Exploring the mechanism of Stille C–C coupling via peptide-capped Pd nanoparticles results in low temperature reagent selectivity
Dennis B.
Pacardo
and
Marc R.
Knecht
*
Department of Chemistry, University of Miami, 1301 Memorial Drive, Coral Gables, Florida 33146, USA. E-mail: knecht@miami.edu; Tel: +1 305 284-9351
First published on 4th December 2012
Abstract
Herein we systematically probed the atom-leaching mechanism of Pd nanoparticle-driven Stille coupling to further elucidate the fate of the highly active Pd0 atoms released in solution. In this regard, initial oxidative addition at the particle surface results in Pd atom abstraction for reactivity in solution. As a result, two reaction sites are present, the particle surface and pre-leached Pd atoms, thus different degrees of reactivity are possible. This effect was probed via aryl halide combinations that varied the halogen identity allowing for oxidative addition of two substrates simultaneously. The results demonstrate that the system was highly reactive for iodo-based compounds in the mixture at room temperature; however, reactivity at bromo-based substrates was only observed at slightly elevated temperatures of 40.0 °C. As such, substrate selectivity was evident from the catalytic materials that can be controlled based upon the aryl halide composition and reaction temperature. Furthermore, both intermolecular and intramolecular selectivity is possible, thus raising the degree of reaction complexity that can be achieved.
Introduction
Recent advances in nanotechnology have enabled the design and fabrication of new materials with unique properties for a wide range of applications. An emerging important application for nanomaterials focuses on their use in catalytic technologies.1 For instance, a variety of different materials have been realized for reactivity in C–C coupling,2–11 olefin hydrogenation,12–15 nitro-group reduction,16–18 and remediation of trichloroethene.19–21 This reactivity typically arises from the inorganic core; however, catalytic activity can also be achieved via the ligands bound to the particle surface.22,23 While the basic catalytic properties of these materials have been demonstrated, enhancing and/or expanding their functionality is highly desirable. Typically, this can be achieved in two ways: by optimizing for reactivity under ambient/green conditions or by increasing the degree of catalytic selectivity based upon the reagents.24–27 Together, such approaches are important as they lower the environmental impact of catalysis, minimize energy consumption required to drive the reaction, as well as limit the number of separation steps required to purify the final product. The latter point of separation is critically linked to catalyst selectivity that minimizes byproduct formation, thus also enhancing the carbon efficiency of the reaction. To achieve these capabilities, it is important to fully understand and exploit the mechanism employed by the catalytic nanomaterials, which can be challenging to study due to the complexity of the system and the limited analytical techniques available. Through this understanding, individual steps of the catalytic cycle could be exploited to enhance the system and/or realize reagent selectivity.We have previously reported the biomimetic synthesis of nanocatalysts designed using Pd-specific peptides that bind directly to the nanoparticle surface.3,28 In this regard, the Pd4 peptide (TSNAVHPTLRHL) caps growing Pd nanoparticles upon recognition of the face centered cubic (fcc) metallic structure through the strong interactions of the histidine residues.3,28–32 Using this approach, ∼2 nm Pd nanoparticles can be generated that are highly reactive for the Stille C–C coupling reaction.3 As such, the reaction can be employed to elucidate important surface structural effects of the biotic/abiotic interface of the materials that would be difficult to study. Using the Pd4-capped Pd nanoparticles for Stille coupling under non-traditional conditions of an aqueous solvent at room temperature, quantitative product yields were observed at low catalyst loadings (>0.001 mol% Pd).3,28 Furthermore, both the particle size and catalytic reactivity can be tuned as a function of the peptide sequence, where minor structural changes can result in significant alterations in the catalytic functionality.29–31 Taken together, these materials represent a model system for studying ligand-capped catalysts that could address environmental and energy concerns.3,28
Given the advantages of generating efficient catalytic systems, it is important to understand the underlying principles driving the reaction. Recent studies of the Stille coupling reaction using the Pd4-capped Pd nanoparticles suggest that an atom-leaching mechanism is possible (Scheme 1).28 In this mechanism, the particles act as catalytic reservoirs containing the active metal species, which has been similarly proposed for other C–C coupling reactions.1,4,28,33,34 During the initial oxidative addition step, Pd atoms are abstracted from the nanoparticles, from which the catalytic cycle can then occur in solution to produce the final product and regenerate the Pd0 species. These free Pd0 atoms can then be recycled through the catalytic process until reaction completion, upon which the atoms are quenched by the remaining particles.1,28,31 These Pd nanoparticles also demonstrated varying degrees of reactivity with aryl halides as a function of the halogen group. To that end, iodo-derivatives are typically reactive for Stille coupling at room temperature, while bromo-based species require a slight increase in temperature to 40.0 °C for reactivity.28 This temperature effect could be directly related to the bond dissociation energies of the carbon–halogen bond wherein C–Br (285 kJ mol−1) > C–I (232 kJ mol−1).35 While it is clear that aryl bromides are unable to abstract Pd0 atoms from the nanoparticle surface at room temperature,28 it is unclear if the abstracted Pd0 atoms can insert across the C–Br bond to drive the reaction under such conditions. As such, to more fully understand and elucidate the fundamental catalytic properties of the system and the overall mechanism, additional studies are required that could be used to enhance the reactivity of the materials.
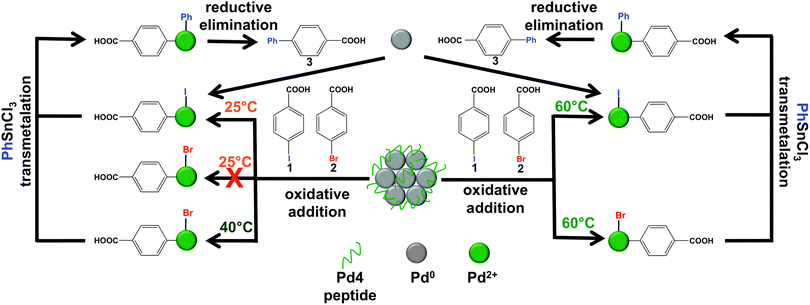 | ||
| Scheme 1 Representative scheme for reagent selectivity of the peptide-capped Pd nanoparticles for two aryl halide substrates at different reaction temperatures based upon the catalytic mechanism. | ||
Here we report the reactivity and substrate selectivity of peptide-capped Pd nanoparticles in a mixture of reagents. This study provides important information concerning the catalytic reaction mechanism and degree of reactivity. By employing different aryl halide substrates, Stille coupling with an organostannane reagent can be achieved, where selectivity is observed as a function of the reaction temperature, aryl halide composition, and the catalytic mechanism. As summarized in Scheme 1, the Pd nanoparticles selectively couple aryl iodide substrates over bromo- or chloro-derivatives in the same reaction mixture at room temperature generating the anticipated product; however, a mild increase in system temperature to 40.0 °C enables reactivity at the bromo substrate. These results indicate functional group catalytic selectivity that is inversely related to the strength of the carbon–halogen bond. By increasing the reaction temperature to 60.0 °C, catalytic selectivity is lost wherein reactivity at both iodo and bromo substituents is observed. Furthermore both intermolecular (separate reagents) and intramolecular (two reactive sites in a single reagent) selectivity is observed, raising the level of reaction complexity possible under low-temperature conditions using the peptide-capped materials. This selectivity is achieved based upon the catalytic leaching mechanism, where an apparent energy barrier is present that prevents bromo-based reactivity at room temperature. This suggests that oxidative insertion of the Pd0 materials across the C–Br bond is prevented, regardless of whether the metallic component is constrained within the zerovalent particle or pre-leached into solution. These results are important for advancing catalytic nanotechnologies to achieve energy- and carbon-efficient systems.
Experimental
Chemicals
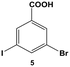
4-Iodobenzoic acid (1), NaBH4, CDCl3, NaCl, 4-tert-butylphenol, KOH, N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA), and dichloromethane were purchased from Acros Organics. 4-Bromobenzoic acid (2) was acquired from Alfa Aesar. 4-Chlorobenzoic acid (4) and 3-bromo-5-iodobenzoic acid (5) were purchased from TCI America. 3-Chloro-5-iodobenzoic acid (8), 3-bromo-5-chlorobenzoic acid (10), and 2-chloro-5-iodobenzoic acid (19) were obtained from Oakwood Products, while 3,5-diiodobenzoic acid (11) was acquired from Spectra Group Limited. PhSnCl3, K2PdCl4, 3,5-dibromobenzoic acid (13), 2-bromo-5-iodobenzoic acid (17), and 2-chloro-4-iodobenzoic acid (21) were obtained from Sigma-Aldrich. Finally, 2,5-diiodobenzoic acid (14) was purchased from MP Biomedicals. Diethyl ether was obtained from VWR and 18 mΩ cm water (Millipore) was employed throughout. All reagents were used as received without additional purification.Characterization
The reaction products were characterized using GC-MS and 1H NMR. For GC-MS analysis, ∼1 mg of the product was dissolved in 200 μL of MSTFA, which was allowed to react for 2.00 h.3 The sample was then diluted with 2.00 mL of dichloromethane before analysis using an Agilent 5975C GC-MS system. All products, including those generated using both the mono- and di-substituted aryl halides, were processed using this approach. For 1H NMR analysis, a Bruker 400 MHz spectrometer with an autotuning multinuclear probe was employed. The percent yields of the products were determined by comparing the peaks of the internal standard (t-butyl phenol) with the product peak.3 For the reactions using 1 and 2, the peak at 8.17 ppm corresponds to the biphenylcarboxylic acid (3) product while the peak at 6.7 ppm corresponds to the internal standard. The ratio of the integration of these two peaks was used to calculate the percent yield of the reaction as previously demonstrated.3 Furthermore, the amount of unreacted starting materials was determined by comparing the ratio of the peaks at 7.8 ppm (1), 7.93 ppm (2), and 8.0 ppm (4) with that of t-butyl phenol. For the di-substituted aryl halides, identical methods were employed where peaks associated with the mono- and di-substituted products were used to determine the product yield.Catalytic reactions with multiple aryl halides
For all reactions, Pd nanoparticles capped with the Pd4 peptide were prepared at a Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :peptide ratio of 3.3 using previously described methods.3 For the catalytic reactions involving two separate aryl halide substrates, a system composed of 1 and 2 is described; however, identical procedures and conditions were employed for reactions with two reagents. In a reaction flask at room temperature, 5.00 mmol of 1 and 5.00 mmol of 2 were co-dissolved in 160 mL of 2.25 M KOH. To this solution, 12.0 mmol of PhSnCl3 was added, followed by the direct injection of 10.0 mL of the freshly prepared Pd4-capped Pd nanoparticles. Under these conditions, a reaction with 0.05 mol% Pd was generated with respect to the total aryl halide concentration in solution. Immediately upon nanoparticle addition, 8.00 mL aliquots were extracted, separated into vials, reacted while stirring, and subsequently quenched with 50.0 mL of 5% HCl at specified time intervals. After 2.0 h at room temperature, the remaining reaction vials were placed in an oil bath at 40.0 °C and then quenched at selected time points. For all of the quenched reactions, the product was extracted, characterized, and quantitated.
:peptide ratio of 3.3 using previously described methods.3 For the catalytic reactions involving two separate aryl halide substrates, a system composed of 1 and 2 is described; however, identical procedures and conditions were employed for reactions with two reagents. In a reaction flask at room temperature, 5.00 mmol of 1 and 5.00 mmol of 2 were co-dissolved in 160 mL of 2.25 M KOH. To this solution, 12.0 mmol of PhSnCl3 was added, followed by the direct injection of 10.0 mL of the freshly prepared Pd4-capped Pd nanoparticles. Under these conditions, a reaction with 0.05 mol% Pd was generated with respect to the total aryl halide concentration in solution. Immediately upon nanoparticle addition, 8.00 mL aliquots were extracted, separated into vials, reacted while stirring, and subsequently quenched with 50.0 mL of 5% HCl at specified time intervals. After 2.0 h at room temperature, the remaining reaction vials were placed in an oil bath at 40.0 °C and then quenched at selected time points. For all of the quenched reactions, the product was extracted, characterized, and quantitated.
This same system was also studied at 60.0 °C throughout the reaction to determine the effect of higher temperature. In this setup, the reaction mixture was prepared and mixed, from which aliquots were transferred to separate vials and immediately placed in an oil bath heated to 60.0 °C. At selected time points, the reactions were quenched and the products were extracted and quantitated using the standard approach.
Aryl dihalide reactivity
The effects of multiple halogen functional groups within a single molecule on both the catalytic mechanism and the reaction selectivity were initially studied using 5. In a reaction vial, 0.25 mmol of 5 and 0.6 mmol of PhSnCl3 were dissolved in 6.00 mL of 1.5 M KOH. To this mixture, 0.25 mL of the freshly prepared Pd nanoparticles (0.05 mol% Pd) was added and the reaction was stirred at room temperature for 24.0 h. Once complete, the reaction was quenched using 50.0 mL of 5% HCl. The products of the reaction were extracted and quantitated as described above. Additional variations to this basic procedure were also studied by changing the Pd catalyst concentration and/or reaction temperature while holding all other conditions constant. The same procedure was employed for the following aryl dihalides: 8, 10, 14, 17, 19, and 21. For 11 and 13, identical procedures were also used; however, the starting materials were dissolved using 15.0 mL of 1.0 M KOH and 7.0 mL of 1.5 M KOH, respectively, due to reagent solubility.The time-resolved formation of the coupling products between 5 and PhSnCl3 was monitored by scaling up the reaction and extracting aliquots at selected time intervals. Specifically, 3.75 mmol of 5 and 9.0 mmol of PhSnCl3 were co-dissolved in 90.0 mL of 1.50 M KOH. When the reagents were dissolved, 5.00 mL of the freshly prepared Pd nanoparticles (0.05 mol% Pd) was added. 6.00 mL aliquots were then taken and quenched every 30.0 min for 5.0 h followed by product extraction and quantitation. A similar procedure was used for 11; however, product formation was monitored every 10.0 min for 1.00 h, followed by 30.0 min intervals over an additional 2.00 h.
Results and discussion
Based upon the proposed Pd leaching mechanism, aryl halide oxidative addition occurs at two specific locations: at the nanoparticle surface and at free Pd0 atoms in solution.1,4,28,33,34 Initially, oxidative addition at the nanoparticle surface occurs to leach the Pd atoms into solution, from which subsequent oxidative addition processes can occur with additional aryl halide reagents.28 Intrinsically, the reactivity for these two steps could be significantly different based upon the aryl halide substrate. For instance, from previous studies using the peptide-capped materials, it is known that aryl iodides can oxidatively add to the Pd metal at the nanoparticle surface at room temperature, while aryl bromides cannot;3,28 however, different reactivity for the leached Pd0 atoms could be observed as a function of the halogen group. In this regard, while no reaction is observed for aryl bromides at room temperature with the Pd nanoparticles, Stille coupling could be noted from such reagents using the leached Pd0 atoms. Such mechanistic information could be quite useful in the design of selective catalytic materials. As such, equal amounts of aryl halides that varied the halogen only were co-reacted with the peptide-capped Pd nanoparticles to monitor product formation. Initially, 1, 2, and 4 were used since the substrates exhibited different levels of reactivity: 1 is known to readily react for Stille coupling at room temperature, slightly higher temperatures of 40.0 °C are required to drive the same reaction with 2, while 4 does not typically react.3,28Fig. 1a schematically presents the process employed for using two different aryl halide substrates in the reaction. For this, 1 and 2 are commixed in the reaction medium with PhSnCl3 and sufficient Pd nanoparticles to reach a concentration of 0.05 mol% Pd. The reaction is initially monitored at room temperature (∼25.0 °C), followed by heating to 40.0 °C after 2.00 h. The progress of the reaction, monitored at selected time points, is shown in Fig. 1b. The plot represents the amount, in mmol, of 1 (blue triangles), 2 (red circles), and the product, 3 (green squares), present in the reaction at the given time. Immediately after catalyst addition at room temperature, rapid consumption of 1 was observed as shown by the exponential decrease in the amount of reagent in the reaction. After 1.0 h, only trace amounts of this reagent were present in the mixture, which reached complete consumption at 2.0 h. Consequently, the quantity of product 3 rapidly increased over the initial 1.0 h time frame to eventually saturate. Conversely, the amount of 2 remained constant for the first 2.0 h of the reaction at room temperature. A minor decrease may be present; however, this is within the statistical noise of the study. After 2.0 h at room temperature, the reaction mixture was mildly heated to 40.0 °C, where previous results demonstrated maximal reactivity for 2.28 At the elevated temperature, the bromo-based substrate is consumed, concomitant with an increase in the product quantity. Interestingly, the rate of consumption of 2 at 40.0 °C was slower than the consumption of 1 at room temperature, suggesting a change in the reaction rate as a function of halogen identity.
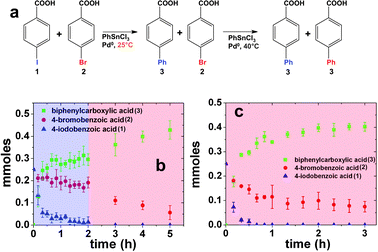 | ||
| Fig. 1 Effects of aryl halide mixtures for the Stille coupling reaction using peptide-capped Pd nanoparticle catalysts. Part (a) presents the reaction between 1 and 2 at the indicated temperatures. Part (b) displays the time based analysis of the reactions using 1 and 2 at 25.0 °C (blue region) for 2.0 h, followed by heating the reaction mixture to 40.0 °C (red region), while part (c) presents the Stille coupling analysis over time using 1 and 2 at 60.0 °C throughout the study. | ||
These results indicate that the peptide-capped Pd nanoparticles preferentially drive coupling at the iodo-derivative over the bromo-species at room temperature, regardless of whether Pd0 atoms are pre-leached into solution. Pd0 atoms are abstracted from the nanoparticle surface by aryl iodide oxidative addition, thus they would be available for reactivity with the aryl bromide compound upon completion of the first Stille coupling cycle. As such, this suggests that an energy barrier exists for aryl bromide oxidative addition at both the nanoparticle surface and the highly reactive Pd0 atoms in solution.
To probe this mechanistic observation, additional studies of different aryl halide combinations and selected reaction temperatures were employed. Initially, an identical coupling system was studied with 1 and 2; however, the reaction was processed at 60.0 °C throughout the experiment. At this temperature, both substrates should readily undergo coupling to generate common product 3, as shown in Fig. 1c. In this study, rapid consumption of 1 is observed, faster than at room temperature, resulting in complete reagent consumption within 40.0 min. Additionally, immediate coupling is observed for 2; however, the rate of the reaction for the bromo-derivative is slower compared to the iodo-derivative. Complete exhaustion of 2 is not observed, resulting in ∼70% of the substrate generating product, consistent with previous studies.28 Further co-reactivity studies were also conducted using a mixture of 1 and 4, as presented in Fig. 2. Fig. 2a specifically presents the study of the reaction at room temperature, while Fig. 2b presents the analysis of the reaction at 60.0 °C. As anticipated, when the reaction was processed at room temperature, rapid formation of product 3 (green squares) was observed from 1 (blue triangles), with complete reagent consumption at 2.00 h, while no coupling was noted from 4 (black diamonds) throughout the process. When the reaction was studied at the elevated temperature of 60.0 °C (Fig. 2b), rapid consumption of 1 was noted over a 40.0 min time frame with no product formation arising from the chloro-derivative. Additional Stille coupling studies of mixtures of 2 and 4 were performed using the peptide-capped Pd nanoparticle catalysts. Fig. 2c presents the catalytic analysis for the Stille coupling reaction using these two reagents at room temperature with a 0.05 mol% Pd catalyst concentration. Under these conditions, no coupling product is observed after 5.00 h. When the reaction was studied at a temperature of 60.0 °C, Fig. 2d, consumption of 2 is noted (red circles) along with an increase in the formation of 3 (green squares). Again, no reactivity of 4 is observed even at the higher temperature with leached Pd0 present in the medium. Fig. 2e presents the scheme for the competition reaction between 2 and 4 at different temperatures.
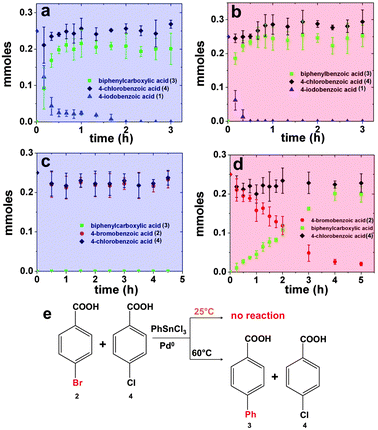 | ||
| Fig. 2 Parts (a and b) present the time resolved analysis of the Stille coupling process employing mixtures of 1 and 4 at room temperature and at 60.0 °C, respectively, while parts (c and d) display the same temperature analysis for mixtures of 2 and 4 at the same temperatures. The scheme for the reaction of 2 and 4 at different temperatures is shown in part (e). | ||
Taken together, these studies demonstrate important results concerning the reactivity of Pd nanoparticles for C–C coupling reactions based upon the catalytic mechanism. In the proposed leaching mechanism (Scheme 1), aryl halide oxidative addition occurs at the nanoparticle surface, which abstracts reactive Pd species. After completion of the catalytic cycle, highly reactive Pd0 components are released in the reaction medium, from which secondary oxidative addition with additional substrates can occur to recycle through the process. Eventually, these reactive Pd0 species are quenched by the remaining particles in the mixture; however, two different oxidative addition steps are present that are likely to be both thermodynamically and kinetically different. These results indicate that while the two oxidative additions are indeed different, the reactivity for the separate processes remains similar. To that end, aryl iodides are reactive for oxidative addition at room temperature at both the nanoparticle surface and the leached Pd0 atoms, while elevated temperatures are required for reactivity in both processes for aryl bromides. Regardless of the oxidative addition step, aryl chlorides remain unreactive. Such results are likely to be related to the relative values of the C–X bond dissociation energies associated wherein C–I (232 kJ mol−1) < C–Br (285 kJ mol−1) < C–Cl (338 kJ mol−1).35 As such, in the Stille coupling reaction, the aryl iodide substrate reacts rapidly at room temperature, indicating that both oxidative addition steps readily occur without the need for thermal activation. On the other hand, the bromo-species require mild heating to 40.0 °C for the coupling reaction to occur due to the stronger C–Br bond. The aryl chloride, however, does not undergo coupling even at 60.0 °C due to the greater energy required for activation of the C–Cl bond. Mechanistically, these results provide a greater understanding of the different factors that affect the oxidative addition processes and could potentially be applied to different C–C coupling reactions. Furthermore, these capabilities could also be exploited to engender the biomimetic system with catalytic selectivity. In that regard, these studies suggest selectivity for iodo-based substrates over bromo- and chloro-based reagents as a function of temperature. This selective reactivity is based upon the mechanism of the reaction and the thermodynamics of the system. While this was observed for intermolecular reaction systems, intramolecular reactivity and selectivity may also be possible; however, the electronics of the system may change the degree of reactivity.
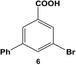
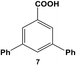
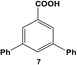
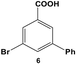
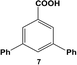
The reactivity of aryl dihalide substrates was initially studied using a benzoic acid derivative containing two different halide groups, 5 (Table 1, entry 1). In this reaction, the effect of having two reaction centers on the aryl ring was investigated through the formation of two likely products, 3-bromo-5-phenylbenzoic acid (6) and 3,5-diphenylbenzoic acid (7); the former is generated from the reaction of the iodo-component of the substrate, while the latter from the reaction of both the iodo- and bromo-components. A third product is possible, 3-iodo-5-phenylbenzoic acid; however, this product is unlikely based upon the observed reactivity of iodo over bromo groups. Using 0.05 mol% Pd at 25.0 °C, 70.8 ± 0.9% of 6 and 22.1 ± 2.5% of 7 were produced after 24.0 h. These results suggest that the iodo-component of the aryl dihalide preferentially undergoes Stille coupling at room temperature, leading to the formation of the mono-substituted product. Interestingly, the generation of the di-substituted product at room temperature indicates that the C–Br bond in the mono-substituted product is potentially activated to undergo oxidative addition and eventual Stille coupling (discussed below).
| Entry | Aryl dihalides | Possible products | |||
|---|---|---|---|---|---|
| Mono-substituted | Percent yield | Di-substituted | Percent yield | ||
| Reaction conditions:a 0.05 mol% Pd, 25 °C, 24 h.b 0.10 mol% Pd, 25 °C, 24 h.c 0.10 mol% Pd, 40 °C 24 h.d 0.05 mol% Pd, 60 °C 24 h. | |||||
| 1 |
|
|
70.8 ± 0.9a
33.6 ± 1.9b 19.3 ± 6.0c 12.9 ± 11.2d |
|
22.1 ± 2.5a
60.9 ± 1.3b 80.6 ± 6.4c 69.8 ± 11.5d |
| 2 |
|
|
88.2 ± 4.6a
89.2 ± 9.0b 97.2 ± 3.5c 95.5 ± 7.6d |
|
0.0a
0.0b 0.0c 0.0d |
| 3 |
|
|
0.0a
0.0b 0.0c 0.0d |
|
0.0a
0.0b 0.0c 0.0d |
| 4 |
|
|
0.0a
0.0b 0.0c 0.0d |
|
88.6 ± 9.0a
87.5 ± 7.4b 96.6 ± 3.8c 82.4 ± 13.9d |
| 5 |
|
|
0.0a
0.0b 32.5 ± 3.0c 30.9 ± 3.2d |
|
0.0a
0.0b 24.6 ± 3.7c 39.5 ± 1.4d |
To further understand the dynamics of the system, time-based analysis of the reaction was employed to monitor product formation (Fig. 3). Over the first 5.00 h of the reaction at room temperature, formation of mono-substituted product 6 (blue circles) is observed from coupling at the iodo-group of 5 (green squares). Minor formation of the di-substituted product, 7 (red triangles), is observed 1.50 h after reaction initiation. Note that the formation of the mono-substituted product is noted first, followed by production of the di-substituted species. Furthermore, the rates of C–C coupling are also quite slower as compared to 1 and 2 above. As such, these results suggest that coupling at aryl dihalides provides changes to the electronics of the system that impact the coupling reactions; however, the general selectivity of the catalyst remains. This results in slower coupling reactions, as well as activation of the second halogen group within the ring system. To that end, the electron-donating phenyl ring in 6 possibly activates the bromo-component at room temperature. As a result, weakening of the C–Br bond could occur, thus making it more susceptible to oxidative addition. The C–Br species from 6 could then participate in the atom abstraction process or react with the free Pd0 atoms available in solution after the initial catalytic cycle at the iodo-component; however, only minimal di-substituted product yields are observed.
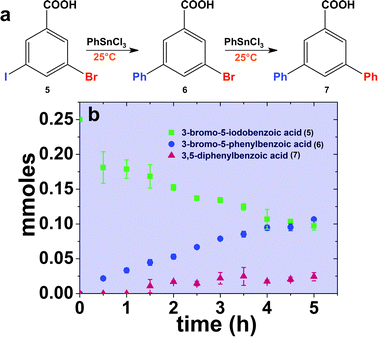 | ||
| Fig. 3 Part (a) presents the likely reaction scheme for the Stille coupling process using the peptide-capped Pd nanoparticles employing 5 as the aryl dihalide reagent, while part (b) displays the time-based results for the reaction at room temperature. | ||
When the same reaction was performed at a higher catalyst loading of 0.10 mol% Pd, the amount of di-substituted product 7 generated increased to 60.9 ± 1.3%, while that of mono-substituted product 6 decreased to 33.6 ± 1.9% (Table 1, entry 1). Such changes are likely due to more rapid product formation rates based upon the higher catalyst loading. Further shifting of the reaction towards the di-substituted product (7) was observed by heating the reaction system. When studying the process at 40.0 °C using 0.10 mol% Pd, the yields of 7 reached 80.6 ± 6.4%, while the yields for 6 were 19.3 ± 6.0%. When the reaction mixture was heated to 60.0 °C, the yields of the two products were statistically similar to the results at 40.0 °C, indicating a potential saturation point for product formation at higher temperatures. These results suggest that at elevated temperatures, the formation of di-substituted product 7 increases due to the additional thermal energy where both the C–I and C–Br bonds can now undergo oxidative addition with both the particle surface and leached Pd0 atoms. This process, however, could still occur in a step-wise fashion due to the more rapid reaction event at the iodo group over the bromo group, giving rise to the formation of intermediate 6. Furthermore, no evidence of the 3-iodo-5-phenylbenzoic acid intermediate was ever observed. These results demonstrate that intramolecular selectivity is possible for the peptide-capped Pd nanocatalyst under ambient/low temperature conditions using low catalyst concentrations based upon the reaction mechanism and system thermodynamics.
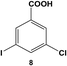
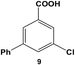
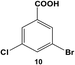
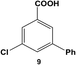
Expanding the scope of the intramolecular selectivity of the nanocatalyst was explored using different derivatives of benzoic acid dihalides (Table 1). For the reaction using 8, only the mono-substituted product, 3-chloro-5-phenylbenzoic acid (9), was generated at an 88.2 ± 4.6% yield after 24.0 h at room temperature using 0.05 mol% Pd (Table 1, entry 2). Increasing the catalyst loading and/or raising the reaction temperature generated increasing amounts of mono-substituted product 9 to nearly quantitative yields. As expected, the chloro-component of the reagent was not reactive, thereby the formation of di-substituted product 7 was not observed; the formation of intermediate 9 did not significantly activate the C–Cl bond to facilitate coupling. Interestingly, when employing 10 as the substrate, no coupling was observed for either the bromo or chloro groups under any reaction conditions (Table 1, entry 3). Such results may arise from inductive effects of the electronegative chloro-component, which could cause deactivation of the bromo-component, even at higher temperatures. In general, the different halide groups of the aryl dihalide systems follow a similar trend observed above with the mono-substituted reagents. In this regard, the Pd nanoparticles generally react at the iodo-component first, followed by the bromo-component, with no reactivity for the chloro-component under the selected conditions. Activation of the second functional group is also possible, but the low coupling rates at these positions still allow for selective coupling at the initial reactive site (i.e. iodo over bromo).
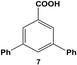
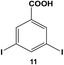
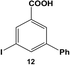
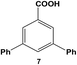
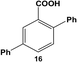
Reactions of aryl dihalide substrates with the same halogen group at the 3 and 5 positions were also studied. Initially, 11 was employed for Stille coupling using 0.05 mol% Pd at room temperature (Table 1, entry 4). The two highly reactive iodo-components generated di-substituted product 7 in 88.6 ± 9.0% yield, after 24.0 h. A general increase in the formation of the di-phenyl product was observed proportional to the catalyst loading and reaction temperature, as anticipated. Interestingly, the mono-substituted intermediate, 3-iodo-5-phenylbenzoic acid (12), was never observed under any conditions. To probe the reaction mechanism for 11, a time-based analysis of the reaction at room temperature with a catalyst loading of 0.05 mol% Pd was studied, as shown in Fig. 4. The results indicate that after 10.0 min, the di-substituted product, 7 (blue squares), was the only isolated product of the coupling reaction; an increase in formation of 7 over the time frame of the study was observed with a complementary consumption of the starting material (11, red circles). Throughout the study, mono-substituted intermediate 12 from coupling at a single iodo group was never observed. To produce such results two likely reaction mechanisms are possible: (1) simultaneous coupling at both iodo groups using separate catalytic Pd materials or (2) coupling at a single group to produce intermediate 12 that activates the second iodo-group to substantially increase the second-step reaction rate. Based upon the results of the coupling reaction for 5, the latter mechanism for the di-iodo reagent is more likely. In this regard, the initial attachment of the phenyl group from the coupling reaction of one of the iodo-species produces the bi-aryl ring system that activates the second C–I bond. This then results in exceedingly rapid coupling, generating the di-substituted product (7). In this regard, the coupling at the first iodo-group is likely to be the rate-limiting step, otherwise mono-substituted product 12 should have been observed. This step-wise reactivity is more probable due to the kinetic consideration of the atom-leaching mechanism wherein there is a higher probability of a single halogen site reacting with a Pd atom, rather than the two halogens simultaneously coupling with separate catalysts.
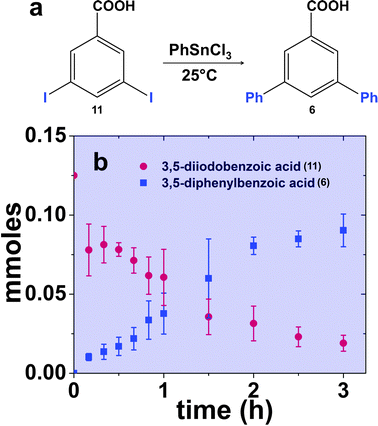 | ||
| Fig. 4 Part (a) presents the likely reaction scheme for the Stille coupling reaction using the peptide-capped Pd nanoparticles employing 11 as the aryl dihalide reagent, while part (b) displays the time-based results for the reaction at room temperature. | ||
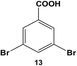
Changing the halogen groups to bromides such as in 13 demonstrated an additional change in reactivity. For this system, the two possible products, 6 and 7, from coupling at either one or both of the halogens, respectively, were not obtained at room temperature using either 0.05 or 0.10 mol% Pd (Table 1, entry 5). Such results were expected based upon the energy required for oxidative addition using bromo-based reagents. When the reaction mixture was heated to 40.0 °C with 0.10 mol% Pd, however, the reactivity of the di-bromo substrate was increased, generating both the mono-substituted intermediate 6 and the di-substituted final product 7 with yields of 32.5 ± 3.0% and 24.6 ± 3.7%, respectively, after 24.0 h. When the reaction was performed at 60.0 °C with 0.05 mol% Pd, the yield of the di-substituted product increased to 39.5 ± 1.4%, with a mono-substituted product yield of 30.9 ± 3.2%. The increase in the amount of 7 at 60.0 °C was attributed to the activating effect of the biphenyl moiety in the mono-substituted intermediate on the C–Br bond and the increased thermal energy, driving the catalytic reaction to produce more of the di-substituted product.
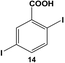
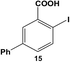
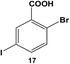
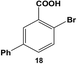
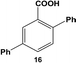
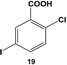
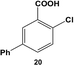
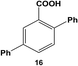
The reactivity of the Pd nanoparticles towards aryl dihalides was further explored using substrates containing halogen groups at different positions in the benzoic acid ring. Initially, the 2,5-orientation of halide groups was studied using the Stille coupling reaction of 14 with PhSnCl3; however, no reactivity was observed at either iodo group (Table 2, entry 1). This is likely due to irreversible binding of the carboxylic acid moiety to the nanoparticle surface during the oxidative addition of the iodide in the 2-position.3,4 This binding event allows for the additional interaction of the acid group with the Pd surface leading to catalyst deactivation. When the halide at the 2-position was changed to bromide in 17, no reactivity was observed for coupling at either position after 24.0 h at room temperature using 0.05 mol% Pd (Table 2, entry 2). Interestingly, when the catalyst loading was increased to 0.10 mol% Pd, a 6.8 ± 0.9% yield of the mono-substituted product, 2-bromo-5-phenylbenzoic acid (18), was obtained. Increasing the temperature of the reaction to 40.0 °C with a Pd loading of 0.10 mol% resulted in a substantial increase in the formation of 18, generating a 23.3 ± 1.6% yield after 24.0 h. When the reaction was performed at 60.0 °C with a catalyst loading of 0.05 mol% Pd, a further increase in the yield of 18 (41.3 ± 1.4%) was obtained after 24.0 h. No indication of reactivity towards the bromo-substituent was noted under any of the reaction conditions. Catalytic poisoning of the nanoparticle surface was not observed due to the required thermal activation of the C–Br bond for reactivity and due to the more reactive C–I bond driving oxidative addition. As a result, the interaction between the carboxylic acid functional group and the Pd surface was minimized. When 19 was used in the reaction (Table 2, entry 3), no detectable product was generated at room temperature with 0.05 mol% Pd; however, when the catalyst loading was increased to 0.10 mol% Pd, the mono-substituted product, 2-chloro-5-phenylbenzoic acid (20), was generated with a yield of 70.1 ± 15.8% over 24.0 h. Increasing the temperature of the reaction generated an increasing amount of 20 with 83.1 ± 6.0% (0.10 mol% Pd) and 88.0 ± 8.3% (0.05 mol% Pd) yields at 40.0 °C and 60.0 °C, respectively. Since the chloro-component was not reactive under the given conditions, catalyst surface poisoning was not observed. The reactivity differences between the iodo-component in these two compounds likely arise from changes in the electronics of the rings structure.
| Entry | Aryl dihalides | Possible products | |||
|---|---|---|---|---|---|
| Mono-substituted | Percent yield | Di-substituted | Percent yield | ||
| Reaction conditions:a 0.05 mol% Pd, 25 °C, 24 h.b 0.10 mol% Pd, 25 °C, 24 h.c 0.10 mol% Pd, 40 °C 24 h.d 0.05 mol% Pd, 60 °C 24 h. | |||||
| 1 |
|
|
0.0a
0.0b 0.0c 0.0d |
|
0.0a
0.0b 0.0c 0.0d |
| 2 |
|
|
0.0a
6.8 ± 0.9b 23.3 ± 1.6c 41.3 ± 1.4d |
|
0.0a
0.0b 0.0c 0.0d |
| 3 |
|
|
0.0a
70.1 ± 15.8b 83.1 ± 6.0c 88.0 ± 8.3d |
|
0.0a
0.0b 0.0c 0.0d |
| 4 |
|
|
73.9 ± 4.0a
92.8 ± 4.3b 97.9 ± 2.4c 95.5 ± 5.5d |
|
0.0a
0.0b 0.0c 0.0d |
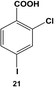
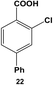
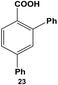
The 2,4-position of the halide groups in benzoic acid was also examined using 21 (Table 2, entry 4). The initial reaction was performed at room temperature using 0.05 mol% Pd, generating 2-chloro-4-phenylbenzoic acid (22) with 73.9 ± 4.0% yield after 24.0 h. When the catalyst loading was increased to 0.10 mol% Pd, the yield increased to 92.8 ± 4.3% after 24.0 h, while at elevated temperatures, almost quantitative yields were obtained for mono-substituted product 22 at the selected catalyst loadings. The increase in the product yield for the reaction of iodo-component 21 compared with 19 suggests that the coupling reaction was accelerated by the combined effect of the chloro-component in the 2-position and electron-withdrawing carboxylic acid moiety, which is para to the iodo group.
The results of the aryl dihalide reactions demonstrate the degree of intramolecular selectivity of the peptide-capped Pd nanocatalysts given two different halide groups in the same substrate. Similar to the results of the intermolecular reactivity, the observed trend for the catalyst selectivity towards aryl dihalides is directly related to the increasing bond strength of the C–X species; however, the orientation of the two halogens within the ring structure can lead to activating and/or deactivating effects. The reactivity of the aryl dihalide substrates likely follows a step-wise process wherein the mono-substituted intermediate was generated first by the reaction of the more active C–I component. To this end, the orientation of the halide groups in the aryl dihalide substrate affects the reactivity of the halide components, but not the selectivity of the nanocatalyst. The 1,4-position of the halogens tends to deactivate the carbon–halogen bond such that higher Pd loadings are required to drive the reaction at room temperature; however, the 1,3-orientation likely activates the bond for the coupling reaction using less Pd. The presence of activating or deactivating groups in the aryl ring did not alter the selectivity of the Pd nanocatalyst with respect to halide identity.
Conclusions
We have studied the reactivity of different aryl halide substrates in a single reaction system to probe the reactivity, catalytic mechanism, and selectivity of peptide-capped Pd nanoparticles. While a variety of catalytic nanoparticles have been studied for C–C coupling reactions,4,9–11 the peptide-based materials are interesting as they react in an aqueous-based solvent, with very low Pd concentrations and reaction temperatures. To further such materials for enhanced reactivity, a fundamental understanding of the reaction mechanism is required. In this sense, the release of free Pd0 atoms into the solution after the initial catalytic cycle of highly active iodo-components does not allow for coupling at less active bromo-groups. The results indicate that the oxidative addition of the C–Br bond requires thermal activation at 40.0 °C, suggesting that an energy barrier must be overcome to enable coupling, thereby allowing for a degree of thermal-control for reaction selectivity at low temperatures. In both intermolecular and intramolecular systems, the selectivity trend for the Pd nanoparticles directly correlates with the strength of the C-halide bond dissociation energy wherein the weakest C–I bond readily reacts at room temperature, while the C–Br bond requires mild heating and the C–Cl bond does not react. Consequently, in aryl dihalide systems, the presence of activating/deactivating groups in the ring alters the electronics and reactivity of the halide components, but the selectivity of the catalyst is generally retained with regards to the halide identity. These results present a deeper understanding of the catalytic mechanism for nanoparticle-driven C–C coupling reactions, which could be useful for the design of optimized systems.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. CBET-1157431. Further support from the University of Miami is acknowledged. The authors wish to thank Dr James Wilson for helpful discussions.Notes and references
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- D. B. Pacardo, M. Sethi, S. E. Jones, R. R. Naik and M. R. Knecht, ACS Nano, 2009, 3, 1288–1296 CrossRef CAS.
- J. C. Garcia-Martinez, R. Lezutekong and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 5097–5103 CrossRef CAS.
- R. Tatumi, T. Akita and H. Fujihara, Chem. Commun., 2006, 3349–3351 RSC.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS.
- J. C. Park, E. Heo, A. Kim, M. Kim, K. H. Park and H. Song, J. Phys. Chem. C, 2011, 115, 15772–15777 CAS.
- V. S. Coker, J. A. Bennett, N. D. Telling, T. Henkel, J. M. Charnock, G. van der Laan, R. A. D. Pattrick, C. I. Pearce, R. S. Cutting, I. J. Shannon, J. Wood, E. Arenholz, I. C. Lyon and J. R. Lloyd, ACS Nano, 2010, 4, 2577–2584 CrossRef CAS.
- J.-H. Li, B.-X. Tang, L.-M. Tao, Y.-X. Xie, Y. Liang and M.-B. Zhang, J. Org. Chem., 2006, 71, 7488–7490 CrossRef CAS.
- V. Calo, A. Nacci, A. Monopoli and F. Montingelli, J. Org. Chem., 2005, 70, 6040–6044 CrossRef CAS.
- P. Dutta and A. Sarkar, Adv. Synth. Catal., 2011, 353, 2814–2822 CrossRef CAS.
- S. Kidambi, J. Dai, J. Li and M. L. Bruening, J. Am. Chem. Soc., 2004, 126, 2658–2659 CrossRef CAS.
- T. Mizugaki, M. Murata, S. Fukubayashi, T. Mitsudome, K. Jitsukawa and K. Kaneda, Chem. Commun., 2008, 241–243 RSC.
- T. Ueno, M. Suzuki, T. Goto, T. Matsumoto, K. Nagayama and Y. Watanabe, Angew. Chem., Int. Ed., 2004, 43, 2527–2530 CrossRef CAS.
- O. M. Wilson, M. R. Knecht, J. C. Garcia-Martinez and R. M. Crooks, J. Am. Chem. Soc., 2006, 128, 4510–4511 CrossRef CAS.
- R. Bhandari and M. R. Knecht, ACS Catal., 2011, 1, 89–98 CrossRef CAS.
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237–243 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- M. O. Nutt, J. B. Hughes and M. S. Wong, Environ. Sci. Technol., 2005, 39, 1346–1353 CrossRef CAS.
- F. He and D. Zhao, Environ. Sci. Technol., 2005, 39, 3314–3320 CrossRef CAS.
- S. Li, Y.-L. Fang, C. D. Romanczuk, Z. Jin, T. Li and M. S. Wong, Appl. Catal., B, 2012, 125, 95–102 CrossRef CAS.
- Z. Zhang, Q. Fu, X. Li, X. Huang, J. Xu, J. Shen and J. Liu, J. Biol. Inorg. Chem., 2009, 14, 653–662 CrossRef CAS.
- R. Bhandari, R. Coppage and M. R. Knecht, Catal. Sci. Technol., 2012, 2, 256–266 CAS.
- V. L. Budarin, J. H. Clark, R. Luque, D. J. Macquarrie and R. J. White, Green Chem., 2008, 10, 382–387 RSC.
- Z. Chen, Z.-M. Cui, P. Li, C.-Y. Cao, Y.-L. Hong, Z. Wu and W.-G. Song, J. Phys. Chem. C, 2012, 116, 14986–14991 CAS.
- C. Yang, A. K. Manocchi, B. Lee and H. Yi, J. Mater. Chem., 2011, 21, 187–194 RSC.
- Y. B. Malysheva, A. V. Gushchin, Y. Mei, Y. Lu, M. Ballauff, S. Proch and R. Kempe, Eur. J. Inorg. Chem, 2008, 379–383 CrossRef CAS.
- D. B. Pacardo, J. M. Slocik, K. C. Kirk, R. R. Naik and M. R. Knecht, Nanoscale, 2011, 3, 2194–2201 RSC.
- R. Coppage, J. M. Slocik, B. D. Briggs, A. I. Frenkel, H. Heinz, R. R. Naik and M. R. Knecht, J. Am. Chem. Soc., 2011, 133, 12346–12349 CrossRef CAS.
- R. Coppage, J. M. Slocik, B. D. Briggs, A. I. Frenkel, R. R. Naik and M. R. Knecht, ACS Nano, 2012, 6, 1625–1636 CrossRef CAS.
- R. Coppage, J. M. Slocik, M. Sethi, D. B. Pacardo, R. R. Naik and M. R. Knecht, Angew. Chem., Int. Ed., 2010, 49, 3767–3770 CrossRef CAS.
- R. B. Pandey, H. Heinz, J. Feng, B. L. Farmer, J. M. Slocik, L. F. Drummy and R. R. Naik, Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 RSC.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- J. Xu, A. R. Wilson, A. R. Rathmell, J. Howe, M. Chi and B. J. Wiley, ACS Nano, 2011, 5, 6119–6127 CrossRef CAS.
- B. d. Darwent, Bond Dissociation Energies in Simple Molecules, U.S. Department of Commerce, National Bureau of Standards, Washington, DC, 1970 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |