 Open Access Article
Open Access ArticleChiral MOP-phosphonite ligands: synthesis, characterisation and interconversion of η1,η6-(σ-P, π-arene) chelated rhodium(I) complexes†
Arne
Ficks
,
Ross W.
Harrington
and
Lee J.
Higham
*
School of Chemistry, Newcastle University, Bedson Building, Newcastle upon Tyne, NE1 7RU, UK. E-mail: lee.higham@ncl.ac.uk; Fax: +44 (0)191 222 6929; Tel: +44 (0)191 222 5542
First published on 25th March 2013
Abstract
The synthesis of rhodium(I) and iridium(I) complexes of chiral MOP-phosphonite ligands is reported. The full characterisation of η1,η6-(σ-P, π-arene) chelated 18VE rhodium(I) complexes reveals hemilabile binding on the arene which has been quantitatively analysed.
Monodentate phosphorus ligands based on the MOP architecture are proven as valuable ligands in asymmetric catalysis.1 It is widely recognised that these ligands can act as P,arene bidentate chelates to stabilise coordinatively unsaturated electron deficient metal centres.2 As such, hemilabile arene interactions are anticipated to be present, or indeed imperative, in catalytic processes. Whilst palladium3 and ruthenium4 complexes of this class have been investigated in detail, studies on rhodium-MOP complexes remain scarce, and some controversy persists about the exact nature of the ligand's coordination via its aryl backbone, as no X-ray crystal structures have been reported.5,6 This is despite the demonstrated catalytic ability of Rh-MOPs in a number of important asymmetric carbon–carbon bond forming reactions of biologically relevant targets,7 and the presence of an additional spectroscopic probe in the form of the NMR active rhodium nucleus (103Rh, I = 1/2),8 which better facilitates the study of these metal bonding modes.
As part of our on-going research on the stability and reactivity of MOP-type primary phosphines,9 we recently reported the synthesis of MOP-phosphonite ligands 1a,b and 2a,b (Fig. 1), and elucidated their coordination behaviour towards palladium by extensive NMR and X-ray crystallographic studies.10
 | ||
| Fig. 1 MOP-phosphonite ligands utilised in this study with numbering. | ||
We have established that changing the stereochemistry of the binol fragment from (S)- to (R)- shuttles the position of the palladium from the front towards the back of the lower naphthyl ring in [PdCl(η3-C4H7)(1/2a,b)] complexes. Coordination to the 1′-aryl carbon was observed in the [Pd(η3-C4H7)(2a,b)]+ derivatives, indicating that these ligands were acting as an η1,η1-chelate. As a result we were able to probe the ramifications of this behaviour on the asymmetric hydrosilylation of styrene, a classic benchmark reaction for MOP ligands.1,11 Herein, we investigate rhodium(I) and iridium(I) complexes of MOP-phosphonites 1a,b and 2a,b and examine their metal–ligand binding modes.
For an initial evaluation of their coordination behaviour, two equivalents of the respective ligands 2a,b were reacted with [RhCl(η4-cod)]2. The resulting complexes [RhCl(2a)(η4-cod)] (3a) and [RhCl(2b)(η4-cod)] (3b) were both formed quantitatively; the 31P{1H} NMR spectra show a doublet caused by coupling to the rhodium nucleus (3a: 162.9 ppm, 1JPRh = 223 Hz; 3b: δ = 161.5 ppm, 1JPRh = 224 Hz, Fig. S1†). In the case of 3a, single crystals suitable for X-ray analysis were obtained from slow diffusion of hexane into a dichloromethane solution (Fig. 2). Typical bond lengths are found within the coordination sphere of the metal. As expected, the Rh–P distance of this phosphonite donor (2.2112(7) Å) is shorter than the bond lengths typically observed for aryl phosphine ligands (2.308(2)–2.3607(14) Å)12 due to their stronger π-acceptor character.10,13 The η4-cod ligand shows the dominant trans effect of the phosphorus donor compared to the chloride; the alkene bond coordinated in the cis position is longer and closer to the rhodium compared to the alkene bound trans to the phosphorus atom.
![View of the molecular structure of [RhCl(2a)(η4-cod)] (3a) (50% probability thermal ellipsoids). Hydrogen atoms are omitted for clarity.](/image/article/2013/DT/c3dt50482d/c3dt50482d-f2.gif) | ||
| Fig. 2 View of the molecular structure of [RhCl(2a)(η4-cod)] (3a) (50% probability thermal ellipsoids). Hydrogen atoms are omitted for clarity. | ||
In order to investigate the possibility of a hemilabile aryl coordination of the MOP-type ligand, an anion exchange from chloride to the non-coordinating tetrafluoroborate ion was carried out on 3a,b. Initial attempts produced large amounts of oxidation and no pure product could be isolated, however when another equivalent of the appropriate ligand 2a or 2b was added to the reaction mixture a clean, quantitative conversion was achieved to yield [Rh(2a)2]BF4 (4a) or [Rh(2b)2]BF4 (4b). The η4-cod ligand was thus replaced by a second phosphorus donor during the course of the reaction. Alternatively, the two compounds could also be obtained from the reaction of two equivalents of either 2a or 2b with [Rh(η4-cod)2]BF4, although in some cases oxidised by-products were formed.
Crystals of 4b suitable for crystallographic analysis were obtained from slow diffusion of diethyl ether into a dichloromethane solution (Fig. 3). The complex contains two phosphorus ligands, one of which is coordinated in the anticipated η1 binding mode via the phosphorus atom (Rh–P bond length: 2.2145(14) Å). The second ligand fills the coordination sphere of the rhodium metal by acting as an η1,η6 chelate; in addition to the η1-phosphorus donor (Rh–P bond length: 2.1882(14) Å), the lower naphthyl ring coordinates side-on via its π-system in an η6-fashion. Selected Rh–C bond lengths of the coordinated aryl group are given in Table 1. The plane of the η6-arene is only slightly distorted; the distance from its centre to the rhodium is 1.85 Å. To the best of our knowledge this is the first time that such a bonding motif of a MOP-type ligand has been unveiled in the solid-state.
![View of the molecular structure of [Rh(2b)2]BF4 (4b) (50% probability thermal ellipsoids). Hydrogen atoms, the BF4− anion and co-crystallised solvent are omitted for clarity.](/image/article/2013/DT/c3dt50482d/c3dt50482d-f3.gif) | ||
| Fig. 3 View of the molecular structure of [Rh(2b)2]BF4 (4b) (50% probability thermal ellipsoids). Hydrogen atoms, the BF4− anion and co-crystallised solvent are omitted for clarity. | ||
NMR studies confirmed that the coordination environment retains these characteristics even in solution. The 31P{1H} NMR spectra show two doublets of doublets (4a: 181.3, 179.6 ppm; 4b: δ = 183.5, 178.4 ppm), caused by the two inequivalent phosphorus atoms coupling to each other (4a: 2JPP = 22.3 Hz; 4b: 2JPP = 23.5 Hz) and to the rhodium nucleus (4a: 1JPRh = 290 Hz, 300 Hz; 4b: 1JPRh = 277 Hz, 309 Hz, Fig. S1†). We attribute the smaller Rh–P coupling to the η1-bound ligand, based on 1H–31P correlations and 1H NOE contacts. In the 13C NMR spectra the η6-aryl binding situation of the coordinated carbon atoms is accompanied by a change in chemical shift to upper field relative to their counterparts in the η1-bound ligand, by a magnitude of 4.7 to 32.3 ppm in 4a (Table S1†) and 10.5 to 36.8 ppm in 4b (Table 1). Sections of the 13C–1H HSQC and HMBC spectra of 4b are shown in Fig. S2 and S3 respectively.†
The proton NMR spectra show the expected 48 independent aromatic resonances, from which 24 originate from each ligand. At room temperature, exchange of all 24 pairs of signals is observed in the 1H-NOESY of 4a and 4b (Fig. 4); at −50 °C the NOESY spectrum of 4b showed strong positive NOE peaks without exchange (Fig. S4†). Combining the information from variable temperature NOESY experiments allowed for the unambiguous assignment of all 48 proton resonances in 4a,b. NOE contacts confirmed the solid-state structure of 4b in solution; the solution structure of 4a was also analysed, and the NOE signals in this case revealed a rotation of the η1-ligand about its C2–P bond in comparison to 4b (further details are given in Fig. S5†).
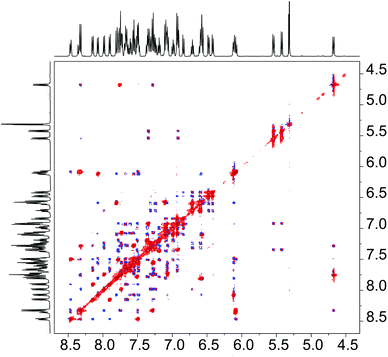 | ||
| Fig. 4 Aromatic resonances of 4b in the 1H-NOESY spectrum (mixing time of 500 ms) in CD2Cl2 at 21 °C. Negative NOE correlations are shown in blue, positive exchange correlations are shown in red. | ||
The dynamic behaviour in 4a,b is a result of the hemilabile binding of the aryl group; the side-on coordination of the η1,η6 chelating ligand is released, while in the same instance the η1-bound ligand coordinates as a chelate, ultimately reproducing the complex (Fig. S6†). Quantitative analysis of the 1H-NOESY spectra yielded exchange rate constants of k294K = 1.2 s−1 and k273K = 0.12 s−1 for 4b in CD2Cl2. The values only changed slightly when the experiments were carried out in CDCl3 (k294K = 1.3 s−1) or THF-D8 (k294K = 0.9 s−1). Thus, we propose a concerted reaction mechanism as the rate of exchange showed no increase in coordinating solvent. Comparable exchange rate values were also found for 4a (in THF-d8: k294K = 0.9 s−1). The free energies of activation of 4b in CD2Cl2 were calculated from the appropriate rate constants and gave values of ΔG‡294K = 71.2 kJ mol−1 and ΔG‡273K = 71.5 kJ mol−1. Related studies by Mirkin and co-workers gave free energies of activation of similar magnitude for their system.6f
In order to clarify whether the phenomenon of η6 side-on coordination to rhodium is exclusive to our bulky MOP-phosphonite ligands 2a,b or is valid for complexes of other MOP type ligands too, we utilised Hayashi's OMe-MOP ligand to synthesise the analogous [Rh(OMe-MOP)2]BF4 (5) complex. Full characterisation by NMR spectroscopy revealed a similar (σ-P, π-arene)-binding situation as observed for 4a,b. Its two 31P NMR resonances are observed at 50.0 and 37.2 ppm (1JPRh = 217 Hz, 197 Hz; 2JPP = 32.1 Hz). The 13C NMR resonances of the six coordinated carbon atoms show the characteristic upfield shift (shifted by 8.4 to 38.4 ppm) which is slightly less pronounced for C9′ and C10′ (Table S1†). In contrast to 4a,b we detected no dynamic exchange in the NOESY NMR spectrum at room temperature, suggesting the arene-coordination is stronger in this case. Reaction of [Rh(acac)(η2-C2H4)2] with two equivalents of OMe-MOP in CDCl3 also gave characteristic upfield peaks in the 13C NMR spectrum indicative of some degree of π-arene bonding, which could have implications in the aforementioned catalysis,7b–d however phosphine oxidation precluded a full characterisation.
Under adapted experimental conditions (to prevent hydrolysis of our phosphonites) we found 2a performed best in the asymmetric addition of phenylboronic acid to 1-napthaldehyde, with a conversion of 85% and an enantioselectivity of 34% (compared to MOP: conversion of 78%, enantioselectivity of 41%).7d
To further understand the coordination behaviour of MOP ligands with the catalytically important group nine transition metals, we also reacted, in an analogous manner, MOP-phosphonites 2a,b with [IrCl(η4-cod)]2 and were able to synthesise (to the best of our knowledge) the first iridium-MOP complexes [IrCl(2a)(η4-cod)] (6a) and [IrCl(2b)(η4-cod)] (6b); the 31P NMR spectra show a resonance at 140.4 (6a) or 139.6 ppm (6b). The crystal structure of 6a is depicted in Fig. 5; bond lengths and angles are very similar to the corresponding rhodium complex 3a (Ir–P distance: 2.2242(8) Å).
![View of the molecular structure of [IrCl(2a)(η4-cod)] (6a) (50% probability thermal ellipsoids). Hydrogen atoms are omitted for clarity.](/image/article/2013/DT/c3dt50482d/c3dt50482d-f5.gif) | ||
| Fig. 5 View of the molecular structure of [IrCl(2a)(η4-cod)] (6a) (50% probability thermal ellipsoids). Hydrogen atoms are omitted for clarity. | ||
In contrast to the bonding situation found for rhodium, treatment of 6a with silver tetrafluoroborate and an additional equivalent of 2a gave [Ir(2a)2(η4-cod)]BF4 (7a). The 31P NMR exhibits a single resonance at 156.3 ppm; rather than side-on coordination of the arene, the coordination sphere of the metal accommodates two equivalently bound η1-phosphines and the η4-cod ligand (Fig. S7†). When a solution of complex 7a in CDCl3 was reacted with hydrogen, we observed an immediate colour change from green to orange. The 1H NMR spectrum showed the disappearance of the cyclooctadiene resonances and the formation of a singlet at 1.53 ppm (indicative of cyclooctane formation) – an assignment of the aryl signals was not possible due to broadened and overlapping resonances. The 31P NMR spectrum revealed the formation of multiple products, with the starting material being completely consumed. Two major product peaks were observed as broadened singlets at 167.4 and 135.0 ppm, and the spectrum also showed a pair of doublets at 130.7 and 125.7 ppm with an associated coupling constant of 43.1 Hz (in addition to a number of other minor peaks between 155 and 142 ppm). These doublets indicate that two inequivalent phosphorus nuclei are coupled to each other, which might suggest the formation of an iridium analogue of the rhodium complex 4b. However, we were unable to isolate any of the products for the in-depth analysis which would be required to prove the existence of such a complex.
In summary, we have reported the first structural confirmation of a η1,η6-(σ-P, π-arene) chelated MOP-type ligand on rhodium(I) and the extent of the bonding has been analysed quantitatively by NOESY NMR. The fine tuning between metal-stabilisation and catalytic activity will be the focus of future research.
We thank the EPSRC for a Career Acceleration Fellowship (L.J.H.), Studentship (A.F.), an Equipment Grant (R.W.H.) and its National Mass Spectrometry Service Centre, Swansea, UK and Dr Jimmy Muldoon (University College Dublin) for valuable NMR advice. We also thank Johnson Matthey for the loan of precious metal salts.
Notes and references
- (a) J. W. Han and T. Hayashi, Tetrahedron: Asymmetry, 2010, 21, 2193 CrossRef CAS; (b) T. Hayashi, Acc. Chem. Res., 2000, 33, 354 CrossRef CAS.
- (a) Y. Canac and R. Chauvin, Eur. J. Inorg. Chem., 2010, 2325 CAS; (b) P. S. Pregosin, Chem. Commun., 2008, 4875 RSC; (c) P. S. Pregosin, Coord. Chem. Rev., 2008, 252, 2156 CrossRef CAS.
- See for example: (a) T.-K. Zhang, D.-L. Mo, L.-X. Dai and X.-L. Hou, Org. Lett., 2008, 10, 3689 Search PubMed; (b) P. Dotta, P. G. A. Kumar, P. S. Pregosin, A. Albinati and S. Rizzato, Organometallics, 2004, 23, 4247 CrossRef CAS.
- (a) T. J. Geldbach, P. S. Pregosin, S. Rizzato and A. Albinati, Inorg. Chim. Acta, 2006, 359, 962 Search PubMed; (b) T. J. Geldbach, F. Breher, V. Gramlich, P. G. A. Kumar and P. S. Pregosin, Inorg. Chem., 2004, 43, 1920 CAS; (c) T. J. Geldbach, P. S. Pregosin and A. Albinati, Organometallics, 2003, 22, 1443 CrossRef CAS; (d) T. J. Geldbach, D. Drago and P. S. Pregosin, J. Organomet. Chem., 2002, 643–644, 214 CrossRef CAS; (e) T. J. Geldbach, P. S. Pregosin, A. Albinati and F. Rominger, Organometallics, 2001, 20, 1932 CrossRef CAS; (f) T. J. Geldbach, D. Drago and P. S. Pregosin, Chem. Commun., 2000, 1629 RSC.
- (a) E. F. Clarke, E. Rafter, H. Müller-Bunz, L. J. Higham and D. G. Gilheany, J. Organomet. Chem., 2011, 696, 3608 Search PubMed; (b) M. Soleilhavoup, L. Viau, G. Commenges, C. Lepetit and R. Chauvin, Eur. J. Inorg. Chem., 2003, 207 CrossRef CAS.
- Selected examples of other phosphorus-alkene ligands on rhodium(I): (a) R. Shintani, R. Narui, Y. Tsutsumi, S. Hayashi and T. Hayashi, Chem. Commun., 2011, 47, 6123 RSC; (b) A. R. O'Connor, W. Kaminsky, D. M. Heinekey and K. I. Goldberg, Organometallics, 2011, 30, 2105 Search PubMed; (c) Z. Liu, H. Yamamichi, S. T. Madrahimov and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2772 CrossRef CAS; (d) M. Montag, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2007, 13, 9043 CrossRef CAS; (e) H. Werner, Dalton Trans., 2003, 3829 RSC; (f) E. T. Singewald, C. A. Mirkin, A. D. Levy and C. L. Stern, Angew. Chem., Int. Ed. Engl., 1994, 33, 2473 CrossRef.
- (a) R. Shintani, M. Inoue and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 3353 CrossRef CAS; (b) T. Hayashi and M. Ishigedani, Tetrahedron, 2001, 57, 2589 CrossRef CAS; (c) T. Hayashi and M. Ishigedani, J. Am. Chem. Soc., 2000, 122, 976 CrossRef CAS; (d) M. Sakai, M. Ueda and N. Miyaura, Angew. Chem., Int. Ed., 1998, 37, 3279 CrossRef CAS.
- (a) L. Carlton, Annu. Rep. NMR Spectrosc., 2008, 63, 49 Search PubMed; (b) J. M. Ernsting, S. Gaemers and C. J. Elsevier, Magn. Reson. Chem., 2004, 42, 721 CrossRef CAS.
- (a) A. Ficks, C. Sibbald, S. Ojo, R. W. Harrington, W. Clegg and L. J. Higham, Synthesis, 2013, 45, 265 Search PubMed; (b) B. Stewart, A. Harriman and L. J. Higham, Organometallics, 2011, 30, 5338 CrossRef CAS; (c) A. Ficks, I. Martinez-Botella, B. Stewart, R. W. Harrington, W. Clegg and L. J. Higham, Chem. Commun., 2011, 47, 8274 RSC; (d) R. M. Hiney, L. J. Higham, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2006, 45, 7248 CrossRef CAS.
- A. Ficks, R. M. Hiney, R. W. Harrington, D. G. Gilheany and L. J. Higham, Dalton Trans., 2012, 41, 3515 RSC.
- (a) T. Hayashi, S. Hirate, K. Kitayama, H. Tsuji, A. Torii and Y. Uozumi, J. Org. Chem., 2001, 66, 1441 CrossRef CAS; (b) T. Hayashi, Acta Chem. Scand., 1996, 50, 259 CrossRef CAS.
- (a) J. Tiburcio, S. Bernès and H. Torrens, Polyhedron, 2006, 25, 1549 CrossRef CAS; (b) Q. L. Horn, D. S. Jones, R. N. Evans, C. A. Ogle and T. C. Masterman, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m51 Search PubMed.
- D. Selent, W. Baumann, R. Kempe, A. Spannenberg, D. Röttger, K.-D. Wiese and A. Börner, Organometallics, 2003, 22, 4265 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, full experimental and crystallographic data. CCDC 911753 (3a), 911754 (4b) and 911752 (6a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt50482d |
| This journal is © The Royal Society of Chemistry 2013 |