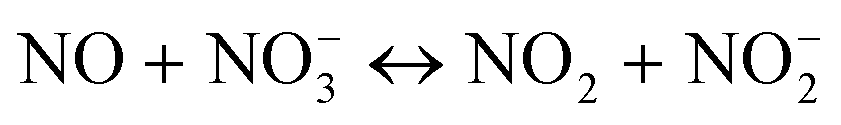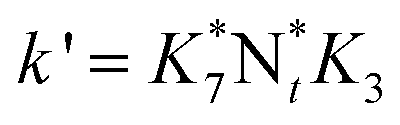A review on selective catalytic reduction of NOx by supported catalysts at 100–300 °C—catalysts, mechanism, kinetics
Received
16th June 2013
, Accepted 25th September 2013
First published on 26th September 2013
Abstract
Removing NOx by supported catalysts is important for industrial flue gases and exhaust fumes of diesel engines at low temperature (100–300 °C). However, it is still a challenge to develop the low-temperature catalyst (LTC) for selective catalytic reduction of NOx, especially at temperatures below 200 °C. This paper reviews the effect of the different carriers with different active centres. Most supported metal oxide catalysts are prepared by impregnation method. But under the same condition, catalysts perform better if they were prepared by sol–gel method. Also, this paper described and compared the research progress of reaction mechanism and kinetics of the supported catalysts which were used for the selective catalytic reduction of NOx at low temperature. There are different mechanisms because of the different reducing agents and active centres. The L–H and E–R mechanisms, or both of them coexisting, can explain the overall mechanism. In order to research the mechanism thoroughly, the exploration of the reactions of the surface chemical process may be a direction to develop low temperature supported catalysts for removing NOx.

Mengfan Fu
| Mengfan Fu received his bachelor's degree of environmental science at Xiangtan University. Now he is studying for a master's degree at Hunan University under the guidance of Prof. Li. His research interests are in air pollution control techniques and environmental catalysis for removing NOx at low temperature. |

Caiting Li
| Caiting Li received his PhD degree at Hunan University in Changsha, China. He is professor and supervisor of doctoral students of Hunan University. He has led more than 30 national and provincial research projects. The technology and equipment of dedusting and desulfurization for coal-fired flue gas have been applied in more than 10 provinces of China. He has published over 90 papers and has over 15 patents in the field of air pollution control. |

Pei Lu
| Pei Lu, bachelor in chemistry and master in environmental engineering, received his PhD degree of environmental science and engineering in 2012 at Hunan University, China. His study interests include air pollution control techniques, applied catalysis for environmental protection and the development of catalysts. Now he is working in the Institute of Chemistry, Chinese Academic Science as a postdoctoral researcher and mainly focusing on the tip enhanced Raman spectroscopy (TERS) and nanomaterials. |

Long Qu
| Long Qu, bachelor in environmental science at Zhengzhou University, is now studying for his master’s degree of environmental science and engineering at Hunan University. His research interests focus on air pollution control techniques and applied industrial catalysts, especially the catalysts used in selective catalytic reduction for NOx with NH3 at low–middle temperature. |
1 Introduction
Nitrogen oxides (NOx, x = 1, 2) are the major causes of photochemical smog, acid rain, ozone depletion and the greenhouse effect.1–4 NOx emits into the atmosphere from natural sources and anthropogenic sources. Furthermore, the combustion of coal and fossil fuels is the primary source of anthropogenic NOx emissions. In China, 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 043000 tons of NOx had been emitted in 2011, which increased by 5.73% compared to that of 2010 (the state of environment situation bulletin in 2011). It is estimated that China's coal resources are to be 5570 billion tons with 1018 billion tons of proven reserves and 4552 billion tons of forecasted reserves, and the production of coal in 2008 ranked first in the world with 42.5% of the world's total.5,6 The NOx emissions are mainly from power stations, cogeneration plants, industrial heaters, and the exhaust gas of gasoline and diesel engines.3,7,8 Therefore, it is necessary to develop advanced technology and seek for better catalysts to control NOx emissions.
043000 tons of NOx had been emitted in 2011, which increased by 5.73% compared to that of 2010 (the state of environment situation bulletin in 2011). It is estimated that China's coal resources are to be 5570 billion tons with 1018 billion tons of proven reserves and 4552 billion tons of forecasted reserves, and the production of coal in 2008 ranked first in the world with 42.5% of the world's total.5,6 The NOx emissions are mainly from power stations, cogeneration plants, industrial heaters, and the exhaust gas of gasoline and diesel engines.3,7,8 Therefore, it is necessary to develop advanced technology and seek for better catalysts to control NOx emissions.
As for the obvious harm of NOx, it is known from legislations and policies that reducing the emission of NOx is getting more and more strict. For example, the NOx emission limits are 100 mg m−3 for most power plants (GB13223-2011) and 150 mg m−3 for gas, oil, and coal-fired industrial boilers (DB11139-2007) in the capital of China, Beijing.
In order to adapt to the stringent regulations, new technologies emerged to displace the old technologies. A number of NOx control technologies in thermal power plants and industry furnaces had been taken, including combustion control and post-combustion control technologies. Combustion controls can reduce NOx emission by altering or modifying the combustion conditions during the combustion process by means of low NOx burners, fuel reburning and flue gas recirculation (FGR). Post-combustion controls mean the selective reduction of NOx to N2. Three major post-combustion processes are available: selective catalytic reduction (SCR), selective non-catalytic reduction (SNCR), and hybrid SNCR–SCR technologies.9–13 The major products of post-combustion processes are harmless nitrogen and water vapour. Post-combustion technologies were first applied in Japan in the 1970's and have now been installed in the US and several European countries.14 SCR, which utilizes NH3 as the reducing agent and heterogeneous reaction of NO on a catalyst, can achieve over 95% of the conversion. For practical implementation of SCR of NOx from coal-fired combustion sources, NH3 slip ranged from 2 to 5 Vppm.5 These technologies are currently experiencing unprecedented growth and development in China. Nowadays, the catalyst of TiO2-supported V2O5, promoted with WO3, is the main catalyst for SCR in industry.15 However, there are some problems with it; the catalyst should be installed upstream of the particulate collector and flue gas desulfurization and it has only a narrow and high working temperature window (300–400 °C)15–17 to ensure a high conversion of NOx, otherwise the effect of particles and sulfur can lead to deactivation and poisoning.8
There are two methods to solve these problems. One is to install the catalyst downstream of the particulate collector and flue gas desulfurization, which requires an additional heat source to heat the catalyst, and that will make the cost increase significantly. The second one is to develop new low temperature catalysts that can work well around 250 °C or even lower.
Up to now, researchers have studied single metal oxide catalysts. Such as the many SCR catalysts containing a transitional metal (Fe, V, Cr, Cu, Co and Mn), which have good low temperature SCR activity, and the nano-MnOx catalyst that has excellent temperature performance.1,18,19 Multi-metal oxide catalysts were also studied. For example, synergetic catalysis of ceria and titania,20 Fe–Ti spinel for the selective catalytic reduction of NO,21 and binary metal oxide solid solutions might be formed between Mn and Cu or Ce, etc.3,8,22–24 Al2O3, TiO2, zeolites, ACF as the main support and metal oxides supported on carbon materials and zeolite catalysts have also been researched.25–30
Some works have been done in researching the low temperature selective catalytic reduction of NOx in our team.25,28,31–37 In this paper, we review the catalysts which were prepared by different carriers, and the performance of the catalysts at low temperature. The developed mechanism and kinetics have also been discussed. It will be useful for future research.
2 Catalysts
Up to now, low temperature catalysts (LTC) have been studied without carriers or using ACF, Al2O3, TiO2, zeolites and SAPO as the carriers. Taking into consideration the influence of components and environmental temperature on the formation of (NH)2SO4, NH4NO3, N2O and other by-products in flue gas, the main goal of these studies is to develop new SCR catalysts with high selectivity and stability, good activity and a broad operating temperature range, which make it possible to place the SCR reactor downstream of the electrostatic precipitator and desulfurizer. However, since most of the catalysts were just researched in the lab, more studies into practical application are needed.
2.1 Catalysts without carriers
According to the literature, many catalysts have been studied to remove NOx. Some catalysts without carriers have been researched.38–42 It showed that the Mn–Ce mixed-oxide catalyst had the best performance. Gongshin Qi and Ralph T. Yang,3,43 researched the performance and mechanism of the MnOx–CeO2 catalyst, and indicated that the best Mn–Ce mixed-oxide catalyst yielded nearly 100% NO conversion at 120 °C at a high space velocity of 42000 h−1. Based on the past investigation, the authors indicated that there may exist a synergistic mechanism between the manganese and cerium oxides by the following chain of reactions: | | | Mn2O3 + 2CeO2 → 2MnO2 + Ce2O3 | (2) |
The SCR reaction of NO by NH3 on the MnOx–CeO2 catalyst most probably takes place according to the following steps.
| | | NH3(a) + O(a) → NH2(a) + OH(a) | (6) |
| | | NO(g) + 1/2O2(g) → NO2(a) | (7) |
| | | NH2(a) + NO(g) → NH2NO(a) → N2(g) + H2O(g) | (8) |
| | | OH(a) + NO2(a) → O(a) + HNO2(a) | (9) |
| | | NH3(a) + HNO2(a) → NH4NO2(a) → NH2NO(a) + H2O → N2(g) + 2H2O(g) | (10) |
The meaning of (g) and (a) are gaseous and adsorbed state, respectively.
The effect of different dopants including tin, niobium, iron, tungsten and zirconium oxide of MnOx–CeO2 catalysts for SCR has been studied.44,45 Although these catalysts can improve NOx reduction, the high cost and difficulty in forming a fixed shape limited their commercial applicability. In order to solve this problem, more research should be done in modifying the catalysts.
2.2 ACF as the carrier
Active carbon fiber (ACF) is widely utilized in water purification and air purification and separation as a superior adsorbent because of its large number of well distributed micropores, high adsorption speed and high surface area (1000–2000 m2 g−1).46,47 As listed in Table 1, coal-tar pitch-based ACF has over 10 times the physical surface area compared to GAC. Some researchers have studied the NO conversions by ACF in the last few decades. Mochida et al.48 reported that the heat treatment of pitch-based activated carbon fibres of moderate surface area markedly increased the conversion of NO to 87% in dry air, 62% in air of 80% relative humidity and 24% in wet air (100% r.h.) at 25 °C and a ratio of fibre mass to gas flow rate of 1.0 × 10−2 g min mL−1. Sudhakar Adapa et al.49 studied the surface characteristics of ACFs synthesized from raw carbonaceous non-activated fibers under varying activation conditions, and analyzed the adsorptive and catalytic properties of ACF for the control of NO only. They indicated that the extent of conversion depended upon the reaction temperature, inlet O2 and NO concentrations, the types and the preparation methods of ACF. They also showed the phenolic resin-based ACF was more effective in the oxidation of NO in comparison to the pitch and viscose rayon-based ACFs.
Table 1 Properties of examined supports46
| Material |
BET surface area (m2 g−1) |
Pore volume (ml g−1) |
Size |
Physical surface area (m2 g−1) |
|
GAC: granular active carbons.
D
1: diameter of active carbon fibers.
D
2: diameter of particles.
L: length of active carbon fibers.
|
| ACF |
740 |
0.342 |
D
1 = 20 μmb |
0.5–1 |
|
L = over 50 mmd |
| GACa |
831 |
0.414 |
D
2 = 1 mmc |
0.01–0.03 |
ACF as the carrier has been generally researched in the laboratory. The large physical surface area can make metal-oxides highly dispersed on ACF during the preparation process of the catalyst. Masaaki Yoshikawa46 researched the three transition-metals, Fe2O3, Co2O3 and Mn2O3, as catalytic components. The result showed that Mn2O3 had the best activity, and CeO2,27,33 V2O5,50 Ni51 have also been researched. It was indicated that Mn and Ce had the best effects as the active component in ACF. Maybe low temperature SCR of NO based on MnOx–CeO2/ACF, or MnOx–CuO–CeO2/ACF and so on can be investigated to achieve higher selectivity and stability, and better activity at low temperature.
2.3 Metal oxide as the carrier
2.3.1 Al2O3 as the carrier.
Al2O3 as an atomic crystal, owns high temperature resistance, withstands wear and tear, is easy to form into a fixed shape, and has a surface area below 200 m2 g−1, so it has attracted some researchers' attention. The performance of removing NOx with the metal oxides and preparation of the catalysts by different methods have been studied. Some research showed a similar reaction mechanism by promoted and unpromoted catalysts, and the sol–gel method can produce the most active catalysts.
A series of Pt/Al2O3 catalysts promoted by metal oxides (Ba, Ce, Co, Cs, Cu, K, La, Mg, Mo, Ti) or noble metals (Ag, Au, Pd, Rh) was prepared and tested for the lean NOx reaction using C3H6 as the reductant. There was a significant effect (beneficial or otherwise) on the activity and the operation temperature range of Pt/Al2O3 (Fig. 1), but a major shortcoming was their high selectivity towards N2O, a currently unregulated but very undesirable oxide of nitrogen.52 Zhang et al.53 tested the catalyst of MnOx/CeO2–ZrO2–Al2O3(MnOx/CZA), almost 90% NO was transformed to N2 in the temperature range of 143–300 °C. Erol Seker54 prepared alumina supported silver catalysts by co-precipitation, impregnation and single step sol–gel methods, to study the selective NOx reduction by propene in the presence of oxygen. The catalysts prepared by sol–gel method had the highest activation for selective reduction of NOx, with almost 100% selectivity to N2. R. Lanza55 researched three catalysts (Pt, Rh and Ag on alumina). The Pt/Al2O3 and Rh/Al2O3 were active at low temperature (T50 = 200–250 °C) but had quite high selectivity towards NO2. Meanwhile, Ag/Al2O3 was active at higher temperature, but it showed very high selectivity towards N2. A strong boosting effect on NO conversion was recorded if H2 was added to the gas mixture. Derek Creaser56 showed that feed concentrations of NO and hydrogen had significant effects on the NOx conversion, indicating that nitrates poisoned the catalytic sites and hydrogen's role was to reduce nitrate species over the Ag–Al2O3 catalyst.
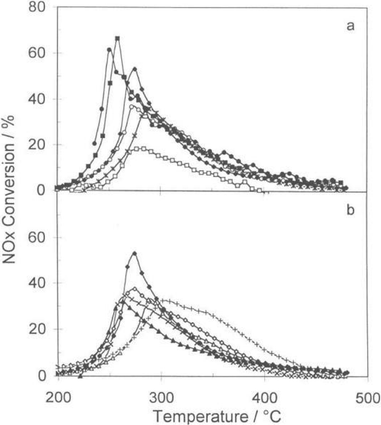 |
| | Fig. 1 The effect of metal oxide promoters on NOx conversion over Pt/Al2O3 based catalysts. (a): ◆ No promoter, ○ Ce, * Co, □ Cs, ● Mo, ■ Ti; (b) ◆ No promoter, ◇ Ba, ▲ Cu, ╳ K, + La, △ Mg.52 | |
2.3.2 TiO2 as the carrier.
Titania has stable physical and chemical properties, it is not a poison and its surface area is under 100 m2 g−1.57 V2O5–WO3/TiO2 is the most prevalent commercial catalyst, exhibiting high activity, but it is active only in a narrow temperature window of 300–400 °C.58,59 Maybe this shortcoming can be made up for by modifying the carriers and catalysts.
Ruihua Gao et al.60 modified the V2O5–WO3/TiO2 catalysts by iron oxides, and these combination metal oxide catalysts show not only high catalytic activities but also good resistance to SO2. According to other research, the catalysts of Mn and Ce oxide supported on TiO2 had better performance for removing NOx. Wu et al.61 prepared the catalyst of MnOx/TiO2 by sol–gel method. Under the optimal conditions, the efficiency of NO removal could exceed 90% at a temperature of 150 °C. It was found that the transition metal had significant effects on the catalytic activity. The temperature could be reduced to 88 °C by using Fe(0.1)–Mn(0.4)/TiO262 to achieve 90% NO conversion. At the same time, a series of cerium modified MnOx/TiO2 catalysts revealed that NO conversion could be improved from 39% to 84% at 80 °C.63 Jin Ruiben et al.64 prepared the catalyst of Mn–Ce/TiO2 by sol–gel method with NH3 as the reducing agent, removing NOx by SCR at different temperatures (100–200 °C). This suggested that the unrecoverable SO2 deactivation can be greatly retarded if the low-temperature SCR process over Mn–Ce/TiO2 is operated at about 100 °C. Shen Boxiong et al.65 found that 96.8% NO conversion was obtained over Fe(0.1)–Mn–Ce/TiO2 at 180 °C at a space velocity of 50000 h−1. Sichem Guerrero66 observed a dramatic improvement was achieved by the addition of Na when the Cu/TiO2 was used as NO adsorbent. The idea may open new possibilities for the use of inexpensive NO adsorbents. Also Ma Zhaoxia16 found that approximately 90% NO conversion could be achieved at a reaction temperature as low as 150 °C by the catalyst of iron–copper oxides supported on TiO2 and carbon nanotubes (CNTs).
Different methods can produce different catalysts which have different performances on activity and SO2 resistance. Zhang Zhixiang et al.67 reported that the optimal catalyst of Pt/TiO2 was 0.5 wt.% Pt and provided excess O2. And bimetallic Pt–Ir/TiO2 exhibited high activity in H2–SCR reaction and NOx conversion exceeding 80%, with N2 selectivity >80%, could be obtained in the temperature range of 140–200 °C.68 Jiang et al.69 prepared the catalysts of MnOx/TiO2 by sol–gel, impregnation, and coprecipitation methods, among these catalysts, the sample prepared by sol–gel method had the best performance in terms of both activity and SO2 resistance.
2.4 Molecular sieves as the carrier
2.4.1 Zeolites as the carrier.
There are different types of zeolites, but ZSM-5 has been widely used because of its thermostability, acid and water resistance and the ease of fixing its shape. The surface area was between 200–500 m2 g−1.70 Katariina Rahkamaa-Tolonen71 verified that the zeolite-based catalysts were very promising for the SCR reaction with ammonia. Especially, the activities at low and high temperature were higher than the activities of commercial vanadia-based catalysts. The first time of using Cu–ZSM-5 to catalyze the decomposition of NO to its elements was in 1989.72 Choong-Kil Seo et al.73 researched the Cu–ZSM-5 catalyst on which ZrO2 acted as the additive to improve the low-temperature activity and durability of the SCR catalyst for diesel vehicles. The authors pointed out the NOx conversion of the Cu–ZSM-5–ZrO2 (2 wt%) catalyst was improved by 10–20%, compared to that of the Cu–ZSM-5 or Fe–zeolite catalyst. Other catalysts were researched. For instance, Liu Huayan et al.74 researched the high silica content Na–ZSM-5 molecular sieves, which exhibited high catalytic activity at ambient temperature. Sandro Brandenberger et al.75 used different methods to prepared the Fe–ZSM-5 catalysts and the research groups showed that the preparation method is not a decisive factor in determining catalytic activity. The available data indicated that both isolated and bridged iron species participate in the NH3-SCR reaction over Fe–ZSM-5, with isolated species being the most active. H-β zeolites modified with calcium oxide (Ca-β) have been developed by Chang Xiaofei et al.76 The results displayed that CaO was located inside the pores of H-β and also indicate that NOx can be easily absorbed on Ca-β samples with the NO−3, NO−2 and NO+ species.
2.4.2 SAPO as the carrier.
Silico-alumino-phosphate (SAPO) is a microporous crystal composed of three kinds of tetrahedral elements, namely PO+4, AlO−4 and SiO2. It can be prepared by introducing the silicon into the skeleton of aluminum phosphate, which is a electronegative framework with small pore structure. SAPO can be used as adsorbent and carrier, has exchangeable cations and enough acid centers (with proton acid) on the surface. Now the research application focuses on the use of its surface acidity; the surface acidity and amount of acid can be regulated by the introduction of various kinds of impurity atoms. The performances of different impurity atoms that were introduced into SAPO have been studied.77–81 They demonstrated that copper can improve the activity and life of the SAPO-34, with great potential for industrial application. Dustin W. Fickel et al.82 showed that the NO conversion reached nearly 100% between 200–400 °C. Jun Wang et al.83 studied the effect of Si content in SAPO-34 on NO selective catalytic reduction over Cu/SAPO-34 catalyst. The Si and Al contents affect the contents of Cu2+ species. The contents of active sites and the number of acid sites affect the activity over Cu/SAPO-34 samples with various Si contents. Although SAPO is a potential catalyst for removing NOx, it needs more attention in terms of synthesis process, cost control of the synthesis and reasonable modifications, which include tunable pore size, high stability and ordered arrangement.
In conclusion, much research has comprehensively studied the carriers and achieved high NOx conversion. But it needs more exploration for industrial applications. Ammonia, as the main reducing agent, has been researched using different kinds of catalysts. Gongshin Qi et al.84 suggested that the initial step was adsorption of NH3 onto the acid sites, and then it further reacts with NOx to produce N2 and H2O. So the number of acid sites is important in catalysts. It is known that ammonia adsorbs on acid catalysts and yields two strong bands near 1600 and 1200 cm−1. And some researchers46,77,85–87 studied the contents in ACF, Al2O3, TiO2, zeolites, SAPO.
3 Effect of reaction conditions
With the deepening of the research, the catalysts with high selectivity and stability, good activity and broad range of operating temperature make great progress. However, there is still a burning question regarding catalyst poisoning.
3.1 Effects of H2O and SO2
Water vapor is one of the main components in flue gases and often leads to catalyst deactivation. Even in dry conditions, the catalysts can be affected by the water vapor produced in the SCR reaction. It causes a reduction in activity for both non-supported metal oxide catalysts3 and carbon based catalysts.88 The effect of H2O can be divided into two results, reversible and irreversible. It is reversible when H2O adsorption competes with that of NH3 and NO. If H2O were removed, this effect will disappear. However, the hydroxyl created by H2O adsorption and decomposition on the surface of the catalysts will result in irreversible deactivation of the catalysts.1
H2O and SO2 have a critical influence on the catalyst for NO reduction at low temperature. Huang et al.89 studied the effect of SO2 and H2O on a catalyst of V2O5/AC, in which the AC was obtained by the industrial semi-coke treatment (Fig. 2). The results indicated that the effect of SO2 was positive in the absence of H2O. The reason for this is the transference from SO2 to SO42− on the surface of the catalyst, which can improve the acidity of the catalyst surface and enhance the adsorption capacity of NH3. The experiment also revealed that the effect of the water vapor was small, which may be attributed to competitive adsorption of H2O and reactants (NO and/or NH3). But the co-existence of H2O and SO2 resulted in an obvious decrease. The phenomenon was possibly caused by sulfate particles formed from H2O and SO2, which deposit on the surface of the catalyst in the reaction process.
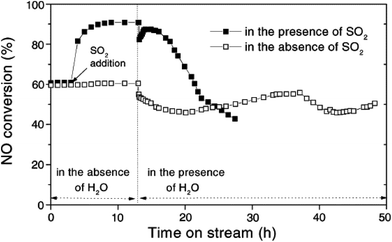 |
| | Fig. 2 The effect of SO2 and H2O on V2O5/AC catalytic performance. Reaction conditions: 500 ppm NO, 600 ppm NH3, 500 ppm SO2 (when used), 3.4% O2, 2.5% H2O (when used), balance Ar, space velocity of 90000 h−1, reaction temperature of 250 °C.89 | |
3.2 Effect of alkali and heavy metal
The fly ash in coal-fired power contains some alkali metal oxide, which will gather in the catalyst surface or react with the active substances in the catalyst and poison the catalyst during the long-term contact with catalyst. In the study of commercial vanadium and tungsten titanium catalyst, a small amount of alkali metal can have a great influence on the activity of the catalyst. Asa Kling et al.90 Wenchao Yu,91 and Liang Chen92 studied the influence of K, Na, Ca, Mg on the catalysts, indicating the influence rank was K > Na > Ca > Mg. Some researchers93–95 suggested that the submicrometer aerosol particles which form in the process of burning alkali and alkaline earth metals, accumulate on the surface of the catalyst, and react with the catalyst causing deactivation. They could decrease the amount and stability of the Brönsted acid sites, and affect the catalytic efficiency.96–98 Maybe an increased resistance to alkali and the regeneration performance of catalysts are better solutions. Researchers97,99,100 studied the resistance to alkali by the catalysts of Cu–zeolite, Ag–Al2O3, vanadia on sulphated-ZrO2 and got an ideal result. Shen Boxiong et al.96 reported that washing with both water and sulfuric acid solution could partly reverse the catalyst deactivation by alkali metals. And washing with water was the best method for regenerating catalysts that were not greatly deactivated by alkali metals.
Heavy metals are easy to volatilize into metallic vapor during the combustion processes because of their high saturation vapor pressure. And they condense to form fine particles after the temperature drops.101 A few studies have investigated the effects of heavy metals on the activity of catalysts. Researchers102–104 attribute the inhibition effect by heavy metals to the formation of alloys from the reaction with active metals.
4 SCR mechanism
At present, many catalysts have been researched, but most of them lack enough clear knowledge of the reaction mechanism. In this paper, the development progress of the mechanisms which remove NOx by different reducing agents was summarized.
4.1 H2 and CO as the reducing agent
Some researchers use H2 and CO as the reducing agent to remove NOx in SCR,105–107 which can get a high conversion rate. According to a study, the effect of CO and NO is to reduce the bond length and separate the bond of N–O. The mechanism is as follows:105| | | NO(a) + CO(a) → N(a) + CO2(a) | (11) |
| | | NO(a) + H(a) → N(a) + OH(a) | (12) |
Another possibility is:| | | CO(a) + NO(a) + H–H(a) → CO–O(a) + NH2(a) | (14) |
4.2 Hydrocarbons as the reducing agent
Some researchers studied the mechanism of a hydrocarbon as the reducing agent, because of the practicability and cost-effectiveness. We can use the unburned hydrocarbons already present in the exhaust gas. In the process of catalytic reduction, “NOy”,108 CH3NO109 or CH3NON2109,110 can be formed from NOx. Due to the effect of the catalysts of Y2O3, the first step is the formation of CH3 radicals from CH4, then these will react with NOx to form CH3NO2, finally, CH3NO2 will become N2via the intermediates of CH2NOH, CN, NCO and N2O.109 At present, the hydrocarbons used as a reducing agent to remove NOx mainly include methane, propane, propylene.
Aylor et al.111 analyzed the mechanism using Mn–ZSM-5 as the catalyst and methane as the reducing agent. First, the NO was oxidized to NO2, then NO2 adsorbed on the surface of the catalyst and reacted with CH4. In the process of the reaction, CN and NCO were formed, and CN would react with NO2 quickly to generate N2 and CO2. They suggested that cyanide is the active intermediate.
Lobree et al.112 studied the mechanism that removed NOx with C3H8 as the reducing agent (Fig. 3), but they did not explain the detailed reaction process between C3H8 and nitrates, and did not reveal the rate controlling steps in the process of the reaction.
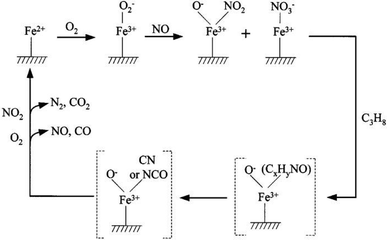 |
| | Fig. 3 Proposed mechanism for the reduction of NO by C3H8 over Fe–ZSM-5.112 | |
Satsuma et al.113 researched the catalysts involving Al2O3 as the carrier, including Cu–Al2O3 and Ag–Al2O3, for the removal of NOx. They reported that the NO was oxidized by O2 to NO2, and then formed a nitrate species which had a stronger oxidizing ability that could be adsorbed on the catalyst's surface. At the same time, C3H6 was partially oxidized to an acetic salt species or other oxide. Then the species will react with nitrate to form NCO and CN, which will continue to react with nitrate to finally form N2 and CO2. The author pointed out that the generated N2 was the rate controlling step. Yu Yunbo et al.114 did similar research and got similar results, but Yu thought the NCO's reaction was the rate controlling step.
In practice, hydrocarbons are composed of many kinds of alkanes, alkenes and alkynes. However, in the laboratory, we generally use one of them to test and analyse. Different possible synergistic effects or antagonistic effects would happen with the hydrocarbons during the catalysis. This can change the path which the catalysts react with NOx, so it needs further research. Of course, it is difficult to completely simulate the physical truth, but it is also important to reveal active intermediates in the process of the reaction and the rate controlling steps, taking into account the key factors which affect the rate controlling step.
4.3 3NH3 as the reducing agent
Many researchers have studied the mechanism of SCR by NH3. Concerning the reaction pathway over metal oxide catalysts, most researchers suggested that NH3 was adsorbed to the Lewis acid center and intermediates like NH2115,116 or adsorbed NH3117,118 formed, then they reacted with aerial NO and NO2 through an E–R (Eley-Rideal) mechanism producing N2 and H2O. They can also react with activated nitrite intermediate adsorbed on the surface of the catalysts through a L–H (Langmuir-Hinshelwood) mechanism. Marban et al.118 proposed an E–R mechanism over carbon-supported Mn3O4 catalyst, in which surface-active NH3 species reacted with NO2, and to a lesser extent NO, from the gas phase (see Fig. 4).
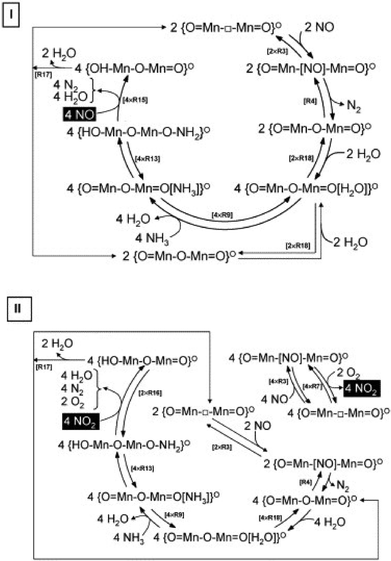 |
| | Fig. 4 Mechanism of the steady-state SCR reaction: (I) in the absence of oxygen; (II) in the presence of oxygen.118 | |
Richter et al.119 supposed symmetric O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O–NO species formed on the MnOx/NaY catalyst after contact with NO. They suggested a “diazotization” mechanism, in which NH3 was protonated to NH4+, NO interacts with the catalyst, and nitrite and nitrate surface species formed in the presence of O2. Followed by NH4+ reacting with NO2− to produce N2 and H2O. However, a parallel “amide/nitrosamide” SCR reaction route was possible since prevailing Lewis acid sites on these catalysts should enable NH3 activation via amide species.
N–O–NO species formed on the MnOx/NaY catalyst after contact with NO. They suggested a “diazotization” mechanism, in which NH3 was protonated to NH4+, NO interacts with the catalyst, and nitrite and nitrate surface species formed in the presence of O2. Followed by NH4+ reacting with NO2− to produce N2 and H2O. However, a parallel “amide/nitrosamide” SCR reaction route was possible since prevailing Lewis acid sites on these catalysts should enable NH3 activation via amide species.
Meanwhile, some researchers investigated the mechanisms of Cu and Fe on zeolite catalysts. For Fe/zeolite catalysts, it is supposed that Fe3+ ions with oxo-Fe3+ sites,29 binuclear ions [HO–Fe–O–Fe–OH]2+120 and others were the active sites for the SCR reaction. Schwidder et al.121 established that mononuclear Fe ions are active sites for SCR reactions, but oligomers contribute as well. Brandenberger et al.122,123 supported and further developed these views that oligomers have high oxidation potential, causing undesired total oxidation of the reductant, particularly in the case of isobutene. Authors suggested that the SCR of NO by NH3 is primarily caused by monomeric iron sites at temperatures below 300 °C. A reaction mechanism which describes NO or NO2 reduction by NH3 over H-form zeolites was most developed and accepted (see Fig. 5).124,125 It suggested that adsorbed NH3 is the most reactive agent when it is bonded to the Brønsted acid sites in zeolites through three hydrogen bonds. An NO2-type intermediate is formed on the zeolite surface during the SCR reaction. Formation of this NO2-type species appears to be a necessary step in the SCR reaction mechanism, since the concentration of adsorbed NH3 does not decrease (i.e., react) until a band corresponding to this NO2-type species appears. Grossale126,127 presented a systematic study of the chemical steps in the NO/NO2–NH3 fast SCR reaction over a commercial Fe–ZSM-5 catalyst.
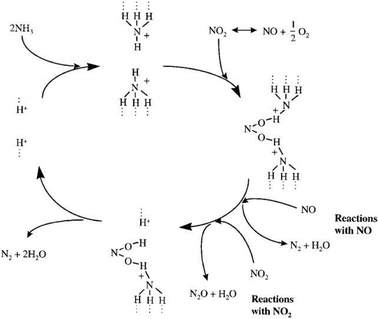 |
| | Fig. 5 Reaction mechanism of SCR of NOx with NH3 over H-form zeolites.124,125 | |
The schematic summary of the fast SCR chemistry in terms of surface species is displayed in Fig. 6. Results were demonstrated via a global sequence involving NH4NO3 or related surface species as intermediates, which is the same as that proposed previously for the fast SCR chemistry over V-based catalysts and other zeolite catalysts and thus is considered a general mechanism. It further showed that the redox reaction between NO and nitrates is the rate-controlling step and is inhibited by ammonia. Remarkably, the same strongly enhanced de-NOx activity observed in the fast SCR reaction was also observed in the absence of gaseous NO2, but in the presence of surface nitrates. Accordingly, they propose a general summary of the fast SCR chemistry over V-based and zeolite catalysts that emphasizes the key role of surface nitrates.
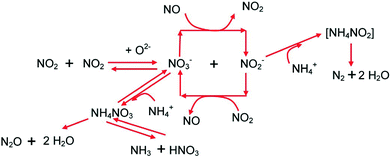 |
| | Fig. 6 The fast SCR reaction proceeds at low temperatures.126,127 | |
The fast SCR chemistry126 basic reaction steps in NO/NO2–NH3 SCR chemistry over V-based and zeolite catalysts are as follows.
Involving NO2 only
| | 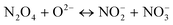 | (16) |
In the presence of NH3
| | 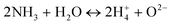 | (17) |
| | 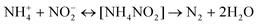 | (18) |
| | 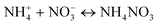 | (19) |
In the presence of NO
| | 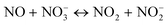 | (20) |
Global reactions observed in this work, and their relationship with the basic reactions above
| | 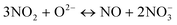 | (21) |
| | | 2NO2 + 2NH3 → NH4NO3 + N2 + H2O | (22) |
| | | NO + NH4NO3 → NO2 + N2 + 2H2O | (23) |
| | | NO + NH3 + 1/2NH4NO3 → 2/3N2 + 5/2H2O | (24) |
| | | 2NH3 + NO + NO2 → 2N2 + 3H2O | (25) |
5 Kinetics
A lot of work had been done for researching the mechanism and kinetics of removing the NOx at low temperature. The research of kinetics for SCR reaction at low temperature was based on empirical equations (as power-law kinetics) and obtained from mechanistic analysis or both of them.
Yang et al.128 studied the kinetics of the catalyst of MnOx–CeO2 based on the kinetic equation of power function in eqn (26). They got some data from the result: the reaction orders of NO, NH3, and O2 ([O2] < 1%) were 1, 0 and 0.5 at 120 °C, respectively. This was consistent with the results from Wu et al.129 The kinetics of MnOx/TiO2 was researched based on the kinetic equation of power function.
| | | rNO = k[NO]x[NH3]y[O2]z | (26) |
But different results were reported by Richter et al.130 using the same equation to obtain the experimental data of MnOx/NaY at low temperature. The reaction orders were 2 and 0.3 for NO and O2, respectively. The reaction order of 2 means that two molecules of NO were involved in the rate controlling step of the reaction. It corresponded to the reaction model that they proposed: it was the oxidation of NO to NO2, and the intermediate was similar to the N2O3 by NO and NO2 formed from reaction with NH3.
Kijlstra et al.115 proved the existence of L–H and E–R mechanisms immediately via some experiments. The authors supposed a low temperature SCR reaction model of the MnOx/Al2O3 catalyst below 230 °C, as in eqn (27–34). They thought that the mechanisms of L–H and E–R happened in parallel, but the formation of N2 was most attributed to the E–R mechanism. This reaction model was supported and proved by subsequent studies.
| | O2 + 2* ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) 2O–* 2O–* | (27) |
| | | NH3 + * NH3–* (Lewis acid Mn3+ site) K3 = K3/K−3 | (28) |
| | | NO + O–* NO–O–* bridged and monodentate nitrites K4 | (29) |
| | | NO–O–* + O–* → NO3–* + * bidentate nitrate K5 | (30) |
| | | NO3–* + O–* → NO2–* + OH–* K6 | (31) |
| | | NO2–* + NO → N2 + H2O + * ER mechanism K7 | (32) |
| | | NO2–* + NO–O–* → N2 + H2O + O–* + * LH mechanism K8 | (33) |
| | | 2OH–* H2O + O–* + * K9 | (34) |
They deduced the kinetic equation in eqn (35) based on the mechanism of L–H and E–R.131
| | 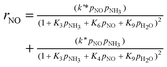 | (35) |
Where
The researchers131 indicated that the reaction order of NO is less than 1, which can be explained by the fact that the NO joining the SCR reaction in a form other than gaseous, may be adsorbed on the catalyst. It means both E–R and L–H mechanisms existed. But if just the E–R mechanism exists, it can also be explained that there exists a rate-controlling step—the activation of NH3. Or maybe a composite exists that is formed from NO and NH3. Further research should be done on this point.
6 Conclusions and perspective
The supported catalysts are crucial in the removal of NOx from the flue gas of power plants and diesel engines at low temperature. A lot of supported catalysts have been researched in the past decade. The catalysts using Mn–Ce as the active sites have a better effect than other metals, with ACF, Al2O3, TiO2, zeolites and SAPO as the carriers or without carriers for SCR of NOx at low temperature. But the high selectivity, stability and resistance of H2O and SO2, alkali and heavy metals require further improvement. Maybe it can be achieved with catalysts by compounding the transition metals and doping rare earth elements into appropriate carriers, such as ACF, γ-Al2O3 and so on. At the same time, due to the development of analytical methods and analog computation, the synergistic effect of supported catalysts is a hot topic in recent years.
Some research has been done to study the mechanism. The phenomena of the E–R, L–H mechanisms or both of them coexisting in the progress of the removal NOx reaction is the main theory. But there are some debates in the research of the reaction order of NO. Different reaction orders will give different analysis results. Some models to study the kinetics of the SCR NOx have been established at low temperature. The main point of the kinetics research is based on empirical equations (as power-law kinetics) and obtained from mechanistic analysis or both of them. Although de-NOx by SCR has been studied for a long time, the low-temperature efficiency, especially practical application efficiency, was undesirable. Besides, the mechanism was also not very clear and cannot effectively control the poisoning of the catalyst. The catalyst was limited for wide applications and cannot lower the huge cost. According to the exploration of the surface chemical process, the mechanism of the de-NOx can be proved, and the root cause of the poisoning and deactivation of the catalyst can be found. Therefore, the surface chemical process is a very worthy subject to study in-depth.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51108169, 51278177), the National High Technology Research and Development Program of China (863 Program, no. 2011AA060803), the Scientific and Technological Major Special Project of Hunan Province in China (no. 2010XK6003), National Undergraduate Innovation Experiment Project (no. 531107061105). All of the authors thank Mr. Zheng Zeng in Arizona State University for his helpful discussion and the direction on the writing of this paper.
References
- J. Li, H. Chang, L. Ma, J. Hao and R. T. Yang, Catal. Today, 2011, 175, 147–156 CrossRef CAS PubMed.
- J. Zhu and A. Thomas, Appl. Catal., B, 2009, 92, 225–233 CrossRef CAS PubMed.
- G. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS PubMed.
- W. Yang, J. Zhou, Z. Zhou, Z. Lu, Z. Wang, J. Liu and K. Cen, Fuel Process. Technol., 2008, 89, 1317–1323 CrossRef CAS PubMed.
- Z. Liang, X. Ma, H. Lin and Y. Tang, Appl. Energy, 2011, 88, 1120–1129 CrossRef CAS PubMed.
- W. Chen and R. Xu, Energy Policy, 2010, 38, 2123–2130 CrossRef PubMed.
- Y. Zhao, L. Duan, T. Larssen, L. Hu and J. Hao, Environ. Sci. Technol., 2007, 41, 1815–1820 CrossRef CAS.
- G. Qi and R. T. Yang, Appl. Catal., B, 2003, 44, 217–225 CrossRef CAS.
- Q. Tang, M. Denison, B. Adams and D. Brown, Proc. Combust. Inst., 2009, 32, 2649–2657 CrossRef CAS PubMed.
- P. Marín, D. Fissore, A. A. Barresi and S. Ordóñez, Chem. Eng. Process.: Process Intensif., 2009, 48, 311–320 CrossRef PubMed.
- A. T. Evulet, A. M. Elkady, A. R. Branda and D. Chinn, Energy Procedia, 2009, 1, 3809–3816 CrossRef CAS PubMed.
- C. Casaca and M. Costa, Proc. Combust. Inst., 2009, 32, 2641–2648 CrossRef CAS PubMed.
- R. Leng, C. Wang, C. Zhang, D. Dai and G. Pu, J. Cleaner Prod., 2008, 16, 374–384 CrossRef PubMed.
- M. Radojevic, Environ. Pollut., 1998, 102, 685–689 CrossRef CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- Z. Ma, H. Yang, Q. Li, J. Zheng and X. Zhang, Appl. Catal., A, 2012, 427–428, 43–48 CrossRef CAS PubMed.
- R. M. Heck, Catal. Today, 1999, 53, 519–523 CrossRef CAS.
- B. Huang, R. Huang, D. Jin and D. Ye, Catal. Today, 2007, 126, 279–283 CrossRef CAS PubMed.
- M. Stanciulescu, G. Caravaggio, A. Dobri, J. Moir, R. Burich, J. P. Charland and P. Bulsink, Appl. Catal., B, 2012, 123–124, 229–240 CrossRef CAS PubMed.
- Y. Shen, D. Zheng, B. Yang, S. Ni and S. Zhu, J. Rare Earths, 2012, 30, 431–436 CrossRef CAS.
- S. Yang, J. Li, C. Wang, J. Chen, L. Ma, H. Chang, L. Chen, Y. Peng and N. Yan, Appl. Catal., B, 2012, 117, 73–80 CrossRef PubMed.
- M. Kang, E. D. Park, J. M. Kim and J. E. Yie, Catal. Today, 2006, 111, 236–241 CrossRef CAS PubMed.
- Z. R. Ma, D. Weng, X. D. Wu, Z. C. Si and B. Wang, Catal. Commun., 2012, 27, 97–100 CrossRef CAS PubMed.
- S. M. Lee, K. H. Park and S. C. Hong, Chem. Eng. J., 2012, 195–196, 323–331 CrossRef CAS PubMed.
- P. Lu, Z. Zeng, C. Li, G. Zeng, J. Guo, X. Jiang, Y. Zhai and X. Fan, Environ. Technol., 2012, 33, 1029–1036 CrossRef CAS.
- X. Fan, C. Li, G. Zeng, X. Zhang, S. Tao, P. Lu, S. Li and Y. Zhao, Fuel Process. Technol., 2012, 104, 325–331 CrossRef CAS PubMed.
- L. Zhu, B. Huang, W. Wang, Z. Wei and D. Ye, Catal. Commun., 2011, 12, 394–398 CrossRef CAS PubMed.
- P. Lu, C. Li, G. Zeng, L. He, D. Peng, H. Cui, S. Li and Y. Zhai, Appl. Catal., B, 2010, 96, 157–161 CrossRef CAS PubMed.
- M. Iwasaki, K. Yamazaki, K. Banno and H. Shinjoh, J. Catal., 2008, 260, 205–216 CrossRef CAS PubMed.
- J. Park, H. Park, J. Baik, I. Nam, C. Shin, J. Lee, B. Cho and S. Oh, J. Catal., 2006, 240, 47–57 CrossRef CAS PubMed.
- Y. Zhao, P. Lu, C. Li, X. Fan, Q. Wen, Q. Zhan, X. Shu, T. Xu and G. Zeng, Environ. Technol., 2013, 34, 201–207 CrossRef CAS.
- P. Lu, C. Li, G. Zeng, Y. Zhao, Q. Zhan, J. Song and X. Fan, Environ. Technol., 2013, 34, 101–111 CrossRef CAS.
- Z. Zeng, P. Lu, C. Li, G. Zeng, X. Jiang, Y. Zhai and X. Fan, Environ. Technol., 2012, 33, 1331–1337 CrossRef CAS.
- Z. Zeng, P. Lu, C. Li, G. Zeng and X. Jiang, J. Coord. Chem., 2012, 65, 1992–1998 CrossRef CAS.
- S. Tao, C. Li, X. Fan, G. Zeng, P. Lu, X. Zhang, Q. Wen, W. Zhao, D. Luo and C. Fan, Chem. Eng. J., 2012, 210, 547–556 CrossRef CAS PubMed.
- X. Jiang, P. Lu, C. Li, Z. Zeng, G. Zeng, L. Hu, L. Mai and Z. Li, Environ. Technol., 2013, 34, 591–598 CrossRef CAS.
- Z. Zeng, P. Lu, C. Li, L. Mai, Z. Li and Y. Zhang, Catal. Sci. Technol., 2012, 2, 2188 CAS.
- M. A. Gomez-Garcia, V. Pitchon and A. Kiennemann, Environ. Int., 2005, 31, 445–467 CrossRef CAS PubMed.
- Q. Zhang, C. Qiu, H. Xu, T. Lin, M. Gong and Y. Chen, Catal. Commun., 2011, 16, 20–24 CrossRef CAS PubMed.
- Z. Chen, Q. Yang, H. Li, X. Li, L. Wang and S. Chi Tsang, J. Catal., 2010, 276, 56–65 CrossRef CAS PubMed.
- Z. Chen, X. Li, X. Gao, Y. Jiang, Y. Lü, F. Wang and L. Wang, Chin. J. Catal., 2009, 30, 4–6 CrossRef.
- X. Tang, J. Li, L. Wei and J. Hao, Chin. J. Catal., 2008, 29, 531–536 CrossRef CAS.
- G. Qi and R. T. Yang, J. Catal., 2003, 217, 434–441 CAS.
- H. Chang, J. Li, X. Chen, L. Ma, S. Yang, J. W. Schwank and J. Hao, Catal. Commun., 2012, 27, 54–57 CrossRef CAS PubMed.
- M. Casapu, O. Kröcher and M. Elsener, Appl. Catal., B, 2009, 88, 413–419 CrossRef CAS PubMed.
- M. Yoshikawa, A. Yasutake, I. Mochida and E. Cabrera, Appl. Catal., A, 1998, 173, 239–245 CrossRef CAS.
- I. Mochida, Y. Korai, M. Shirahama, S. Kawano, T. Hada, Y. Seo, M. Yoshikawa and A. Yasutake, Carbon, 2000, 38, 227–239 CrossRef CAS.
- I. Mochida, Y. Kawabuchi, S. Kawano, Y. Matsumura and M. Yoshikawa, Fuel, 1997, 76, 543–548 CrossRef CAS.
- S. Adapa, V. Gaur and N. Verma, Chem. Eng. J., 2006, 116, 25–37 CrossRef CAS PubMed.
- H. Huang, D. Ye, B. Huang and Z. Wei, Catal. Today, 2008, 139, 100–108 CrossRef CAS PubMed.
- S. J. Park, G. H. Shim and H. Y. Kim, J. Colloid Interface Sci., 2005, 291, 585–587 CrossRef CAS PubMed.
- R. Burch and T. C. Watling, Appl. Catal., B, 1997, 11, 207–216 CrossRef CAS.
- Q. Zhang, C. Qiu, H. Xu, T. Lin, Z. Lin, M. Gong and Y. Chen, Catal. Today, 2011, 175, 171–176 CrossRef CAS PubMed.
- E. Seker, J. Cavataio, E. Gulari, P. Lorpongpaiboon and S. Osuwan, Appl. Catal., A, 1999, 183, 121–134 CrossRef CAS.
- R. Lanza, E. Eriksson and L. J. Pettersson, Catal. Today, 2009, 147, S279–S284 CrossRef CAS PubMed.
- D. Creaser, H. Kannisto, J. Sjöblom and H. H. Ingelsten, Appl. Catal., B, 2009, 90, 18–28 CrossRef CAS PubMed.
- C. Guillard, B. E. C. Dutriez, J.-M. Herrmann, H. Jaffrezic, N. Jaffrezic-Renault and M. Lacroix, Appl. Catal., B, 2002, 39, 331–342 CrossRef CAS.
- L. Lietti, J. L. Alemany, P. Forzatti, G. Busca, G. Ramis, E. Giamello and F. Bregani, Catal. Today, 1996, 29, 143–148 CrossRef CAS.
- L. J. Alemany, L. Lietti, N. Ferlazzo, P. Forzatti, G. Busca, E. Giamello and F. Bregani, J. Catal., 1995, 155, 117–130 CrossRef CAS.
- R. Gao, D. Zhang, X. Liu, L. Shi, P. Maitarad, H. Li, J. Zhang and W. Cao, Catal. Sci. Technol., 2013, 3, 191 CAS.
- Z. Wu, B. Jiang, Y. Liu, W. Zhao and B. Guan, J. Hazard. Mater., 2007, 145, 488–494 CrossRef CAS PubMed.
- Z. Wu, B. Jiang and Y. Liu, Appl. Catal., B, 2008, 79, 347–355 CrossRef CAS PubMed.
- Z. Wu, R. Jin, Y. Liu and H. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS PubMed.
- R. Jin, Y. Liu, Z. Wu, H. Wang and T. Gu, Catal. Today, 2010, 153, 84–89 CrossRef CAS PubMed.
- B. Shen, T. Liu, N. Zhao, X. Yang and L. Deng, J. Environ. Sci., 2010, 22, 1447–1454 CrossRef CAS.
- S. Guerrero, I. Guzmán, G. Aguila and P. Araya, Catal. Commun., 2009, 11, 38–42 CrossRef CAS PubMed.
- Z. Zhang, M. Chen, Z. Jiang and W. Shangguan, J. Environ. Sci., 2010, 22, 1441–1446 CrossRef CAS.
- J. Li, G. Wu, N. Guan and L. Li, Catal. Commun., 2012, 24, 38–43 CrossRef CAS PubMed.
- B. Jiang, Y. Liu and Z. Wu, J. Hazard. Mater., 2009, 162, 1249–1254 CrossRef CAS PubMed.
- U. De La Torre, B. Pereda-Ayo and J. R. González-Velasco, Chem. Eng. J., 2012, 207–208, 10–17 CrossRef CAS PubMed.
- K. Rahkamaa-Tolonen, T. Maunula, M. Lomma, M. Huuhtanen and R. L. Keiski, Catal. Today, 2005, 100, 217–222 CrossRef CAS PubMed.
- K. C. C. Kharas, D.-J. Liu and H. J. Robota, Catal. Today, 1995, 26, 129–145 CrossRef CAS.
- C.-K. Seo, B. Choi, H. Kim, C.-H. Lee and C.-B. Lee, Chem. Eng. J., 2012, 191, 331–340 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, Y. Xu, Y. Chen and X. Li, Chin. J. Catal., 2010, 31, 1233–1241 CrossRef CAS.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev., 2008, 50, 492–531 CAS.
- X. Chang, G. Lu, Y. Guo, Y. Wang and Y. Guo, Microporous Mesoporous Mater., 2013, 165, 113–120 CrossRef CAS PubMed.
- L. Wang, W. Li, G. Qi and D. Weng, J. Catal., 2012, 289, 21–29 CrossRef CAS PubMed.
- R. Martínez-Franco, M. Moliner, C. Franch, A. Kustov and A. Corma, Appl. Catal., B, 2012, 127, 273–280 CrossRef PubMed.
- C. Kladis, S. K. Bhargava and D. B. Akolekar, J. Mol. Catal. A: Chem., 2003, 203, 193–202 CrossRef CAS.
- T. Ishihara, M. Kagawa, F. Hadama and Y. Takita, J. Catal., 1997, 169, 93–102 CrossRef CAS.
- A. Frache, B. I. Palella, M. Cadoni, R. Pirone, H. O. Pastore and L. Marchese, Top. Catal., 2003, 22, 53–57 CrossRef CAS.
- D. W. Fickel, E. D'Addio, J. A. Lauterbach and R. F. Lobo, Appl. Catal., B, 2011, 102, 441–448 CrossRef CAS PubMed.
- J. Wang, T. Yu, X. Wang, G. Qi, J. Xue, M. Shen and W. Li, Appl. Catal., B, 2012, 127, 137–147 CrossRef CAS PubMed.
- G. Qi and R. T. Yang, J. Phys. Chem. B, 2004, 108, 15738–15747 CrossRef CAS.
- W. S. K. D. S. B. H. I. S. E. K. Poels and A. Bliek, J. Catal., 1997, 171, 219–230 CrossRef.
- Y. Zhang, X. Wang, K. Shen, H. Xu, K. Sun and C. Zhou, Chin. J. Catal., 2012, 33, 1523–1531 CrossRef CAS.
- K. Hadjiivanov, D. Klissurski, G. Ramis and G. Busca, Appl. Catal., B, 1996, 7, 251–267 CrossRef CAS.
- M. E. Gálvez, M. J. Lázaro and R. Moliner, Catal. Today, 2005, 102–103, 142–147 CrossRef PubMed.
- Z. Huang, Z. Zhu and Z. Liu, Appl. Catal., B, 2002, 39, 361–368 CrossRef CAS.
- A. Kling, C. Andersson, A. Myringer, D. Eskilsson and S. Jaras, Appl. Catal., B, 2007, 69, 240–251 CrossRef CAS PubMed.
- W. Yu, X. Wu, Z. Si and D. Weng, Appl. Surf. Sci., 2013, 283, 209–214 CrossRef CAS PubMed.
- L. Chen, J. Li and M. Ge, Chem. Eng. J., 2011, 170, 531–537 CrossRef CAS PubMed.
- A.-C. Larsson, J. Einvall, A. Andersson and M. Sanati, Energy Fuels, 2006, 20, 1398–1405 CrossRef CAS.
- X. Lixian, S. Boxiong, Y. Xiaoyan, D. Lidan and Z. Ning, Environ. Chem., 2010, 29, 690–685 Search PubMed.
- Z. Chongbing, J. Baosheng, Z. Zhaoping, L. Feng and Z. Junxia, Environ. Chem., 2007, 26, 783–786 Search PubMed.
- S. Boxiong, Y. Yan, C. Jianhong and Z. Xiaopeng, Microporous Mesoporous Mater., 2013, 180, 262–269 CrossRef PubMed.
- L. Schill, S. S. R. Putluru, C. F. Jacobsen, C. H. Hansen, R. Fehrmann and A. D. Jensen, Appl. Catal., B, 2012, 127, 323–329 CrossRef CAS PubMed.
- L. Lisi, G. Lasorella, S. Malloggi and G. Russo, Appl. Catal., B, 2004, 50, 251–258 CrossRef CAS PubMed.
- S. S. R. Putluru, A. Riisager and R. Fehrmann, Appl. Catal., B, 2011, 101, 183–188 CrossRef CAS PubMed.
- A. L. Kustov, M. Y. Kustova, R. Fehrmann and P. Simonsen, Appl. Catal., B, 2005, 58, 97–104 CrossRef CAS PubMed.
- F.-Y. Chang, J.-C. Chen and M.-Y. Wey, Fuel Process. Technol., 2009, 90, 576–582 CrossRef CAS PubMed.
- M. López Granados, C. Larese, F. Cabello Galisteo, R. Mariscal, J. L. G. Fierro, R. Fernández-Ruíz, R. Sanguino and M. Luna, Catal. Today, 2005, 107–108, 77–85 CrossRef PubMed.
- J.-C. Chen, M.-Y. Wey, C.-L. Yeh and Y.-S. Liang, Appl. Catal., B, 2004, 48, 25–35 CrossRef CAS PubMed.
- C. Larese, Appl. Catal., B, 2003, 40, 305–317 CrossRef CAS.
- G. Qi, R. T. Yang and L. T. Thompson, Appl. Catal., A, 2004, 259, 261–267 CrossRef CAS PubMed.
- C. Costa, J. Catal., 2001, 197, 350–364 CrossRef CAS.
- L. F. Davide Ferri, M. A. P. Dekkers and B. E. Nieuwenhuys, Appl. Catal., B, 1998, 16, 339–345 CrossRef.
- X. Wang, H. Y. Chen and W. M. H. Sachtler, J. Catal., 2001, 197, 281–291 CrossRef CAS.
- M. D. Fokema and J. Y. Ying, J. Catal., 2000, 192, 54–63 CrossRef CAS.
- T. Liese, E. Loffler and W. Grunert, J. Catal., 2001, 197, 123–130 CrossRef CAS.
- A. W. Aylor, L. J. Lobree, J. A. Reimer and A. T. Bell, J. Catal., 1997, 170, 390–401 CrossRef CAS.
- L. J. Lobree, I.-C. Hwang, J. A. Reimer and A. T. Bell, Catal. Lett., 1999, 63, 233–240 CrossRef CAS.
- A. Satsuma and K.-I. Shimizu, Prog. Energy Combust. Sci., 2003, 29, 71–84 CrossRef CAS.
- Y. Yunbo and H. Hong, Chin. J. Catal., 2003, 24, 385–390 Search PubMed.
- W. S. Kijlstra, D. S. Brands, H. I. Smit, E. K. Poels and A. Bliek, J. Catal., 1997, 171, 219–230 CrossRef CAS.
- W. S. Kijlstra, D. S. Brands, E. K. Poels and A. Bliek, J. Catal., 1997, 171, 208–218 CrossRef CAS.
- F. Eigenmann, M. Maciejewski and A. Baiker, Appl. Catal., B, 2006, 62, 311–318 CrossRef CAS PubMed.
- G. Marban, J. Catal., 2004, 226, 138–155 CrossRef CAS PubMed.
- M. Richter, A. Trunschke, U. Bentrup, K. W. Brzezinka, E. Schreier, M. Schneider, M. M. Pohl and R. Fricke, J. Catal., 2002, 206, 98–113 CrossRef CAS.
- T. V. Voskoboinikov, H.-Y. Chen and W. M. H. Sachtler, Appl. Catal., B Environ., 1998, 19, 279–287 CrossRef CAS.
- M. Schwidder, M. Kumar, K. Klementiev, M. Pohl, A. Bruckner and W. Grunert, J. Catal., 2005, 231, 314–330 CrossRef CAS PubMed.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Appl. Catal., B, 2010, 95, 348–357 CrossRef CAS PubMed.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Appl. Catal., A, 2010, 373, 168–175 CrossRef CAS PubMed.
- J. Eng and C. H. Bartholomew, J. Catal., 1997, 171, 27–44 CrossRef CAS.
- R. Long, J. Catal., 2002, 207, 224–231 CrossRef CAS.
- A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, J. Catal., 2008, 256, 312–322 CrossRef CAS PubMed.
- A. Grossale, I. Nova and E. Tronconi, J. Catal., 2009, 265, 141–147 CrossRef CAS PubMed.
- G. Qi and R. T. Yang, J. Catal., 2003, 217, 434–441 CAS.
- Z. B. Wu, B. Q. Jiang, Y. Liu, W. R. Zhao and B. H. Guan, J. Hazard. Mater., 2007, 145, 488–494 CrossRef CAS PubMed.
- M. Richter, A. Trunschke, U. Bentrup, K.-W. Brzezinka, E. Schreier, M. Schneider, M.-M. Pohl and R. Fricke, J. Catal., 2002, 206, 98–113 CrossRef CAS.
- W. S. Kijlstra, D. S. Brands, E. K. Poels and A. Bliek, Catal. Today, 1999, 50, 133–140 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.