−3, trioxidocarbonate(˙1−); CO32−, carbonate, trioxidocarbonate(2−); HCO3−, hydrogencarbonate, hydridooxidodioxidocarbonate(1–); NO2−, nitrite, dioxidonitrate(1−); O2, dioxygen; H2O2, hydrogen peroxide or dioxidane.1,2 |
| ‡ Reactions 1–5 are part of Scheme 1. |
| § Reactions 1–5 are part of Scheme 1 with correspondingly numbered rate constants. |
| This journal is © The Royal Society of Chemistry 2013 |