Highly dispersed CeO2 on carbon nanotubes for selective catalytic reduction of NO with NH3†
Cheng
Fang
abc,
Dengsong
Zhang
*ab,
Liyi
Shi
ac,
Ruihua
Gao
a,
Hongrui
Li
a,
Liping
Ye
d and
Jianping
Zhang
b
aResearch Center of Nano Science and Technology, Shanghai University, Shanghai 200444, China. E-mail: dszhang@shu.edu.cn; Fax: +86 21 66134852
bDepartment of Chemistry, Shanghai University, Shanghai 200444, China
cSchool of Material Science and Engineering, Shanghai University, Shanghai 200072, China
dShanghai Research Institute of Chemical Industry, Shanghai 200062, China
First published on 12th November 2012
Abstract
Highly dispersed CeO2 on carbon nanotubes (CNTs) is successfully prepared by a pyridine-thermal route for selective catalytic reduction (SCR) of NO with NH3. This catalyst is mainly characterized by the techniques of X-ray diffraction (XRD), transmission electron microscopy (TEM), temperature-programmed reduction by hydrogen (H2-TPR), temperature-programmed desorption of ammonia (NH3-TPD) and X-ray photoelectron spectroscopy (XPS). The results of the XRD, TEM and TPR analysis show that the CeO2 particles on the CNTs are highly dispersed with a strong interaction between the particles and the CNTs. The NH3-TPD profiles indicate that this catalyst exhibits abundant strong acid sites. Furthermore, the O 1s XPS spectra show that the Oα/(Oα + Oβ) ratio of this catalyst is very high, which can result in more surface oxygen vacancies and therefore favor the NH3-SCR reaction. Compared with the catalysts prepared by impregnation or physical mixture methods, the catalyst prepared by the pyridine-thermal route presents the best NH3-SCR activity in the temperature range of 150–380 °C as well as favourable stability and good SO2 or H2O resistance. More than 90% of NO can be removed in the range of 250–370 °C with a desirable N2 selectivity. Moreover, the NO conversion can be kept at about 97% with the presence of SO2 or H2O at 300 °C. In addition, this catalyst shows a high catalytic activity with a NO conversion remaining constant at ca. 98% during a 16 h continuous run duration at 300 °C. Highly dispersed CeO2 on the CNTs as well as the strong interaction between the particles and the CNTs, the large amounts of strong acid sites and the high Oα/(Oα + Oβ) ratio could be ascribed to the excellent NH3-SCR performance of the catalyst prepared by the pyridine-thermal route.
1. Introduction
Nitrogen oxides (NOx) from the combustion of fossil fuels are very harmful for the ecosystem and humanity because they can induce acid rain, photochemical smog, the greenhouse effect and ozone depletion. The selective catalytic reduction (SCR) of nitrogen oxides (NOx) with ammonia is a very effective technology for eliminating NOx in the flue gas from stationary sources and motor vehicles.1,2 A V-based catalyst is usually used for NH3-SCR of NOx and V2O5–WO3 (MoO3)/TiO2, when used as a commercial catalyst, exhibits high catalytic activity and selectivity.1,3,4 However, there are still some inevitable problems, such as the high working temperature, the toxicity of vanadium species,5 the high activity for the oxidation of SO2 to SO36 and the low N2 selectivity in a high temperature range.7 Therefore, it is important to develop vanadium-free SCR catalysts with a relatively low working temperature.8,9In the past few years, the unique one-dimensional tubular structure, accessible surface and mechanical characteristics make carbon nanotubes (CNTs) a promising candidate for wide applications in catalysis.10–13 It is reported that CNTs are good sorbents of nitric oxides, ammonia and other gas molecules.14–16 Moreover, the CNTs show abilities for decomposing and reducing NO.17–19 Recently, CNTs have been of interest as a support material for application in the SCR process.19–25 It has been reported that the addition of CNTs could reduce the decomposition temperature of sulfates and bisulfates.21,24 Bai et al.21 found that the formation and accumulation of excess ammonium sulfates can be avoided because NH4HSO4 on the surface of the V2O5/CNT catalyst can react with NO continuously below 250 °C, which indicates that SO2 can promote the catalytic effect at low temperature in the presence of CNTs. Furthermore, Santillan-Jimenez et al.19 reported that H2O did not show any obvious inhibition of the catalytic activity of HC-SCR catalysts when using functionalized multi-walled CNTs as the support. These studies indicate that CNTs are ideal candidates for supports of de-NOx catalysts.
Ceria has been recently studied for its excellent oxygen storage capacity and redox properties via the shift between Ce4+ and Ce3+ under oxidizing and reducing conditions, respectively.26,27 Because of these characteristics, CeO2 could strengthen the ability of oxidizing NO to NO2, resulting in an increase in the SCR activity.28,29 Thus, CeO2 has often been used as an active component to enhance the activity in NH3-SCR.30–34 In previous studies, CeO2 based catalysts for NH3-SCR were mainly prepared by an impregnation method32,35 and other methods, such as coprecipitation and sol–gel routes, have also been reported by several researchers.32 As we know, the synthesis method is a key factor to determine the dispersion of the active components, which could greatly influence the catalytic activity of the SCR catalysts.36 However, it is hard to ensure that CeO2 is uniformly dispersed on the surface of the support in the above mentioned methods. Thus, it still remains a challenge to synthesize SCR catalysts with active components uniformly dispersed.
Considering the advantages of CNTs and CeO2, it is hopeful to obtain good SCR catalysts with CeO2 uniformly dispersed on CNTs. Recently, Chen et al.22 prepared cerium oxide supported on nitric acid-treated CNTs by an impregnation method and a NO conversion of more than 70% could be obtained in a medium temperature range (250–400 °C). Fan et al.37 synthesized Mn–Ce–Ox/TiO2-CNTs by a sol–gel method for NH3-SCR of NO. However, the dispersion of the active components obtained was not desirable, which leaves a lot of room to improve the de-NOx performance. To the best of our knowledge, the in situ synthesis of highly dispersed CeO2 on CNTs has not yet been reported for SCR of NO with NH3.
Previously, we have developed various methods to prepare highly dispersed nanoparticles on CNTs.38–40 Recently, we found that pyridine can be employed to enhance the dispersion of CNTs in various mediums and the obtained suspensions remain stable for a long time. The pyridine molecules are absorbed easily onto the CNTs due to the π–π conjugate role of accepting a proton from the water molecular preferentially to produce OH− around the CNTs. Subsequently, the metal ions in situ react with OH− and the resultant nanoparticles deposit on the surface of the CNTs. Finally, uniformly dispersed metal oxide on CNTs was obtained. Herein, we introduce a pyridine-thermal route to in situ highly disperse CeO2 on CNTs and discuss the performance for NH3-SCR of NO. A comparison of the activity and characterization of the catalysts prepared by impregnation and physical mixture methods were also carried out. It is found that highly dispersed CeO2 on CNTs prepared by a pyridine-thermal route exhibits the best activity for NH3-SCR of NO and excellent resistance to H2O or SO2 poisoning as well as good stability.
2. Experimental
2.1 Catalyst preparation
Multi-wall CNTs were purchased from Qinhuangdao Tai Chi Ring Nano Product Co. Ltd (China) and were 10–30 nm in diameter and 1–10 μm in length. The CNTs were first purified by refluxing in 65% nitric acid at 140 °C for 6 h. After the treatment, the CNTs were separated by filtration, washed fully with deionized water until the pH value reached 6–7 and dried at 100 °C in air overnight. All the other chemicals were purchased from Sinopharm Chemical Regent Company and used without further purification.0.03 g of Ce(NO3)3·6H2O and 0.24 g of purified CNTs were mixed and dispersed in 80 mL of pyridine under ultrasonication for 30 min. The solution was then transferred into a 100 mL PTFE autoclave and subsequently placed in an oven and maintained at 180 °C for 24 h. After the autoclave was naturally cooled to ambient temperature, the precipitates were collected and washed several times with deionized water and absolute ethanol and finally dried at 100 °C overnight.
For comparison, the catalyst was also made by impregnation and physical mixture methods. The impregnation procedure is as follows: 0.03 g of Ce(NO3)3·6H2O was dissolved in 5 mL of deionized water and added to a beaker containing a certain amount of CNTs. The mixture was ultrasonicated for 1 h and then dried at 100 °C for 24 h. In the physical mixture procedure, pure CeO2 nanopaticles, prepared according to the literature,41 and purified CNTs were physically mixed, fully grinded and then dried at 100 °C for 12 h.
The mass percentages of CeO2 in all the samples were selected as 5%. All the as-synthesized catalysts were calcined at 500 °C for 4 h in a nitrogen stream. These samples were labelled as CeO2/CNTs-PT, CeO2/CNTs-IM and CeO2/CNTs-PM, which denotes the catalysts prepared by the pyridine-thermal route, the impregnation and physical mixture methods, respectively. CeO2/CNTs-PT catalysts with a CeO2 amount of 3% and 10% were also prepared to further investigate the effect of ceria loading on the structural properties and catalytic performance of CeO2/CNTs-PT.
2.2 Characterization
Powder X-ray diffraction (XRD) was performed with a Rigaku D/MAX-2200 X-ray diffractometer using Cu–Kα (40 kV, 40 mA) radiation and a secondary beam graphite monochromator. The morphologies were observed by a transmission electron microscope (TEM, JEOL JEM-200CX) and a field emission high resolution transmission electron microscope (HRTEM, JEOL JEM-2100F). Nitrogen adsorption–desorption isotherms of the samples were measured at 77 K using an ASAP 2020 volumetric adsorption analyzer. Before the measurements, all the samples were degassed overnight at 573 K in a vacuum line. The specific surface area and the pore volume of the samples were calculated by the Brunauer–Emmett–Teller (BET) method and the pore size distributions were derived from the adsorption branches of the isotherms using the Barrett–Joyneer–Halenda (BJH) model. X-ray photoelectron spectroscopy (XPS) was recorded on a Perkin–Elmer PHI 5000C ESCA system equipped with a dual X-ray source, using a Mg–Kα (1253.6 eV) anode and a hemispherical energy analyzer. The background pressure during data acquisition was kept below 10−6 Pa. All the binding energies were calibrated using contaminant carbon (C 1s = 284.6 eV) as a reference.Temperature-programmed reduction by hydrogen (H2-TPR) was obtained on a FineSorb-3010D apparatus. 50 mg of the calcined catalyst was outgassed at 300 °C under an Ar flow. After cooling to room temperature under an Ar flow, the flowing gas was switched to 10% H2/Ar and the sample was heated to 750 °C at a ramping rate of 10 °C min−1. The H2 consumption was monitored by a thermal conductivity detector (TCD). Temperature-programmed desorption experiments of ammonia (NH3-TPD) were conducted on a Tianjin XQ TP5080 auto-adsorption apparatus. Before the TPD, each sample was pretreated with high-purity (99.999%) N2 (30 mL min−1) at 300 °C for 0.5 h and then saturated with high-purity anhydrous ammonia at 100 °C for 1 h and subsequently flushed at the same temperature for 1 h to remove physisorbed ammonium. Finally, the TPD operation was carried out from 100 to 750 °C at a heating rate of 10 °C min−1. The amount of NH3 desorbed was monitored by a TCD.
2.3 Catalytic activity measurements
The NH3-SCR activity measurement was carried out in a fixed-bed quartz micro-reactor operating in a steady state flow mode. 0.2 g of the catalysts were sieved with a 20–40 mesh and used in each test. The reactant gas composition was typically: 500 ppm NO, 500 ppm NH3, 3 vol% O2, 100 ppm SO2 (when used), 4 vol% H2O (when used) and balance N2. The total flow rate was 250 mL min−1 and thus a GHSV of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 was obtained. The temperature increased from 150 to 380 °C. At each temperature step the data were recorded when the SCR reaction reached steady state after 15 min. The concentration of NO in the inlet and outlet gas was measured by a KM9106 flue gas analyzer. The concentrations of N2O and NH3 were measured by a Transmitter IR N2O analyzer and IQ350 ammonia analyzer.
000 h−1 was obtained. The temperature increased from 150 to 380 °C. At each temperature step the data were recorded when the SCR reaction reached steady state after 15 min. The concentration of NO in the inlet and outlet gas was measured by a KM9106 flue gas analyzer. The concentrations of N2O and NH3 were measured by a Transmitter IR N2O analyzer and IQ350 ammonia analyzer.
A relative turnover frequency (TOF) value was employed to compare the activities of the different catalysts.42 The relative TOF (s−1) of NO over each Ce atom was calculated by the following equation:
 | (1) |
3. Results and discussion
3.1. Characteristics
Fig. 1 shows the XRD patterns of the catalysts. All the catalysts exhibit three characteristic diffraction peaks from the CNTs, which can be ascribed to the (002), (101) and (004) reflections, as reported.22 For CeO2/CNTs-PT, only the characteristic lines of the CNTs can be observed while small diffraction peaks corresponding to (111), (200), (220), (311) of CeO2 (PDF-ICDD 34-0394) appear in the CeO2/CNTs-IM and CeO2/CNTs-PM spectra, indicating the presence of a CeO2 phase with larger particle sizes in these two catalysts than in CeO2/CNTs-PT. It also suggests that the CeO2 nanoparticles disperse well on the surface of the CNTs in the catalyst prepared by the pyridine-thermal route. The XRD results confirm that the preparation method is a key factor for the dispersion of the active components.32,43 | ||
| Fig. 1 XRD patterns of the catalysts. | ||
TEM was also performed to characterize the microstructures of the samples. Fig. 2 shows the TEM and HRTEM micrographs of different catalysts as well as their ceria particle size distribution histograms. Compared with the purified CNTs (Fig. S1, ESI†), it is clear that the CeO2 particles distribute uniformly on the surface of the CNTs over CeO2/CNTs-PT, while obvious CeO2 agglomerates are observed for CeO2/CNTs-IM and CeO2/CNTs-PM. These observations are in agreement with Fig. 1. As shown in Fig. 2(b), the distinct lattice plane of the CeO2 nanoparticles on CeO2/CNTs-PT corresponds to the fluorite cubic structure of CeO2, which can be attributed to the identified {111} facet with an interplanar distance of 0.31 nm. Other characteristic facets of CeO2 are also found in Fig. S2 (ESI†).41,44 However, the XRD profile of CeO2/CNTs-PT does not show the characteristic diffraction peaks of CeO2, which could be explained by the fact that the CeO2 nanoparticles on the CNTs were too small to be identified by XRD. This also suggests that CeO2 is highly dispersed on the surface of the CNTs. The EDS spectrum of CeO2/CNTs-PT is also shown in Fig. 2. The EDS spectrum of the region circled in red reveals that the sample contains elemental Ce and O, which implies that the nanoparticle is a kind of cerium oxide. Both the XRD and EDS results confirm the formation of the CeO2/CNTs composites. Fig. 2(f) and (g) shows the ceria particle size distribution of CeO2/CNTs-PT and CeO2/CNTs-IM. Clearly, CeO2/CNTs-PT possesses smaller ceria particles and a narrower ceria particle diameter distribution than CeO2/CNTs-IM. It is hard to provide a ceria particle size distribution histogram of CeO2/CNTs-PM due to the presence of obvious ceria agglomerates. Furthermore, we also observed the presence of some ceria particles inside the central channel of the CNTs for CeO2/CNTs-PT as well as a smaller amount of ceria particles for CeO2/CNTs-IM. It has been demonstrated that confinement inside the CNTs can influence the catalytic activity in redox reactions.45 Thus, the confinement effect could also explain the improved catalytic performance of the CeO2/CNTs composites. Additionally, it is found that the interaction between the CeO2 nanoparticles and CNTs in CeO2/CNTs-PT is more stable than in the other two catalysts, which has been confirmed by the fact that the nanoparticles could not be removed under intensive sonication. Generally, as for a metal oxide catalyst, highly dispersed active components are often related to high activity and stability in the catalytic reactions.32,35,46 Thus, there is a deduction that the pyridine-thermal route can create catalysts with highly dispersed active components, which may be one of the reasons for its excellent catalytic performance. The formation process of highly dispersed CeO2 on CNTs prepared by the pyridine-thermal route is explained as follows.38 On the one hand, the CNTs can disperse stably in pyridine due to the noncovalent bond between pyridine and the CNTs. On the other hand, the aromatic ring of pyridine can be absorbed onto the CNTs by the π–π conjugate role.47 The pyridine molecules absorbed onto the CNTs are more easily protonated than those in the bulk solvent due to the presence of a pair of isolated nitrogen electrons that is more easily delocalized over its entire aromatic ring and the CNTs by the π–π stacking interaction,48 and then ionize the water molecular nearby to produce OH− around the CNTs. Subsequently, the OH− reacts in situ with Ce3+ through the electrostatic effect and the resultant nanoparticles deposit on the surface of the CNTs. Finally, highly dispersed CeO2 on the CNTs can be obtained. In our previous work, we investigated the affecting factors of preparing CeO2 uniformly coated CNTs and confirmed the importance of pyridine as the solvent.
 | ||
| Fig. 2 TEM images of (a) CeO2/CNTs-PT, (c) CeO2/CNTs-IM and (d) CeO2/CNTs-PM; (b) HRTEM image of CeO2/CNTs-PT; (e) EDS patterns of the region circled in red in panel (b); (f) Ceria particle size distribution of CeO2/CNTs-PT and (g) ceria particle size distribution of CeO2/CNTs-IM. | ||
The N2 adsorption–desorption isotherms and pore size distributions of the catalysts are shown in Fig. 3. As can be observed, all the adsorption isotherms and hysteresis loops of the samples seem to be of type IV and type H3 according to IUPAC classification,49 which is the characteristic feature of mesoporous solids, and the sharpness of the capillary condensation steps indicates the uniformity of the mespore size distribution.50 On the other hand, the pore size distributions from the desorption branch of the isotherms calculated by the BJH model are presented in the inset of Fig. 3. The sharp peak at around 5 nm might correspond to the internal cavity of the CNTs. Obviously, all the catalysts display a relatively narrow pore size distribution in the mesoporous range. The physical characteristics of the catalysts are summarized in Table 1. The BET surface area and pore structure parameters of these samples are similar to each other.
 | ||
| Fig. 3 N2 adsorption–desorption isotherms and pore size distributions (inset) of the catalysts. The isotherms for CeO2/CNTs-IM and CeO2/CNTs-PT are offset vertically by 100 and 250 cm3 g−1 STP, respectively. | ||
| Catalyst | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | Ce loadinga (wt%) | NO Conversionb (%) | TOFc (×10−3 s−1) |
|---|---|---|---|---|---|---|
| a Ce loading is calculated from the XPS spectra. b NO conversion of the catalyst is obtained at 150 °C. c TOF values are obtained after a 15 min reaction time. | ||||||
| CeO2/CNTs-PT | 152.9 | 0.52 | 13.9 | 3.67 | 25.9 | 3.4 |
| CeO2/CNTs-IM | 156.0 | 0.57 | 13.5 | 4.24 | 26.5 | 3.0 |
| CeO2/CNTs-PM | 151.0 | 0.50 | 12.3 | 3.83 | 16.1 | 2.0 |
XPS was applied to investigate the chemical states of the elements in the near-surface region. The O 1s XPS results of the catalysts are shown in Fig. 4. The O 1s bands are deconvoluted by the curve-fitting procedure. The sub-bands at lower binding energies (528.7–530.9 eV) correspond to the lattice oxygen O2− (denoted as Oβ) and the sub-bands at higher binding energies (531.4–532.5 eV) correspond to the surface adsorbed oxygen (denoted as Oα), such as O− or O22−, belonging to hydroxyl-like or defect-oxide groups.51 The Oα/(Oα + Oβ) on CeO2/CNTs-PT (40.5%) is slightly higher than that on CeO2/CNTs-IM (39.0%). Usually, Oα can result in more surface oxygen vacancies due to its higher mobility than Oβ.52 Thus, Oα is more active in oxidation reactions than Oβ and a high Oα/(Oα + Oβ) ratio is beneficial for NO oxidation to NO2 and thereafter facilitates the “fast SCR” reaction.31 The similar Oα/(Oα + Oβ) ratio on CeO2/CNTs-PT and CeO2/CNTs-IM may lead to a tiny difference in their catalytic activities. However, the Oα/(Oα + Oβ) ratio on CeO2/CNTs-PM (28.0%) is far less than that on the other two catalysts. Furthermore, the Oα/(Oα + Oβ) value on nitric acid-treated CNTs is 27.2% (Fig. S3, ESI†), close to that for CeO2/CNTs-PM. The variation of Oα/(Oα + Oβ) also suggests the differentiation of the interaction between the ceria crystal and CNTs in the catalysts.
 | ||
| Fig. 4 O 1s core level XPS spectra of the catalysts. | ||
The reducibility of the catalysts was investigated by H2-TPR in Fig. 5. As illustrated in Fig. 5, both CeO2/CNTs-PT and CeO2/CNTs-IM show a three-step reduction process of the Ce species. The reduction peaks of CeO2/CNTs-PT centre at 308 °C, 493 °C and 630 °C. As for CeO2/CNTs-IM, the reduction peaks can be observed at 390 °C, 482 °C and 637 °C. In the case of CeO2/CNTs-PM, a broad peak in the temperature range of 300–400 °C and a sharp peak at 658 °C are observed. The redox behavior is strongly dependent on the dispersity of the active components and the differences in the H2-TPR profiles clearly point out that the dispersion of the CeO2 particles in these three samples are not the same. For CeO2/CNTs-PT and CeO2/CNTs-IM, their first reduction peak is attributed to the reduction of highly dispersed isolated CeO2 strongly interacting with the CNTs, the second peak is attributed to the reduction of polymeric CeO2 weakly associated with the CNTs and the last one corresponds to the reduction of bulk CeO2.30,53–55 For CeO2/CNTs-PM, the two reduction peaks are assigned to the reduction of surface and bulk CeO2, respectively.55 The lowest reduction temperatures of CeO2/CNTs-PT can be indicative of the best dispersion of CeO2 on the CNTs with a very small CeO2 particle size, which would lead to an easier reduction of the ceria phase with respect to the unsupported cerium oxide.56 Furthermore, compared with unsupported CeO2, the reduction of CeO2 over the CNTs shifts to much lower temperature regions, also indicating an interaction between CeO2 and the CNTs, with the interaction in CeO2/CNTs-PT being the strongest.
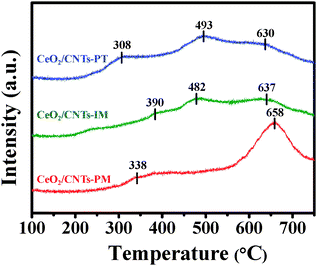 | ||
| Fig. 5 H2-TPR profiles of the catalysts. | ||
The NH3-TPD technique was employed to determine the surface acid amount and strength of the catalysts. The area and position of the desorption peak correlate with the acid amount and the acid strength, respectively. As shown in Fig. 6, the NH3-TPD spectra of all the samples contain three desorption peaks. The peaks observed below 200 °C and between 300 °C and 400 °C can be assigned to the NH3 desorbed by weak and medium acid sites on the catalysts, respectively. The peaks above 500 °C can be attributed to chemisorbed NH3 molecules adsorbed by the strong acid sites.57,58 Compared with CeO2/CNTs-IM and CeO2/CNTs-PM, the desorption peaks of CeO2/CNTs-PT shift to a higher temperature range, suggesting that the strength of the acid sites on CeO2/CNTs-PT is stronger.59 At the same time, for CeO2/CNTs-PT, the area of the desorption peaks is the largest, which means that the amount of NH3 adsorbed on the surface of CeO2/CNTs-PT is obviously greater than that on CeO2/CNTs-IM and CeO2/CNTs-PM.59,60 Furthermore, the NH3-TPD spectrum of the nitric acid-treated CNTs was also investigated (Fig. S4, ESI†) and one peak below 200 °C as well as one peak above 800 °C corresponding to NH3 desorbed by weak and strong acid sites can be observed, respectively. This indicates that, as a support, the purified CNTs can supply strong acid sites, which might be in favour of the NH3-SCR reaction. The above results imply that the preparation method has a significant effect on the amount and the strength of the acidic sites on the catalysts. Dong et al.61 reported that highly dispersed VOx species on the surface of Ti0.5Sn0.5O2 possess a large number of acidic sites and exhibit the best catalytic performance. Thus, the maximum amount of acid sites on CeO2/CNTs-PT should be attributed to the highly dispersed isolated CeO2 on the CNTs while the strongest acid sites could be associated with the strongest interaction between CeO2 and the CNTs.
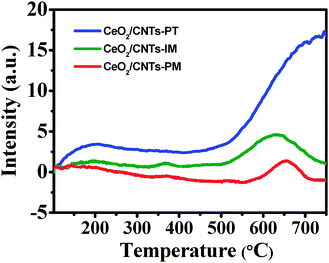 | ||
| Fig. 6 NH3-TPD profiles of the catalysts. | ||
3.2 Catalytic activity
The catalysts were tested in the NH3-SCR of NO and the results are shown in Fig. 7. The activity of the CeO2 nanoparticles and pure CNTs was also investigated. In these experiments, the NO conversion changes on increasing the reaction temperature over all the samples, except for the pure CNTs. For the CeO2 nanoparticles, the maximum NO conversion is only 50% in the whole temperature range. The conversion of NO over pure CNTs is very low, which could be attributed to the adsorption–desorption of NO. When the CNTs are introduced as a support to CeO2, the NO conversion over all the catalysts increases. These phenomena suggest that the active component is CeO2 and the interaction between CeO2 and the CNTs is important for the SCR reaction. The CeO2/CNTs-PT catalyst shows the best activity in a wide temperature range, with NO conversion above 90% from 250 to 370 °C. Compared with CeO2/CNTs-PT, CeO2/CNTs-IM had a similar catalytic activity but its operation temperature window shifts towards the high temperature range to a certain extent. For CeO2/CNTs-PM, the activity is much lower than that of the other two samples and the maximum NO conversion can barely reach 80% below 380 °C.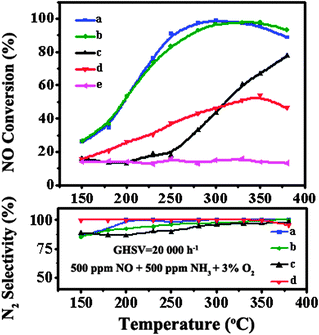 | ||
| Fig. 7 NO conversion and N2 selectivity of (a) CeO2/CNTs-PT, (b) CeO2/CNTs-IM, (c) CeO2/CNTs-PM, (d) CeO2 and (e) CNTs. | ||
According to Table 1, these three catalysts have similar BET surface areas, pore volumes and pore sizes, which should not contribute to the difference in their SCR activities. Thus, these characteristics are not important factors for the catalytic activity. Combined with the results of the XRD patterns and TEM images, the superior dispersion of the active components on the surface of CeO2/CNTs-PT leads to a higher catalytic activity than that of CeO2/CNTs-IM and CeO2/CNTs-PM. Additionally, the highly dispersed CeO2 of CeO2/CNTs-PT possesses smaller diameters and a narrower particle size distribution than that of CeO2/CNTs-IM. Furthermore, more CeO2 particles inside the central channel of the CNTs for CeO2/CNTs-PT than for CeO2/CNTs-IM was found. Su et al.62 prepared a catalyst in which some MnOx was introduced into the CNT channels and found it exhibited higher NOx conversion than that in which all the MnOx was on the outer surface of the CNTs. Their characterization results showed that MnOx confined in the channels had better abilities for supplying oxygen and adsorbing NO, which can be partially attributed to the electronic interaction between MnOx and the inner surface of the CNTs. The small difference in activity between CeO2/CNT-PT and CeO2/CNT-IM might be partially related to the difference in their ceria particle size distribution and the confinement effect. Table 1 also lists the Ce loading calculated from the XPS and TOF values of the catalysts obtained at 150 °C accordingly. The TOF value of CeO2/CNTs-PT is higher than those of CeO2/CNT-IM and CeO2/CNT-PM, indicating the variation in the activities of these samples. Furthermore, the high concentration of surface adsorbed oxygen on CeO2/CNTs-PT also favors the NH3-SCR reaction. From these results, we can see that the catalyst preparation method has a significant influence on the catalytic performance, which is consistent with reports elsewhere.32,43 Moreover, the N2 selectivity of CeO2/CNTs-PT is the highest among the catalysts. Shibata et al.63 reported that the N2 selectivity of Pt components loaded onto various supports for SCR of NO by hydrogen (H2-SCR) was related to the acid strength of the support. They found that more acid sites on the support lead to more surface NH4+, which will react with NO and O2 to produce N2. Here, we could ascribe the high N2 selectivity of CeO2/CNTs-PT to its large number of acidic sites resulting from the highly dispersed isolated CeO2 on the CNTs.
3.3. Effect of CeO2 loadings
A detailed characterization study has been undertaken to investigate the effect of ceria loading on the structural properties of the CeO2/CNTs-PT catalysts and to find a correlation with the catalytic performance in the NH3-SCR reaction. Fig. 8(a) and (b) shows the TEM images of CeO2/CNTs-PT with ceria amounts of 3% and 10%. The CeO2 particles are highly dispersed over the former sample while a small amount of CeO2 agglomerates are observed for the latter one. Their XRD patterns are also shown in Fig. 8(c). From the XRD patterns, it can be seen that the intensity of the peaks due to the CNTs decreases with an increase in the ceria loading. At low ceria loadings, none of the XRD spectra give intense peaks for cerium oxide, indicating that the CeO2 particles are well dispersed on the CNTs. On increasing the ceria loading, the crystalline phase of CeO2 becomes apparent. When the ratio is more than 10%, there are apparent peaks from CeO2 formed in the catalyst. This implies that the agglomeration takes place and then leads to a poor dispersion of CeO2 on the CNTs, which contributes to an adverse effect for NO removal. Therefore, the mass percentage of CeO2 should be smaller than 10% for catalysts synthesized by the pyridine-thermal route and then a higher activity can be obtained. The effect of the ceria amount on the performance of CeO2/CNTs-PT is presented in Fig. 8(d). The catalytic activity of all the catalysts increases monotonously first and then decreases. The NO conversion improves on increasing the ceria amount from 3% to 5% and decreases on further increasing the ceria amount to 10% below 300 °C. At high temperature, the NO conversion decreases on increasing the ceria amount, which can possibly be due to the agglomeration of CeO2 nanoparticles. As for CeO2/CNTs-PT, the suitable loading mass is 5% with the temperature ranging from 150 °C to 380 °C. In addition, the N2 selectivity of all the samples is desirable above 200 °C. | ||
| Fig. 8 TEM images of CeO2/CNTs-PT with ceria amounts of (a) 3% and (b) 10%; (c) XRD patterns and (d) NO conversion and N2 selectivity of CeO2/CNTs-PT. | ||
3.4. Stability and H2O or SO2 tolerance
The long-term stability of the CeO2/CNTs-PT catalyst at 300 °C was examined under the same reaction conditions with activity measurements. In Fig. 9(a), it is clear that the catalyst shows a high catalytic activity with a constant NO conversion at ca. 98% during a 16 h continuous running duration. Therefore, the CeO2/CNTs-PT catalyst not only provides high reactivity but also exhibits good stability, suggesting that CeO2/CNTs-PT is a good candidate for NH3-SCR reactions. Moreover, it is known from previous studies that H2O or SO2 in flue gas have a strong inhibitory effect on the removal of NOx.64–66 Therefore, we further investigated the effect of H2O or SO2 on the SCR activities of the CeO2/CNTs-PT catalyst. Fig. 9(b) and (c) presents the influence of H2O and SO2 on the NO conversion of CeO2/CNTs-PT at 300 °C. Santillan-Jimenez et al.19 reported that when CNTs were used as the support for a SCR catalyst, H2O did not show any obvious inhibition on the catalytic activity. In our experiment, when 4% H2O is added to the system, the NO conversion of CeO2/CNTs-PT has little variation with time and is maintained at about 97%. Upon switching off the H2O, the activity is still flat with that of the fresh catalyst. Likewise, when 100 ppm of SO2 is added to the reaction gas, no obvious decrease is found and the NO conversion also remains at a stable level of about 98% after the SO2 supply is stopped. According to the literature,67–69 the possible reason for SO2 poisoning the SCR catalyst is the deposition of sulfates and bisulfates. Ma et al.24 reported that the addition of CNTs could reduce the decomposition temperature of sulfates and bisulfates. Thus, these observations suggest that the deactivation caused by SO2 could be totally suppressed at 300 °C on CeO2/CNTs-PT. Furthermore, the influence of H2O or SO2 on the N2 selectivity for CeO2/CNTs-PT is negligible (Fig. S5, ESI†). From the above results, it can be seen that the catalyst prepared by the pyridine-thermal route has a good resistance to H2O or SO2 poisoning. Moreover, the thermal stability of CeO2/CNTs-PT and CeO2/CNTs-IM was investigated by TG experiments in an air stream and is shown in Fig. S6 (ESI†), which also confirms that the stability of CeO2/CNTs-PT is improved compared with CeO2/CNTs-IM. | ||
| Fig. 9 (a) Stability test, (b) H2O resistance study and (c) SO2 tolerance study of CeO2/CNTs-PT. | ||
4. Conclusions
Highly-dispersed CeO2 on CNTs was successfully prepared by a pyridine-thermal route for selective catalytic reduction of NO with NH3, which presents excellent activity for NO removal in the temperature range of 150–380 °C and good resistance to SO2 or H2O poisoning alone as well as favourable stability. More than 90% of NO can be removed in the range of 250–370 °C with a desirable N2 selectivity. Moreover, the NO conversion can be kept at about 97% with 100 ppm of SO2 or 4% H2O present at 300 °C. In addition, this catalyst shows a high catalytic activity with a constant NO conversion at ca. 98% during a 16 h continuous running duration at 300 °C. For comparison, the catalyst was also prepared by impregnation and physical mixture methods and then tested in the NH3-SCR reaction. The CeO2/CNTs-PT catalyst exhibits a better catalytic performance than CeO2/CNTs-IM and CeO2/CNTs-PM. From the results of the XRD, TEM, N2 sorption and TPR analyses, the small CeO2 nanoparticles on CeO2/CNTs-PT are highly dispersed and possess a narrow particle size distribution and the interaction between ceria and the CNTs is the strongest. The NH3-TPD profiles indicate that the CeO2/CNTs-PT catalyst exhibits the highest amount and strongest acid sites. Furthermore, the O 1s XPS spectra show that the Oα/(Oα + Oβ) ratio for CeO2/CNTs-PT is higher than that for the catalysts prepared by the other two methods, which can result in more surface oxygen vacancies and therefore favor the NH3-SCR reaction. These features should be attributed as the main reasons for the excellent SCR performance of the CeO2/CNTs-PT catalyst.Acknowledgements
The authors acknowledge the support of the National Natural Science Foundation of China (51108258), the Science and Technology Commission of Shanghai Municipality (11nm0502200 and 10540500100) the and Key Subject of Shanghai Municipal Education Commission (J50102). The authors would like to thank Mr W. J. Yu and Mr P. F. Hu from the Analysis and Test Center of SHU for help with the TEM and HRTEM measurements.Notes and references
- J. H. Li, R. H. Zhu, Y. S. Cheng, C. K. Lambert and R. T. Yang, Environ. Sci. Technol., 2010, 44, 1799–1805 CrossRef CAS.
- A. Tomita, T. Yoshii, S. Teranishi, M. Nagao and T. Hibino, J. Catal., 2007, 247, 137–144 CrossRef CAS.
- N. Y. Topsøe, Science, 1994, 265, 1217–1219 Search PubMed.
- P. G. W. A. Kompio, A. Brückner, F. Hipler, G. Auer, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237–247 CrossRef CAS.
- P. Balle, B. Geiger and S. Kureti, Appl. Catal., B, 2009, 85, 109–119 CrossRef CAS.
- J. P. Dunn, P. R. Koppula, H. G. Stenger and I. E. Wachs, Appl. Catal., B, 1998, 19, 103–117 CrossRef CAS.
- S. Djerad, M. Crocoll, S. Kureti, L. Tifouti and W. Weisweiler, Catal. Today, 2006, 113, 208–214 CrossRef CAS.
- P. Li, Y. Xin, Q. Li, Z. P. Wang, Z. L. Zhang and L. R. Zheng, Environ. Sci. Technol., 2012, 46, 9600–9605 CrossRef CAS.
- H. R. Li, D. S. Zhang, P. Maitarad, L. Y. Shi, R. H. Gao, J. P. Zhang and W. G. Cao, Chem. Commun., 2012, 48, 10645–10647 RSC.
- J. M. Planeix, N. Coustel, B. Coq, V. Brotons, P. S. Kumbhar, R. Dutartre, P. Geneste, P. Bernier and P. M. Ajayan, J. Am. Chem. Soc., 1994, 116, 7935–7936 CrossRef CAS.
- L. L. Li, C. X. Han, X. Y. Han, Y. X. Zhou, L. Yang, B. G. Zhang and J. L. Hu, Environ. Sci. Technol., 2010, 45, 726–731 CrossRef.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS.
- R. Q. Long and R. T. Yang, Ind. Eng. Chem. Res., 2001, 40, 4288–4291 CrossRef CAS.
- H. Chang, J. D. Lee, S. M. Lee and Y. H. Lee, Appl. Phys. Lett., 2001, 79, 3863–3865 CrossRef CAS.
- S. Santucci, S. Picozzi, F. Di Gregorio, L. Lozzi, C. Cantalini, L. Valentini, J. M. Kenny and B. Delley, J. Chem. Phys., 2003, 119, 10904–10910 CrossRef CAS.
- J. Z. Luo, L. Z. Gao, Y. L. Leung and C. T. Au, Catal. Lett., 2000, 66, 91–97 CrossRef CAS.
- S. J. Wang, W. X. Zhu, D. W. Liao, C. F. Ng and C. T. Au, Catal. Today, 2004, 93–95, 711–714 CrossRef CAS.
- E. Santillan-Jimenez, V. Miljković-Kocić, M. Crocker and K. Wilson, Appl. Catal., B, 2011, 102, 1–8 CrossRef CAS.
- B. C. Huang, R. Huang, D. J. Jin and D. Q. Ye, Catal. Today, 2007, 126, 279–283 CrossRef CAS.
- S. L. Bai, J. H. Zhao, L. Wang and Z. P. Zhu, Catal. Today, 2010, 158, 393–400 CrossRef CAS.
- X. B. Chen, S. Gao, H. Q. Wang, Y. Liu and Z. B. Wu, Catal. Commun., 2011, 14, 1–5 CrossRef CAS.
- L. S. Wang, B. C. Huang, Y. X. Su, G. Y. Zhou, K. L. Wang, H. C. Luo and D. Q. Ye, Chem. Eng. J., 2012, 192, 232–241 CrossRef CAS.
- Z. X. Ma, H. S. Yang, Q. Li, J. W. Zheng and X. B. Zhang, Appl. Catal., A, 2012, 427–428, 43–48 CrossRef CAS.
- S. L. Bai, J. H. Zhao, L. Wang and Z. P. Zhu, J. Fuel Chem. Technol., 2009, 37, 583–587 CrossRef CAS.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2003, 107, 5162–5167 CrossRef CAS.
- D. S. Zhang, X. J. Du, L. Y. Shi and R. H. Gao, Dalton Trans., 2012, 41, 14455–14475 RSC.
- R. Q. Long and R. T. Yang, J. Am. Chem. Soc., 1999, 121, 5595–5596 CrossRef CAS.
- R. Q. Long and R. T. Yang, Appl. Catal., B, 2000, 27, 87–95 CrossRef CAS.
- L. Chen, J. H. Li and M. F. Ge, J. Phys. Chem. C, 2009, 113, 21177–21184 CAS.
- F. D. Liu, W. P. Shan, H. He, X. Y. Shi and C. B. Zhang, Chem. Commun., 2011, 47, 8046–8048 RSC.
- X. Gao, Y. Jiang, Y. C. Fu, Y. Zhong, Z. Y. Luo and K. F. Cen, Catal. Commun., 2010, 11, 465–469 CrossRef CAS.
- W. P. Shan, F. D. Liu, H. He, X. Y. Shi and C. B. Zhang, ChemCatChem, 2011, 3, 1286–1289 CrossRef CAS.
- W. P. Shan, F. D. Liu, H. He, X. Y. Shi and C. B. Zhang, Catal. Today, 2011, 184, 160–165 CrossRef.
- W. Q. Xu, Y. B. Yu, C. B. Zhang and H. He, Catal. Commun., 2008, 9, 1453–1457 CrossRef CAS.
- P. R. Ettireddy, N. Ettireddy, S. Mamedov, P. Boolchand and P. G. Smirniotis, Appl. Catal., B, 2007, 76, 123–134 CrossRef CAS.
- X. Y. Fan, F. M. Qiu, H. S. Yang, W. Tian, T. F. Hou and X. B. Zhang, Catal. Commun., 2011, 12, 1298–1301 CrossRef CAS.
- D. S. Zhang, C. S. Pan, L. Y. Shi, L. Huang, J. H. Fang and H. X. Fu, Microporous Mesoporous Mater., 2009, 117, 193–200 CrossRef CAS.
- D. S. Zhang, T. T. Yan, L. Y. Shi, C. S. Pan and J. P. Zhang, Appl. Surf. Sci., 2009, 255, 5789–5794 CrossRef CAS.
- D. S. Zhang, H. L. Mai, L. Huang and L. Y. Shi, Appl. Surf. Sci., 2010, 256, 6795–6800 CrossRef CAS.
- Tana, M. L. Zhang, J. Li, H. J. Li, Y. Li and W. J. Shen, Catal. Today, 2009, 148, 179–183 CrossRef CAS.
- C. Z. Sun, J. Zhu, Y. Y. Lv, L. Qi, B. Liu, F. Gao, K. Q. Sun, L. Dong and Y. Chen, Appl. Catal., B, 2011, 103, 206–220 CrossRef CAS.
- B. Q. Jiang, Y. Liu and Z. B. Wu, J. Hazard. Mater., 2009, 162, 1249–1254 CrossRef CAS.
- Z. L. Wang and X. D. Feng, J. Phys. Chem. B, 2003, 107, 13563–13566 CrossRef CAS.
- X. L. Pan and X. H. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS.
- J. H. Li, J. M. Hao, L. X. Fu, Z. M. Liu and X. Y. Cui, Catal. Today, 2004, 90, 215–221 CrossRef CAS.
- R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef CAS.
- K. Jurewicz, K. Babeł, R. Pietrzak, S. Delpeux and H. Wachowska, Carbon, 2006, 44, 2368–2375 CrossRef CAS.
- P. Kumar, N. Mal, Y. Oumi, K. Yamana and T. Sano, J. Mater. Chem., 2001, 11, 3285–3290 RSC.
- X. Q. Wang, J. S. Lee, C. Tsouris, D. W. DePaoli and S. Dai, J. Mater. Chem., 2010, 20, 4602–4608 RSC.
- F. D. Liu, H. He, Y. Ding and C. B. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS.
- Z. B. Wu, R. B. Jin, Y. Liu and H. Q. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS.
- P. R. Ettireddy, N. Ettireddy, T. Boningari, R. Pardemann and P. G. Smirniotis, J. Catal., 2012, 292, 53–63 CrossRef CAS.
- G. S. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS.
- X. A. Gao, X. S. Du, L. W. Cui, Y. C. Fu, Z. Y. Luo and K. F. Cen, Catal. Commun., 2010, 12, 255–258 CrossRef CAS.
- J. C. Serrano-Ruiz, E. V. Ramos-Fernandez, J. Silvestre-Albero, A. Sepulveda-Escribano and F. Rodriguez-Reinoso, Mater. Res. Bull., 2008, 43, 1850–1857 CrossRef CAS.
- R. B. Jin, Y. Liu, Z. B. Wu, H. Q. Wang and T. T. Gu, Catal. Today, 2010, 153, 84–89 CrossRef CAS.
- H. Wang, X. Chen, X. Weng, Y. Liu, S. Gao and Z. Wu, Catal. Commun., 2011, 12, 1042–1045 CrossRef CAS.
- Y. Shen and S. Zhu, Catal. Sci. Technol., 2012, 2, 1806–1810 CAS.
- S. J. Yang, J. H. Li, C. Z. Wang, J. H. Chen, L. Ma, H. Z. Chang, L. Chen, Y. peng and N. Q. Yan, Appl. Catal., B, 2012, 117–118, 73–80 CrossRef CAS.
- L. H. Dong, C. Z. Sun, C. J. Tang, B. Zhang, J. Zhu, B. Liu, F. Gao, Y. H. Hu, L. Dong and Y. Chen, Appl. Catal., A, 2012, 431–432, 126–136 CrossRef CAS.
- Y. Su, B. Fan, L. Wang, Y. Liu, B. Huang, M. Fu, L. Chen and D. Ye, Catal. Today, 2012, 201, 115–121 CrossRef.
- J. Shibata, M. Hashimoto, K. Shimizu, H. Yoshida, T. Hattori and A. Satsuma, J. Phys. Chem. B, 2004, 108, 18327–18335 CrossRef CAS.
- J. Yu, F. Guo, Y. L. Wang, J. H. Zhu, Y. Y. Liu, F. B. Su, S. Q. Gao and G. W. Xu, Appl. Catal., B, 2010, 95, 160–168 CrossRef CAS.
- M. Casapu, O. Krocher and M. Elsener, Appl. Catal., B, 2009, 88, 413–419 CrossRef CAS.
- P. P. Hu, Z. W. Huang, W. M. Hua, X. Gu and X. F. Tang, Appl. Catal., A, 2012, 437–438, 139–148 CAS.
- H. H. Phil, M. P. Reddy, P. A. Kumar, L. K. Ju and J. S. Hyo, Appl. Catal., B, 2008, 78, 301–308 CrossRef CAS.
- N. Tang, Y. Liu, H. Q. Wang and Z. B. Wu, J. Phys. Chem. C, 2011, 115, 8214–8220 CAS.
- Q. Li, H. S. Yang, Z. X. Ma and X. B. Zhang, Catal. Commun., 2012, 17, 8–12 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM image, HRTEM image, XPS spectrum, NH3-TPD profiles, N2 selectivity and TG curves of the samples. See DOI: 10.1039/c2cy20670f |
| This journal is © The Royal Society of Chemistry 2013 |