The role of the support properties in the catalytic performance of an anchored copper(II) aza-bis(oxazoline) in mesoporous silicas and their carbon replicas†
Ana Rosa
Silva
*a,
Vanessa
Guimarães
a,
Ana Paula
Carvalho
b and
João
Pires
b
aDepartamento de Química, CICECO, Universidade de Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: ana.rosa.silva@ua.pt; Fax: +351 234 401 470; Tel: +351 234 370 604
bCQB, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade de Lisboa, 1749-016 Lisboa, Portugal
First published on 23rd October 2012
Abstract
A copper(II) chiral aza-bis(oxazoline) catalyst (CuazaBox) was anchored onto ordered mesoporous silicas and their carbon replicas. The materials were characterized by elemental analysis (C, N, H, S), ICP-AES, FTIR, XPS, thermogravimetry and isotherms of N2 adsorption at −196 °C. The materials were tested as heterogeneous catalysts in the reaction of cyclopropanation of styrene to check the effect of porous material type on the catalytic parameters, as well as on their reutilization. Generally, the composites were more active and enantioselective in the cyclopropanation of styrene than the corresponding homogeneous phase reaction run under similar experimental conditions. The materials pHpzc proved to be an important factor not only in the CuazaBox anchoring yields, but also in their catalytic performance. Less acidic surfaces (SPSi and CMK-3) yielded heterogeneous catalysts with higher styrene conversion and enantioselectivity. The materials could also be recycled with comparable enantioselectivities or generally a slight decrease in the enantioselectivity.
1. Introduction
The synthesis of chiral cyclopropanes remains a considerable challenge, especially due to the fact that cyclopropane rings are often found in a variety of natural products and biologically active compounds.1,2 Therefore, it is crucial to develop useful methods for the synthesis of cyclopropanes. The catalytic asymmetric cyclopropanation of alkenes with diazoacetates has been intensively studied over the last few years by copper complexes with chiral nitrogen ligands such as bis(oxazoline) (1 and 2, Scheme 1)2 and aza-bis(oxazoline) (3, Scheme 1).3,4 These homogeneous catalysts have also demonstrated excellent results in several other asymmetric catalytic reactions, including asymmetric aziridination,5 Diels–Alder,6 Henry reactions,7etc.8 Chiral aza-bis(oxazoline) ligands (3) are obtained in several steps from derivatives from chiral amino alcohols.3,8,9 | ||
| Scheme 1 Commercial bis(oxazoline) (1 and 2) and aza-bis(oxazoline) (3) chiral ligands. | ||
There are different advantages associated with the choice between homogeneous and heterogeneous catalysts. Homogeneous catalysts exhibit high activity and selectivity in several organic reactions. Nevertheless, there is the possibility of product contamination due to incomplete separation of the catalyst and problems with expensive catalyst recovery are frequent.10 Heterogeneous catalysts have obvious advantages over homogeneous catalysts with respect to easy separation of the catalyst from the reaction mixture at the end of the reaction, efficient recycling, improved handling and process control, minimization of metal leaching and low cost.10–13 However, this type of catalysts often present limited activity and selectivity, and hence it is important to find additional value information to overcome their disadvantages and convince the organic synthesis community and industry of the advantages of heterogeneous catalysts.11
In recent years, the interest in ordered mesoporous materials has been growing due to their advantageous properties over traditional porous materials such as high dispersion of catalytic sites, resulting in a well-defined surface and uniform pore size distribution, and large specific surface area.14,15 Hence, these materials are recognized as being an excellent option for support of homogeneous catalysts.15
Ryoo et al. reported the first synthesis of ordered mesoporous carbon (OMC) CMK-3 via a nanocasting route using SBA-15 mesoporous silica as the template in 1999.16 This method consisted of the use of a regular rigid template (SBA-15), filled with a precursor, and posterior polymerization, carbonization and removal of the template. On the other hand, Ting et al. described a method that allows the one pot synthesis of mesoporous materials with different types of porous regularity.17 In this case the silica material (SPSi) and its replicate (SPC) are synthesized at the same time.15,17 Carbonization and removal of the silica template with HF yields mesoporous carbon, whereas heating in air leads to mesoporous silica.15,17
We already reported the asymmetric cyclopropanation of styrene by copper(II) bis(oxazoline) encapsulated onto zeolites18 and anchored onto mesoporous silicas19,20 and carbons.20 The best results were obtained using the anchored copper complex with ligand 2 (2Cu catalyst, Scheme 1) onto mesoporous silica, but generally low to moderate enantioselectivities were obtained with low recyclability.20
It has been reported that in ligands 3 (Scheme 1), the presence of a nitrogen atom instead of a carbon atom at the center leads to a stronger interaction between the chiral ligand and the copper than in the case of bis(oxazoline) (Box, Scheme 1) and hence higher stability of the copper complex formed.4 Therefore better enantioselectivities in the asymmetric cyclopropanation of alkenes and stability upon reuse have been observed for these immobilized homogeneous catalysts onto siliceous mesocellular silica foams and organic polymers.3,4,21,22 At the same time, more flexibility after immobilization is obtained with these catalysts than with bis(oxazoline), 1 and 2 type ligands (Scheme 1).3
Therefore, herein, we report the anchoring of copper(II) aza-bis(oxazoline) (3) onto several unexplored mesoporous silicas (SBA-15, SPSi, HMS) and carbon materials (CMK-3, SPC), in order to evaluate the effect of the type of structure and properties of the materials on the activity, enantioselectivity and recyclability of the heterogeneous catalysts (Scheme 2).
 | ||
| Scheme 2 Anchoring procedure for the copper(II) aza-bis(oxazoline) complex onto the mesoporous materials (MM). | ||
2. Experimental section
2.1. Materials
Tetraethyl orthosilicate (TEOS, 98%), (EO)20(PO)70(EO)20, sucrose (≥99.5%), HF (≥48%), 1-dodecylamine (98%), copper(II) trifluoromethanosulfonate (copper triflate, [Cu(CF3SO3)2] or [Cu(Otf)2], 98%), (S)-tert-leucinol (98%), sodium cyanide (≥97.0%), bromine (reagent grade), ammonia solution 2.0 M in methanol, diethyl carbonate (≥98.0%), sodium ethoxide (≥95%), triethyloxonium tetrafluoroborate solution 1.0 M in dichloromethane, p-toluenesulfonic acid monohydrate (≥98.5%), sodium hydroxide (p.a.), sodium bicarbonate (p.a.), sodium sulphate (≥99.0%), sand (50–70 mesh particle size), triethylamine (Et3N, ≥99%), 3-bromopropyltrimethoxysilane (BrPS, ≥97.0%), deuterated chloroform (99.8 atom% D, 0.03% (v/v) TMS), dry toluene (99.8%), dry tetrahydrofuran (THF, ≥99.9%), methanol (p.a.), styrene (≥99%), n-undecane (≥99%), ethyldiazoacetate (EDA, ≤15% dichloromethane), phenylhydrazine (97%) and potassium bromide (FT-IR grade, ≥99%) were purchased from Aldrich and used as received. Sodium hydride (55–65%) was from Fluka. Ammonium chloride (p.a.), ethanol (p.a.) and HCl (37%) were from Panreac. H2SO4 (95–97%) from José Manuel Gomes dos Santos Lda. Ethyl acetate (>99.9%), n-hexane (95%) and dichloromethane (DCM, ≥99.9%) were from Romil. Silica gel 60 (63–200 μm) and TLC silica gel 60 F254 were from Merck.2.2. Preparation of SBA-15
SBA-15 synthesis was based on the literature.23,24 About 4.0 g of the co-polymer (EO)20(PO)70(EO)20 was dissolved in a concentrated HCl solution. Then tetraethyl orthosilicate (TEOS) was added to this solution with constant stirring. The obtained solution was stirred at 35 °C for 24 hours and subsequently heated at 100 °C for 24 hours, under static conditions. Afterwards, the solution was filtered and the filtrate was set aside for drying in air. The dried up filtrate was calcinated at 550 °C for 5 h with a ramp of 1 °C min−1.2.3. Preparation of CMK-3
The preparation of CMK-3 was based on previous works.24 Briefly, solid sucrose was dissolved in a concentrated H2SO4 solution to which the SBA-15 sample was added under mixing. The as obtained paste was heated in an oven at 100 °C for 6 hours and subsequently at 160 °C for 6 hours. The resulted black powder was then mixed again with sucrose solution (in H2SO4) and reheated with a similar heating program. The obtained black powder was further carbonized, at 875 °C for 1 hour with a ramp of 10 °C min−1, under vacuum. The silica template was then removed by treatment with HF acid followed by repeated washing with distilled water (until neutral pH). The so obtained carbon (CMK-3) was then dried at 95 °C and stored in a dry place.2.4. Preparation of SPSi and SPC in one step
In this methodology a silica–carbon composite material is firstly formed. If heated, the composite will produce a silica material (SPSi) or, if carbonized, will produce the SPC carbon. The preparation of the porous silica SPSi and the corresponding carbon replica SPC was described in detail elsewhere.15 In short, the co-polymer (EO)20(PO)70(EO)20 was dissolved in water and a 2 M HCl solution was added under stirring. To the stirred mixture concentrated H2SO4, sucrose and tetraethylorthosilicate were added. The solution was kept at 50 °C, and stirred for 24 hours, then it was aged hydrothermally in an autoclave at 100 °C for a further 24 hours. The obtained composite was dried in an oven at 100 °C and then in a furnace at 160 °C for 6 hours. When heated in air under static conditions at 550 °C for 6 hours, the synthesized composite generated the silica sample (SPSi). The corresponding carbon sample (SPC) was formed via carbonization of the composite under a dry nitrogen flow at 900 °C for 12 hours, and then the silica being removed with a 40% HF. The solid was finally filtered, washed with water and ethanol, and finally dried in a oven at 70 °C.2.5. Preparation of HMS
HMS was made according to the literature.25 A solution of 10.3 mL of 1-dodecylamine in 88.1 mL of ethanol, and 88.5 mL of deionized water, was stirred for a few minutes. Then 37 mL of tetraethyl orthosilicate (TEOS) was added. The mixture was kept under stirring at room temperature for 24 hours. The white product was then filtered and washed with deionized water until the pH is neutral and then with ethanol. The final product was calcinated at 600 °C for 6 hours with a ramp of 1 °C min−1.2.6. Preparation and characterization of aza-bis(oxazoline) (3)
The synthesis of aza-bis(oxazoline) (3, Scheme 1) was performed in four steps, by adapting reported procedures.3,9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as the eluent to yield an oil. Colorless crystals could be obtained by recrystallization from acetone. Yield: 0.69 g (68%); 1H NMR (300 MHz, CDCl3), δ/ppm: 4.27–4.33 (t, J = 9.2 Hz, 2H), 4.12–4.17 (dd, J = 6.7and 8.9 Hz, 2H), 3.78–3.84 (dd, J = 6.7 and 9.4 Hz, 2H), 0.90 (s, 18H); MS (ESI), m/z: 268.20 [M + H]+; FTIR, ν/cm−1: 3438 m, 2958 m, 1637 s, 1585 s, 1387 m, 1092 m.
:1 as the eluent to yield an oil. Colorless crystals could be obtained by recrystallization from acetone. Yield: 0.69 g (68%); 1H NMR (300 MHz, CDCl3), δ/ppm: 4.27–4.33 (t, J = 9.2 Hz, 2H), 4.12–4.17 (dd, J = 6.7and 8.9 Hz, 2H), 3.78–3.84 (dd, J = 6.7 and 9.4 Hz, 2H), 0.90 (s, 18H); MS (ESI), m/z: 268.20 [M + H]+; FTIR, ν/cm−1: 3438 m, 2958 m, 1637 s, 1585 s, 1387 m, 1092 m.
2.7. Preparation and characterization of the homogeneous catalyst (3Cu)
The copper(II) complex with ligand 3 was prepared by dissolution in DCM of equimolar quantities of [Cu(OTf)2] (0.012 mmol) and azaBox (0.012 mmol) and stirring for 4 hours at room temperature. The solution was filtered and upon evaporation of the solvent a green oil was obtained. FTIR, ν/cm−1: 2970 m, 1728 m, 1626 s, 1261 vs, 1174 s, 1036 s, 648 s.2.8. Anchoring of the CuazaBox (3Cu) onto the mesoporous materials
This method was adapted from a reported procedure21 and has three main steps: (i) functionalization of ligand 3, (ii) coordination of copper(II) onto ligand 3 and (iii) anchoring of 3Cu onto the surface of the material (Scheme 2). Typically, 0.112 mmol of 3 were reacted with 0.124 mmol of sodium hydride (1.1 equiv.) in 2.5 mL of dry THF and the mixture was stirred at 60 °C for 6 hours. Then, 0.112 mmol of 3-bromopropyltrimethoxysilane were added to the clear solution and further stirred overnight at 60 °C. The solvent was evaporated under reduced pressure and 15 mL of DCM were added, followed by 0.112 mmol of [Cu(OTf)2]. After stirring for 24 hours at room temperature, 0.50 g of mesoporous material was added to the green solution and the mixture was stirred for 5 days at room temperature. The resulting solid was filtered off, washed with DCM and then further stirred with DCM overnight, at room temperature, to remove untethered species. Finally, after filtration the solid was dried under vacuum at 60 °C for 2 days, yielding green solids in the case of the mesoporous silicas and obviously black in the case of mesoporous carbon.3Cu@SBA-15: elemental analysis (%) N 0.51, C 3.12, H 0.90, S 0.94; ICP-AES Cu 1.0%; loading of 3 121 μmol g−1 and loading of Cu 157 μmol g−1.
3Cu @CMK-3: elemental analysis (%) N 0.50, C 80.71, H 1.12, S 0.94; ICP-AES Cu 0.9%; loading of 3 119 μmol g−1 and loading of Cu 142 μmol g−1.
3Cu @SPSi: elemental analysis (%) N 0.64, C 3.92, H 0.70, S 0.89; ICP-AES Cu 1.0%; loading of 3 151 μmol g−1 and loading of Cu 157 μmol g−1.
3Cu @SPC: elemental analysis (%) N 0.40, C 79.95, H 0.74; ICP-AES Cu 0.57%; loading of 3 94 μmol g−1 and loading of Cu 90 μmol g−1.
3Cu @HMS: elemental analysis (%) N 0.35, C 2.61, H 1.13, S 0.78; ICP-AES Cu 0.88%; loading of 3 83 μmol g−1 and loading of Cu: 138 μmol g−1.
2.9. Physical and chemical methods
Elemental analysis (C, N, H and S) was performed in duplicate at Servicio de Análisis Instrumental, CACTI Vigo, Universidade de Vigo, Spain, or at Department of Chemistry of the University of Aveiro, Portugal. The copper ICP-AES was performed at “Laboratório Central de Análises” of the University of Aveiro, Portugal.FTIR spectra of the materials were obtained as KBr pellets (2.5–3 mg of sample diluted with 200 mg of KBr or for oils a drop on the surface of a KBr pellet), in the range of 400–4000 cm−1, with a FT Mattson 7000 galaxy series spectrophotometer or by ATR (mesoporous silicas) with a Bruker Tensor 27 spectrophotometer; all spectra were collected at room temperature, after drying the pellets in an oven at 75 °C overnight or the mesoporous silicas at 100 °C for 6 hours, using a resolution of 4 cm−1 and 256 scans.
X-ray photoelectron spectroscopy was performed at Centro de Materiais da Universidade do Porto (Portugal), using a VG Scientific ESCALAB 200A spectrometer using non-monochromatized Mg Kα radiation (1253.6 eV). All the materials were compressed into pellets prior to the XPS studies. In order to correct possible deviations caused by electric charge of the samples, the C 1s line at 285.0 eV was taken as the internal standard.
The pH measurements for determination of the pHpzc (pH at which the material has a net zero surface charge) were made with a SympHony SP70P VWR pH meter. The assays were made by reverse mass titration following the method proposed by Noh and Schwarz.26 Thermogravimetry was performed under air flux with a ramp of 5 °C min−1 in a TG-DSC apparatus, model 111 from Setaram.
Nitrogen adsorption isotherms at −196 °C were measured in an automatic apparatus (Asap 2010; Micromeritics). Before the adsorption experiments the samples were outgassed under vacuum for 2.5 h at 150 °C.
2.10. Catalysis experiments
The catalytic activity of the silica based materials, in the cyclopropanation of styrene, was tested at room temperature in batch reactors at atmospheric pressure and with constant stirring. In the experiments, typically, 2.4 mmol of styrene, 0.65 mmol of n-undecane, 70 mg of heterogeneous catalyst in 10.0 mL of dichloromethane and 2 μL of phenylhydrazine were used. Finally, 2.75 mmol of ethyldiazoacetate (EDA) was added to the reaction mixture over 2 hours using a syringe pump. When the addition of ethyldiazoacetate was completed, aliquots (0.05 mL) were periodically withdrawn from the reaction mixture with a hypodermic syringe, filtered through PTFE 0.2 μm syringe filters, and analyzed by GC-FID by the internal standard method. At the end of the reactions the heterogeneous catalyst was recovered by filtration, washed extensively with DCM, dried under vacuum and reused in another cycle using the same experimental procedure.Control experiments were also performed using this experimental procedure in a homogeneous phase with 3Cu or equimolar quantities of [Cu(OTf)2] plus 3 in order to compare with the heterogeneous ones (Table 3).
The reaction mixture was analyzed by GC-FID (using the internal standard method) on a Varian 450 GC gas chromatograph equipped with a fused silica Varian Chrompack capillary column CP-Sil 8 CB Low Bleed/MS (15 m × 0.25 mm id; 0.15 μm film thickness), using helium as a carrier gas. The enantiomeric excesses (%ee) of cyclopropanes were determined in the same chromatograph but using a fused silica Varian Chrompack capillary column CP-Chiralsil-Dex CB (25 m × 0.15 mm i.d. × 0.25 μm film thickness). Conditions used: 60 °C (3 min), 5 °C min−1, 170 °C (2 min), 20 °C min−1, 200 °C (5 min); injector temperature, 200 °C; detector temperature, 230 °C. The several chromatographic peaks were identified against commercially available samples and/or by GC-MS (Finnigan Trace).
3. Results and discussion
3.1. Materials characterization
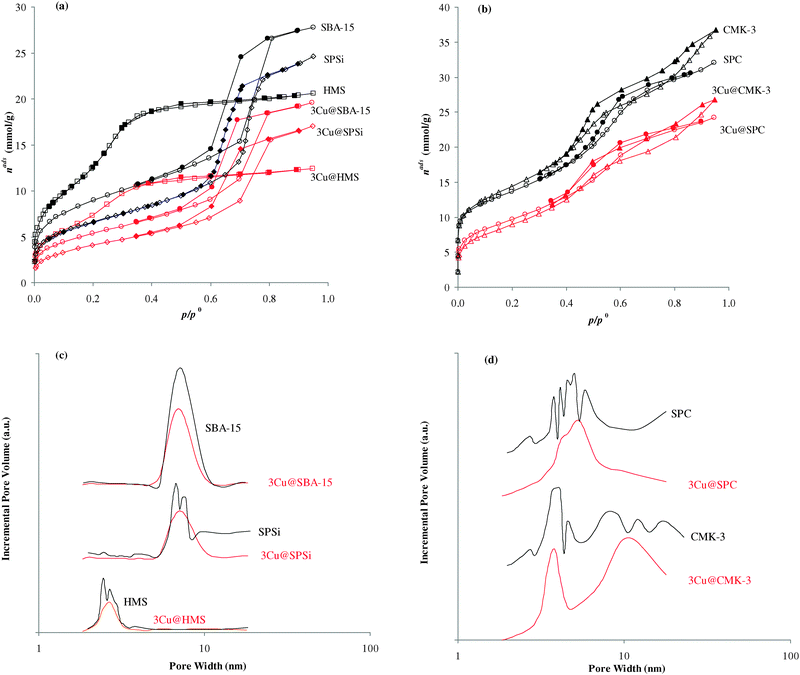 | ||
| Fig. 1 (a) and (b) Nitrogen adsorption–desorption isotherms, at −196 °C, and mesopore size distributions for the indicated samples (open points adsorption; closed points desorption). (c) and (d) Mesopore size distributions from the BdB method. | ||
| Q 2 | Q 3 | Q 4 | pHpzcb | Textural properties | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % Area | ppm (width) | % Area | ppm (width) | % Area | ppm (width) | A BET /m2 g−1 | V total /cm3 g−1 | w /nm | ||
| a Deconvoluted spectra in Fig. S1 of ESI. b pH at which the material has a net zero surface charge. c Specific surface area. d Total porous volume. e Maxima in the widths (w) of the mesopore size distributions for the various samples. | ||||||||||
| HMS | 4.5 | −91.9 (4.2) | 29.4 | −101.0 (6.4) | 66.1 | −110.0 (8.4) | 4.1 | 994 | 0.72 | 2.4; 2.7 |
| SBA-15 | 3.5 | −92.0 (4.8) | 21.5 | −100.9 (6.3) | 75.0 | −110.1 (8.6) | 3.5 | 756 | 0.99 | 7.1 |
| SPSi | 2.5 | −90.7 (4.6) | 18.2 | −100.7 (6.1) | 79.3 | −110.2 (9.1) | 5.0 | 571 | 0.84 | 6.6; 7.4 |
| CMK-3 | 4.7 | 1396 | 1.26 | 4.0; 7.7 | ||||||
| SPC | 3.3 | 1140 | 1.23 | 3.7–5.8 | ||||||
 | ||
| Fig. 2 Solid state NMR of the mesoporous silicas: (a) 29Si MAS and (b) 29Si CPMAS. | ||
Nevertheless, the amount of surface silanols might be correlated with the acidity, which might influence the course of asymmetric organic transformations and/or the stability of the immobilized homogeneous catalysts. The pHpzc values for SBA-15 and SPSi were 3.5 and 5, respectively, which is in accordance with the amount of silanols on the surface. But for the HMS sample the pHpzc value was 4.1, higher than that for SBA-15 which possesses lower amounts of silanols. Hence there is no linear correlation between the amount of silanols of mesoporous silicas and their pHpzc. However this result shows that materials with similar pore size, SBA-15 and SPSi, prepared using the same structuring agent, but by different procedures, yield different surface acidities.
These last materials, SBA-15 and SPSi, were also used as templates for the preparation of two carbon mesoporous materials, CMK-3 and SPC, but using different methodologies. Both materials possess higher areas than the parent mesoporous silicas, due to the presence of micropores also, with mesopores of 4.0 and 7.7 nm for CMK-3 and in the range of 3.7–5.8 for SPC. The pHpzc values are 4.7 and 3.3, for CMK-3 and SPC, respectively. Hence lower surface acidity was obtained for CMK-3 when compared to its parent SBA-15, whereas the SPC possesses higher surface acidity than SPSi. Again by using two different synthesis methodologies two mesoporous carbon materials were obtained with different surface and textural properties.
The elemental analysis of all the materials, compiled in Table 2, shows the presence of nitrogen which indicates that the anchoring of the chiral ligand 3 onto the materials was successful. Taking into consideration that a 3 molecule contains three atoms of nitrogen (Scheme 2), the amount of anchored 3 can be calculated, which is also compiled in Table 2. Hence, the order of the chiral ligand loading is SPSi > SBA-15 ≈ CMK3 > SPC > HMS. Although SBA-15 and its carbon replica possess similar amounts of anchored 3, SPSi has a higher amount of 3 than its carbon replica (SPC). The highest amount of ligand loading found for SPSi is most probably related to the nature of its surface since, as indicated above, the pHpzc values showed that the SPSi material has the least acidic surface. In fact, considering on one hand the silicas and, on the other hand, the replicas, ligand loadings are in general highest for the least acidic materials.
| Sample | Elemental analysis | ICP | μmol g−1 | Cu/3 | 1/2 × OTf/3 | Textural properties | Density/μmol m−2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| %N | %C | %H | %S | %Cu | 3 | OTfb | Cuc | A BET /m2 g−1 | V total /cm3 g−1 | Cu | 3 | |||
| a Calculated from the EA nitrogen content. b Calculated from the EA sulfur content. c Calculated from the ICP copper content by using the formula: %Cu × 10−6/(100 × 63.456). d Below the detection limit: 0.30%. e Below the detection limit: 94 μmol g−1. f Specific surface area. g Total porous volume. | ||||||||||||||
| 3Cu@SBA-15 | 0.51 | 3.12 | 0.90 | 0.94 | 1.0 | 121 | 293 | 157 | 1.3 | 1.2 | 438 | 0.68 | 0.21 | 0.16 |
| 3Cu@CMK-3 | 0.50 | 80.71 | 1.12 | 0.94 | 0.9 | 119 | 292 | 142 | 1.2 | 1.2 | 717 | 0.94 | 0.10 | 0.09 |
| 3Cu@SPSi | 0.64 | 3.92 | 0.70 | 0.89 | 1.0 | 151 | 278 | 157 | 1.0 | 0.9 | 339 | 0.60 | 0.28 | 0.26 |
| 3Cu@SPC | 0.40 | 79.95 | 0.74 | 0.57 | 94 | 90 | 1.0 | 772 | 0.85 | 0.08 | 0.08 | |||
| 3Cu@HMS | 0.35 | 2.61 | 1.13 | 0.78 | 0.88 | 83 | 242 | 138 | 1.7 | 1.5 | 584 | 0.44 | 0.14 | 0.08 |
From the elemental analysis the presence of sulphur which is from the triflate (OTf) counter-anion of copper(II) can also be observed, showing that copper(II) coordination took place. Taking into consideration that each triflate contains one sulphur atom, the amounts of triflate anion can be calculated (Table 2). Thus, the order of the triflate anion content is SBA-15 ≈ CMK-3 > SPSi > HMS. Hence the order of loading of triflate is different from that of ligand 3 suggesting that the amount of copper will also be different from that of 3 and generally slightly higher as can be seen by the 1/2 × OTf/3 ratio, also compiled in Table 2.
To confirm this, the copper content of the materials was also obtained by ICP-AES in order to determine the amount of copper coordinated to ligand 3. As can be seen in Table 2, the amounts of copper are higher than those of 3 from the Cu/3 ratio, for 3Cu@SBA-15, 3Cu@CMK-3 and 3Cu@HMS, which are similar to the 1/2 × OTf/3 ratio, indicating that the copper content can be roughly determined through its counter-anion. However the order of copper loading onto the materials is slightly different from that of the OTf anion: SBA-15 = SPSi > CMK-3 > HMS > SPC. The ordered mesoporous silicas (SBA-15 and SPSi) possess more copper than their corresponding carbon replicas (CMK-3 and SPC).
The N 1s peaks of the materials are centered between 399.6 and 400.8 eV and are large indicating the presence of the chiral ligand 3 with imine nitrogen29,30 as well as tertiary amine (Table S2, ESI†). The Cu 2p3/2 peaks are centered between 932.8 and 934.2 eV, which are typical of copper(II) complexes in a mixed N,O coordination sphere.29–31 However for the SPC materials the Cu 2p3/2 peak is larger and higher in energy than for the other two analyzed silica materials. The O 1s spectra of 3Cu@SBA-15 and 3Cu@HMS can be deconvoluted into a single peak centered at 532.8 eV, whereas the one for 3Cu@SPC is larger and can be deconvoluted into 3 peaks at 531.8, 533.7 and 536.8 eV (Table S2, ESI†). Thus the O 1s XPS spectra of the mesoporous silicas are typical of the Si–O bonds of the framework, whereas a rich oxygen surface chemistry can be deducted for the SPC material.32 This last conclusion is supported by the lowest pHpzc value observed for the SPC mesoporous carbon (Table 1). Although the signal to noise of the Cu 2p spectra of 3Cu@SPC is low, the higher energy and peak width observed for Cu 2p3/2 of 3Cu@SPC suggests that there must be copper(II) in different environments, when compared to 3Cu@SBA-15 and 3Cu@HMS. In the C 1s high resolution spectra besides the bands due to the carbon backbone of oxazoline and the propyl linker at lower energy, for the 3Cu@SBA-15 which contains a higher surface amount of copper, a band at 292 eV due to the CF3 carbon of the triflate anion can be clearly observed (Table S2, ESI†). This anion also gives rise to an intense peak in the F 1s region, with a band at 688 eV (Table S2, ESI†), whereas sulphonate sulfur can sometimes be detected at 168 eV (Table S2, ESI†).
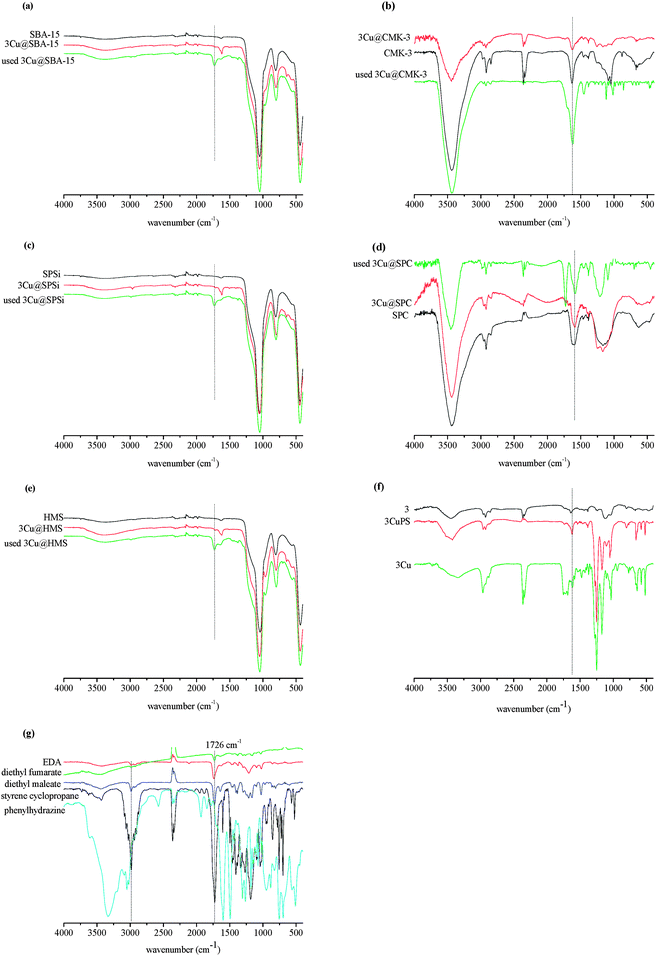 | ||
| Fig. 3 FTIR spectra for the mesoporous materials (MM) and 3Cu@MM materials, before and after catalysis: (a) SBA-15, (b) CMK-3, (c) SPSi, (d) SPC and (e) HMS, (f) azaBox (3), CuazaBox (3Cu) and CuazaBoxPS (3CuPS) and (g) EDA, styrene cyclopropane, diethyl fumarate and diethyl maleate. | ||
In the materials containing the anchored 3Cu, represented in Fig. 3, besides the characteristic bands of the materials very low intensity bands at 2976, between 1560 and 1360 and about 640 cm−1 due, respectively, to the C–H stretching vibrations of the 3tert-butyl groups, vibrations characteristic of ligand 3 and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of the sulfonate group of the triflate counter anion of the copper(II) can be further seen (Fig. 3f and Scheme 2). The characteristic CN stretching vibration of ligand 3 (Fig. 3f and Scheme 2) can be clearly seen in the ATR spectra at 1619 cm−1, but some extent of superimposition of the O–H bending vibration at 1637 cm−1 of the water adsorbed on the surface of the materials can also be observed. Furthermore the low intensity band at 3746 cm−1, due to the stretching of the O–H groups from the isolated silanols, disappeared confirming that grafting between this group and the propylsilane functionalized 3Cu took place according to Scheme 2.33
O vibration of the sulfonate group of the triflate counter anion of the copper(II) can be further seen (Fig. 3f and Scheme 2). The characteristic CN stretching vibration of ligand 3 (Fig. 3f and Scheme 2) can be clearly seen in the ATR spectra at 1619 cm−1, but some extent of superimposition of the O–H bending vibration at 1637 cm−1 of the water adsorbed on the surface of the materials can also be observed. Furthermore the low intensity band at 3746 cm−1, due to the stretching of the O–H groups from the isolated silanols, disappeared confirming that grafting between this group and the propylsilane functionalized 3Cu took place according to Scheme 2.33
It is worth mentioning that copper(II) coordination to the functionalized ligand 3 can be detected as shown in Fig. 3f by the shift of the CN stretching band from 1641 cm−1 in ligand 3 to 1620 cm−1 in the functionalized ligand 3, before addition of the materials to the reaction mixture (see Section 2.4 and Scheme 3).
 | ||
| Scheme 3 Cyclopropanation of styrene with ethyldiazoacetate. | ||
 | ||
| Fig. 4 Thermogravimetric curves for the various samples (a) SBA-15, (b) CMK-3, (c) SPSi, (d) SPC, (e) HMS. | ||
3.2. Catalysis experiments
The copper(II) complexes with aza-bis(oxazoline) ligands are efficient homogeneous catalysts in the asymmetric cyclopropanation of alkenes.3,4,21,22 Therefore, the nanostructured materials with anchored 3Cu were tested as heterogeneous catalysts in the asymmetric cyclopropanation of styrene with ethyldiazoacetate at room temperature (Scheme 3) and the results are compiled in Table 3.| Catalyst | t/h | Run | mol%b | %Cc | 4 trans/cisd | %ee (4)e | TONf | ||
|---|---|---|---|---|---|---|---|---|---|
| Cu | 3 | cis | trans | ||||||
| a Reactions performed at room temperature using 2.40 mmol styrene, 0.65 mmol n-undecane (internal standard), 70 mg of heterogeneous catalyst, 2 μL phenylhydrazine and 2.75 mmol of EDA in 10.0 mL of CH2Cl2. b % of copper and 3 in the catalyst in relation to styrene (see Table 1); for the recycling experiments corrected for the loss of heterogeneous catalyst weight. c Conversion of styrene determined by CG-FID. d trans/cis ratio of 4 (Scheme 3). e cis and trans4 (Scheme 3) enantiomeric excesses determined by chiral GC-FID. f TON = moles of styrene converted/moles of Cu. | |||||||||
| 3 + [Cu(OTf)2] | 24 | 1st | 0.27 | 0.26 | 19 | 66/34 | 41 | 44 | 70 |
| 3 + [Cu(OTf)2] | 24 | 1st | 0.46 | 0.48 | 42 | 73/27 | 63 | 73 | 90 |
| 3 + [Cu(OTf)2] | 3 | 1st | 1.0 | 1.0 | 56 | 73/27 | 76 | 84 | 56 |
| 3Cu @SBA-15 | 24 | 1st | 0.46 | 0.35 | 22 | 70/30 | 62 | 72 | 47 |
| 24 | 2nd | 0.36 | 0.28 | 15 | 65/35 | 41 | 51 | 43 | |
| 3Cu @CMK-3 | 24 | 1st | 0.37 | 0.32 | 47 | 72/28 | 70 | 79 | 126 |
| 24 | 2nd | 0.37 | 0.31 | 39 | 65/35 | 38 | 45 | 104 | |
| 3Cu @SPSi | 24 | 1st | 0.45 | 0.43 | 53 | 71/29 | 72 | 80 | 118 |
| 24 | 2nd | 0.40 | 0.38 | 26 | 70/30 | 64 | 72 | 65 | |
| 3Cu @SPC | 24 | 1st | 0.26 | 0.27 | 37 | 70/30 | 63 | 75 | 130 |
| 24 | 2nd | 0.26 | 0.27 | 41 | 67/33 | 52 | 59 | 158 | |
| 3Cu @HMS | 24 | 1st | 0.40 | 0.24 | 51 | 70/30 | 36 | 47 | 125 |
| 24 | 2nd | 0.38 | 0.23 | 10 | 64/36 | 40 | 49 | 28 | |
The catalyst densities for both Cu and azaBox were also calculated and can be found in Table 3. However the catalyst density and type of material could not explain the differences observed for the enantioselectivity of the several heterogeneous catalysts. The catalytic performance in the first cycle of 3Cu@CMK-3 with 0.1 μmol m−2 is comparable to the best catalyst 3Cu@SPSi with the highest density of 0.3 μmol m−2. Comparing 3Cu@CMK-3 and 3Cu@SPC, both with 0.1 μmol m−2 of active phase, the first is found to be a better heterogeneous catalyst than the second. Nevertheless, in the case of the mesoporous silicas the catalyst density might be an important factor as 3Cu@SPSi, with 0.3 μmol m−2 of active phase, presents better enantioselectivity than 3Cu@SBA-15 (0.2 μmol m−2) and 3Cu@HMS (0.1 μmol m−2). Hence we were driven to search for another support property that could explain the different enantioselectivities observed between all the heterogeneous catalysts. It is difficult to correlate the results from enantioselectivity with the textural and surface chemistry properties of the materials. Nevertheless an attempt can be made for instance considering the SBA-15 and the SPSi samples. For these samples the mesopore size distributions are comparable, although the porous volume of SBA-15 is 18% higher (Table 1). Therefore, the most important parameter in this case seems to be the surface chemistry since, as discussed above, the SPSi surface is less acidic than the one of SBA-15 allowing as well a more uniform distribution of the complex in the SPSi matrix, as suggested earlier by the DSC results (Fig. 4). Moreover, the SPSi carbon replica (SPC) yields a least enantioselective heterogeneous catalyst, whereas CMK-3 yields a more enantioselective heterogeneous catalyst than SBA-15. Despite the differences in the framework composition, textural and surface chemistry properties the same dependence on surface acidity can be inferred. SPC presents a more acidic surface than its parent SPSi and lower enantioselectivities were obtained, whereas the lower acidity of the CMK-3 surface yields a more enantioselective heterogeneous catalyst than its parent SBA-15 (Tables 1 and 3). It is also clear from the results in this work that small mesopore sizes, in the range of those presented by the HMS material, might be disadvantageous in the present context since this solid presented the lowest chiral ligand loading and the lowest enantioselectivity values.
It is noteworthy that in our previous report, upon heterogenization of 2Cu (Scheme 1) onto mesoporous silicas and their carbon replicas, the SPSi material also led to the heterogeneous catalyst with the highest enantioselectivity.20 Therefore this material seems to be superior as a support than conventional SBA-15, and their carbon replicas.
On the other hand, immobilization of the 3Cu catalyst on these mesoporous silica materials also leads to better styrene conversions and TON than the corresponding homogeneous phase reactions (0.46 or 0.26 mol%, Table 3). Therefore positive effects on the catalytic activity of the 3Cu catalyst can also be observed upon immobilization for these silica materials, probably due to the introduction of the propyl groups on the acidic 3 nitrogen (Scheme 2).3 The order for the styrene conversions is SPSi (53%) > HMS (51%) > CMK-3 (47%) > SPC (34%) > SBA-15 (22%). These values do not follow the materials' copper contents (Table 2, see Section 3.1.1). The SPSi and HMS mesoporous silicas were the ones which convert more styrene followed by the mesoporous carbons and finally SBA-15. The TON of the heterogeneous phase reactions are high and in the order: SPC (130) > CMK-3 (126) ≈ HMS (125) > SPSi (118) ≫ SBA-15 (47). It can be concluded that the ordered mesoporous carbons, SPC and CMK-3, yield more active heterogeneous catalysts than when the corresponding mesoporous silicas are used as 3Cu catalyst supports. This is the opposite of that observed for the heterogenized 2Cu on the same supports20 and could be due to a different effect of the type of matrix support, i.e. carbon vs. silica on the heterogenized homogeneous catalyst. Since the heterogeneous catalyst with the HMS material as a support, bearing a pore size near to 2.5 nm,19,20 has a comparable TON as the ones with the mesoporous carbon materials, their higher activity can also be due to the pore size of the materials, since they possess a pore size of 4.0 nm, whereas their parent ordered mesoporous silicas possess pore sizes of around 7 nm.20 The rate determining step in the cyclopropanation of styrene with copper bis(oxazoline) ligands is the formation of a copper(I) carbene complex by coordination of ethyl diazoacetate to the copper centre.10,31 By influence of the spatial restrictions the change in the orientation of the reacting alkene with respect to the catalytic center on the surface of the material, the activity of the system will change, as in the case of enzymes. In smaller pores the trajectory of the reacting alkene might be shorter and directed by the constraints imposed by the pore, yielding therefore an improved activity.
3Cu immobilized onto siliceous mesocellular foam (MCF) using the same anchoring procedure as the one reported herein gave 75% yield of cyclopropane, 86% ee for the trans and 78% ee for the cis cyclopropane with a diastereoselectivity of 62% trans to 38% of cis.21 The enantioselectivities are comparable to our best heterogeneous catalyst, 3Cu@SPSi, but the yields are higher probably because this reaction was performed in an inert atmosphere and using an excess of styrene in relation to EDA. The reported diastereoselectivity is however lower. Nevertheless, by capping the surface silanols of the MCF Ying et al. could further increase the cyclopropane yield and enantioselectivity to 69 and 88% ee for the trans and 83% ee for cis.213Cu has also been immobilized onto a polymeric support3,22,35 methoxypolyethylene glycol and polystyrene; using the first polymer in the cyclopropanation of styrene but with methyl diazoacetate gave 69% yield of cyclopropanes, with 71% trans to 29% cis cyclopropane diastereoselectivity and 91 and 87% enantioselectivity, respectively;3 whereas with polystyrene and again methyl diazoacetate, 28% yield, 70% trans to 30% cis cyclopropane and 88% trans enantioselectivity was obtained.22,35 The enantioselectivities and diastereoselectivities are comparable to our best heterogeneous catalyst, 3Cu@SPSi. Hence a hydrophobic surface seems to be an important factor in designing new robust materials for being used as supports in asymmetric catalysis. On the other hand, 3Cu immobilized onto a Laponite clay and a Nafion silica through electrostatic interactions showed slightly higher enantioselectivities (76% cis, 83% trans and 81% cis, 88% trans, respectively), but lower yields (46 and 30%, respectively) than our best heterogeneous catalyst, 3Cu@SPSi, under comparable experimental conditions.4 Also in comparison with our previous reports on the immobilization of commercial bis(oxazoline) catalysts, 1Cu18,19 and 2Cu20 (Scheme 1), better enantioselectivities and styrene conversions were generally achieved herein with the 3Cu catalysts indicating that their copper(II) complexes must be more stable than with the former homogeneous catalysts, as described in the literature.4
It is noteworthy that in the present reaction no by-products other than diethyl fumarate and diethyl maleate, which are from the dimerization of EDA, were detected. Hence the styrene conversions can be taken as the yields of cyclopropanes.
Nonetheless, the heterogeneous catalysts using the carbon materials as supports remain quite active upon reuse, when compared with the mesoporous silicas, and the SPC retains most of its enantioselectivity upon reuse.
Deactivation upon reuse was also observed for 3Cu immobilized onto a Laponite clay and a Nafion silica through electrostatic interactions,4 as well as polystyrene.22 It is also commonly observed for commercial bis(oxazoline) catalysts immobilized onto mesoporous silica materials.19,20,37 Only when immobilized onto siliceous mesocellular foam,21 which possesses ultra-large pores of 25 nm, and methoxypolyethylene glycol3 that these systems present high enantioselective activity and recyclability. It is noteworthy, however, that the catalytic studies on these reports were performed using 1–2 mol% of Cu21,22 in relation to the substrate, double than the necessary in homogeneous phase reactions,2 whereas in our catalytic experiments the Cu and azaBox contents were between 0.26–0.51 and 0.11–0.43 mol%, respectively. Thus deactivation is more difficult to detect than in our case.
4. Conclusions
A copper(II) chiral aza-bis(oxazoline) homogeneous catalyst (3Cu) could be anchored onto ordered silica mesoporous materials and their carbon replicas. The materials pHpzc proved to be an important factor in the 3Cu anchoring yields, as well as in the uniform distribution of the complex in the SPSi matrix. All the composites prepared were more active and enantioselective in the cyclopropanation of styrene than in the corresponding homogeneous phase reactions, run under the same experimental conditions, suggesting positive immobilization effects, with the only exception of 3Cu@HMS that presented lower enantioselectivity. The most enantioselective heterogeneous catalyst, with %ee cis and %ee trans of 72 and 80, respectively, was the one using the ordered mesoporous silica SPSi. Since CMK-3 was the second most enantioselective catalyst with %ee cis and %ee trans of 70 and 79, surface chemistry seems to be an important parameter as these two materials present the highest pHpzc values of the studied supports. The ordered mesoporous carbons, CMK-3 and SPC, yield higher reaction TON than when the parent mesoporous silicas, SBA-15 and SPSi, are used as catalyst supports. This could be due to the pore size of the materials as the HMS material, with a pore size much smaller than SBA-15 and SPSi, is also a very active catalyst.The heterogeneous catalysts with HMS, SPSi and SPC can be reused with comparable enantioselectivities, or decrease in the enantioselectivity, and with higher activity in the case of the SPC. But for most of the catalysts there is loss of activity upon reuse which could be due to partial deactivation, probably by coordination of the EDA dimerization by-products.36
The results presented herein are comparable to the ones of the same 3Cu homogeneous catalyst immobilized onto mesocellular foams,21 despite different experimental conditions, and superior to the same catalysts immobilized by electrostatic interactions into clays.4
Herein, with this comprehensive study, we were able to show that when preparing asymmetric heterogeneous catalysts the acidity of the support surfaces can have a detrimental role on the enantioselection of the immobilized chiral homogeneous catalyst. Therefore the preparation of new robust porous materials bearing less acidic surfaces is needed in order to design superior asymmetric heterogeneous catalysts, without the need for post-hydrophobization of the surface.
Acknowledgements
This work was funded by Fundação para a Ciência e a Tecnologia (FCT) through the project PTDC/QUI/64770/2006, which was co-financed by EU under the programs COMPETE, QREN and FEDER. FCT is also acknowledged for financing under the Projetos Estratégicos Pest-C/CTM/LA0011/2011 (CICECO) and PEst-OE/QUI/UI0612/2011 (CQB/FC/UL). ARS thanks FCT, Fundo Social Europeu and Programa Operacional Potencial Humano for the contract under the program Ciência 2008.References
- H. Pellissier, Tetrahedron, 2008, 64, 7041–7095 CrossRef CAS.
- D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726–728 CrossRef CAS.
- M. Glos and O. Reiser, Org. Lett., 2000, 2, 2045–2048 CrossRef CAS.
- J. M. Fraile, J. I. Garcia, C. I. Herrerias, J. A. Mayoral, O. Reiser, A. Socuellamos and H. Werner, Chem.–Eur. J., 2004, 10, 2997–3005 CrossRef CAS.
- D. A. Evans, M. M. Faul, M. T. Bilodeau, B. A. Anderson and D. M. Barnes, J. Am. Chem. Soc., 1993, 115, 5328–5329 CrossRef CAS.
- D. A. Evans, S. J. Miller and T. Lectka, J. Am. Chem. Soc., 1993, 115, 6460–6461 CrossRef CAS.
- D. A. Evans, D. Seidel, M. Rueping, H. W. Lam, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2003, 125, 12692–12693 CrossRef CAS.
- G. Desimoni, G. Faita and K. A. Jorgensen, Chem. Rev., 2011, 111, PR284–PR437 CrossRef.
- H. Werner, R. Vicha, A. Gissibl and O. Reiser, J. Org. Chem., 2003, 68, 10166–10168 CrossRef CAS.
- A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS.
- D. Rechavi and M. Lemaire, Chem. Rev., 2002, 102, 3467–3493 CrossRef CAS.
- J. M. Fraile, J. I. Garcia, C. I. Herrerias, J. A. Mayoral and E. Pires, Chem. Soc. Rev., 2009, 38, 695–706 RSC.
- J. M. Fraile, J. I. Garcia and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS.
- A. Taguchi and F. Schuth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- J. Pires, S. Borges, A. P. Carvalho and A. R. Silva, Adsorpt. Sci. Technol., 2010, 28, 717–726 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- C.-C. Ting, H.-Y. Wu, S. Vetrivel, D. Saikia, Y.-C. Pan, G. T. K. Fey and H.-M. Kao, Microporous Mesoporous Mater., 2010, 128, 1–11 CrossRef CAS.
- A. R. Silva, H. Albuquerque, A. Fontes, S. Borges, A. Martins, A. P. Carvalho and J. Pires, Ind. Eng. Chem. Res., 2011, 50, 11495–11501 CrossRef CAS.
- A. R. Silva, H. Albuquerque, S. Borges, R. Siegel, L. Mafra, A. P. Carvalho and J. Pires, Microporous Mesoporous Mater., 2012, 158, 26–38 CrossRef CAS.
- A. R. Silva, L. Carneiro, A. P. Carvalho and J. Pires, unpublished results.
- J. Lim, S. N. Riduan, S. S. Lee and J. Y. Ying, Adv. Synth. Catal., 2008, 350, 1295–1308 CrossRef CAS.
- H. Werner, C. I. Herrerias, M. Glos, A. Gissibl, J. M. Fraile, I. Perez, J. A. Mayoral and O. Reiser, Adv. Synth. Catal., 2006, 348, 125–132 CrossRef CAS.
- J. Pires, M. Pinto, J. Estella and J. C. Echeverria, J. Colloid Interface Sci., 2008, 317, 206–213 CrossRef CAS.
- V. K. Saini, M. Andrade, M. L. Pinto, A. P. Carvalho and J. Pires, Sep. Purif. Technol., 2010, 75, 366–376 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CAS.
- J. S. Noh and J. A. Schwarz, J. Colloid Interface Sci., 1989, 130, 157–164 CrossRef CAS.
- B. Jarrais, A. R. Silva and C. Freire, Eur. J. Inorg. Chem., 2005, 4582–4589 CrossRef CAS.
- A. R. Silva, V. Budarin, J. H. Clark, C. Freire and B. de Castro, Carbon, 2007, 45, 1951–1964 CrossRef CAS.
- A. P. Carvalho, C. Castanheira, B. Cardoso, J. Pires, A. R. Silva, C. Freire, B. de Castro and M. B. de Carvalho, J. Mater. Chem., 2004, 14, 374–379 RSC.
- C. Pereira, S. Patricio, A. R. Silva, A. L. Magalhaes, A. P. Carvalho, J. Pires and C. Freire, J. Colloid Interface Sci., 2007, 316, 570–579 CrossRef CAS.
- J. Pires, J. Francisco, A. Carvalho, M. B. de Carvalho, A. R. Silva, C. Freire and B. de Castro, Langmuir, 2004, 20, 2861–2866 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- A. R. Silva, K. Wilson, A. C. Whitwood, J. H. Clark and C. Freire, Eur. J. Inorg. Chem., 2006, 1275–1283 CrossRef CAS.
- W. W. Lukens Jr., P. Schmidt-Winkel, D. Y. Zhao, J. L. Feng and G. D. Stucky, Langmuir, 1999, 15, 5403–5409 CrossRef.
- J. M. Fraile, L. Perez, J. A. Mayoral and O. Reiser, Adv. Synth. Catal., 2006, 348, 1680–1688 CrossRef CAS.
- S. S. Lee and J. Y. Ying, J. Mol. Catal. A: Chem., 2006, 256, 219–224 CrossRef CAS.
- J. M. Fraile, J. I. Garcia, C. I. Herrerias, J. A. Mayoral, E. Pires and L. Salvatella, Catal. Today, 2009, 140, 44–50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20638b |
| This journal is © The Royal Society of Chemistry 2013 |