Halide bridged trinuclear rhodium complexes and their inhibiting influence on catalysis†
Angelika
Preetz
a,
Christina
Kohrt
a,
Antje
Meißner
a,
Siping
Wei
a,
Hans-Joachim
Drexler
a,
Helmut
Buschmann
b and
Detlef
Heller
*a
aLeibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059, Rostock, Germany. E-mail: detlef.heller@catalysis.de; Fax: +49 381-1281-51183; Tel: +49 381-1281-183
bPharmaconsulting Aachen, Ludwigsallee 21, 052062 Aachen, Germany. E-mail: h.buschmann@pharma-consulting-aachen.de
First published on 26th September 2012
Abstract
The addition of halides to the cationic solvate complexes of the type [Rh(PP)(solvent)2][anion] leads to the formation of trinuclear μ3-halide-bridged complexes. The corresponding complexes [Rh3(PP)3(μ3-X)2][BF4] with X = Cl or Br and diphosphines PP Me-DuPHOS, Et-DuPHOS, DIPAMP, t-Bu-BisP* and Tangphos were characterized – in most cases – also by X-ray analysis. By reducing the concentration of the active catalyst, the in situ formation of these μ-halide-bridged multinuclear complexes in catalytic reactions leads to a decrease in activity or even to a total inactivity. The required halides – in most cases chloride – are usually present as impurities in the substrates (also when produced industrially). The extent of deactivation, known from enzyme catalysis as competitive inhibition, depends on several factors: the type of halide, the ratio of stability constants of multinuclear halide complexes and of substrate complexes, and the concentration of halide and substrate in solution.
Introduction
The influence of basic additives on the selectivity of rhodium-complex-catalyzed reactions such as e.g. asymmetric homogeneous hydrogenation has often been described in the literature.1 Halpern et al. were the first to succeed in characterizing a reaction product formed by treatment of a cationic rhodium complex with a base. The reaction of [Rh(DPPE)(MeOH)2][BF4] (DPPE = bis(diphenylphosphino)-ethane) with triethylamine led to the trinuclear complex [Rh3(DPPE)3(μ3-OMe)2][BF4]2 Recently, we reported the synthesis and characterization of further rhodium complexes of this kind.3 Structurally similar trinuclear rhodium complexes are described in ref. 4. With ligands Me-DuPhos, DIPAMP, DPPE, DPPP as well as BINAP several complexes with μ3-bridging methoxy and/or hydroxy anions were characterized by X-ray analysis.3,5 They share a common structural motif: the three rhodium centers define a regular triangle, each is coordinated by one bidentate diphosphine ligand and they are held in place by two μ3-bridging anions. The P–Rh–P plane is perpendicular to the Rh3 plane, and the μ3-bridging anions are located above and below the Rh3 plane.†A possible reaction sequence for the irreversible formation of such trinuclear complexes has been described by Saito et al. for BINAP as the ligand.5 The base generates an anion from the solvent which initially reacts with the solvate complex6 to form a dinuclear complex. In a consecutive step the dinuclear complex reacts with another solvate complex forming the trinuclear complex.
The rather stable trinuclear Rh(I)-complexes can be applied as ‘diolefin-free precatalysts’, e.g. in asymmetric homogeneous hydrogenations although, in such cases, characteristic induction periods are observed in the corresponding hydrogen consumption curves.3 The trinuclear complexes themselves are in fact catalytically inactive due to the steric hindrance which hampers access to the [Rh]3 core. However, hydrogenations of prochiral olefins with such trinuclear complexes result in the same activity and selectivity as with the corresponding solvate complex if an acid is added to the reaction mixture, which supports the idea that the solvate complex is the actual catalytically active species.3
Very interesting and of practical relevance is the fact that certain substrates can be basic enough to trigger the formation of such trinuclear complexes. An example is provided by the prochiral olefin (E)-1-(2-methyl-3-phenylallyl)piperidine: at a substrate to catalyst ratio of 10 already 40% of the overall rhodium complex concentration can be assigned to a trinuclear complex as detected by 31P-NMR spectroscopy.3
Although Saito et al. excluded the possibility of trinuclear methoxy-bridged complex formation with BINAP, i.e. [Rh3(BINAP)3(μ3-OMe)2][ClO4], for steric reasons,5 the question remains whether such trinuclear complexes might be formed in the presence of even bulkier anions such as halides. This is especially important considering the fact that, following substrate synthesis, e.g. of prochiral olefins, halides of alkali metals such as NaCl may remain in the substrate as an impurity due to insufficient removal during work up – also on an industrial scale. For example, it could be shown that in the hydrogenation of 1-methylenesuccinamic acid7 with a rhodium-DuPhos catalyst activity might be increased by a factor of 26 if traces of chloride remaining from substrate synthesis were thoroughly removed from the substrate.8 Unfortunately, the findings could not be explained at that time.
Due to the relevance of deactivation phenomena in industrial catalytic applications,9 we set out to investigate whether (a) halides may lead to the formation of hitherto unknown trinuclear complexes of the type [Rh3(PP)3(μ3-X)2][anion] (PP = diphosphine, X = halide) and if so (b) whether their formation negatively affects rhodium catalysed processes.
Results and discussion
So far methoxo- or hydroxo-bridged trinuclear complexes have been reported only with the chiral ligands BINAP, DIPAMP and Me-DuPhos. By using the ligand t-Bu-BisP* we have successfully shown that such interesting complexes might be formed also in the presence of very electron rich ligands: the X-ray structure of the corresponding hydroxo-bridged trinuclear complex is shown in Fig. 1.![Molecular structure of the cation of [Rh3(t-Bu-BisP*)3(μ3-OH)2][BF4] (only one of the two cations in the unit cell is shown); ORTEP, 30% probability ellipsoids. The hydrogen atoms of the ligand were omitted for clarity. Selected distances [Å] and angles [°]: Rh–Rh 3.074–3.084(2), Rh–P 2.177–2.185(2), Rh–O 2.146–2.163(4), P–Rh–P 85.0–85.2(1), O–Rh–O 68.6–68.9(2). 31P{1H}-NMR (MeOH-d4): δ = 85.1 ppm , JP–Rh = 196.9 Hz.](/image/article/2013/CY/c2cy20591b/c2cy20591b-f1.gif) | ||
| Fig. 1 Molecular structure of the cation of [Rh3(t-Bu-BisP*)3(μ3-OH)2][BF4] (only one of the two cations in the unit cell is shown); ORTEP, 30% probability ellipsoids. The hydrogen atoms of the ligand were omitted for clarity. Selected distances [Å] and angles [°]: Rh–Rh 3.074–3.084(2), Rh–P 2.177–2.185(2), Rh–O 2.146–2.163(4), P–Rh–P 85.0–85.2(1), O–Rh–O 68.6–68.9(2). 31P{1H}-NMR (MeOH-d4): δ = 85.1 ppm , JP–Rh = 196.9 Hz. | ||
As already mentioned, certain substrates are basic enough to trigger the formation of such trinuclear complexes. One further example, so far undescribed, is provided by [2-(3-methoxy-phenyl)-cyclohex-1-enylmethyl]-dimethylamine:10 when this substrate is hydrogenated in MeOH with [Rh(DIPAMP)(MeOH)2][BF4] at room temperature up to 75% of the rhodium content is unavailable for hydrogenation due to the formation of inactive trinuclear complexes, see Fig. S1 (ESI†).
μ-Chloro-bridged multinuclear complexes
Addition of a solution of NaCl in methanol11 to the solvate complex [Rh(Me-DuPHOS)(MeOH)2][BF4]12 in the same solvent at room temperature led to the spontaneous formation of a precipitate which is insoluble even at higher temperatures. The isolated solid dissolves in acetone and the corresponding 31P-NMR spectrum in acetone-d6 exhibits two doublets, Fig. 2, indicative of the presence of two compounds. From a dichloromethane solution of the solid that was layered with diethyl ether, crystals of one of the compounds were grown suitable for X-ray analysis. Surprisingly, the trinuclear μ3-chloro-bridged complex [Rh3(MeDuPHOS)3(μ3-Cl)2][BF4] was obtained, its molecular structure is shown in Fig. 3 (left).![31P{1H}-NMR spectrum of the precipitate resulting from the addition of NaCl to [Rh(Me-DuPHOS)(MeOH)2][BF4] dissolved in acetone-d6.](/image/article/2013/CY/c2cy20591b/c2cy20591b-f2.gif) | ||
| Fig. 2 31P{1H}-NMR spectrum of the precipitate resulting from the addition of NaCl to [Rh(Me-DuPHOS)(MeOH)2][BF4] dissolved in acetone-d6. | ||
![Molecular structure of the cations of [Rh3((R,R)-Me-DuPHOS)3(μ3-Cl)2][BF4] (left) as well as [Rh3((R,R)-t-Bu-BisP*)3(μ3-Cl)2][BF4], (right); ORTEP 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances [Å] and angles [°] for [Rh3((R,R)-Me-DuPHOS)3(μ3-Cl)2][BF4]/[Rh3((R,R)-t-Bu-BisP*)3(μ3-Cl)2][BF4]: Rh–Rh 3.175–3.275 (2)/3.230–3.267 (2), Rh–P 2.172–2.179 (2)/2.181–2.187 (2), Rh–Cl 2.460–2.478 (4)/2.442–2.475 (4), P–Rh–P 85.4–85.7 (1)/84.9–85.7 (1), Cl–Rh–Cl 81.5–81.6 (1)/80.2–80.7 (1).](/image/article/2013/CY/c2cy20591b/c2cy20591b-f3.gif) | ||
| Fig. 3 Molecular structure of the cations of [Rh3((R,R)-Me-DuPHOS)3(μ3-Cl)2][BF4] (left) as well as [Rh3((R,R)-t-Bu-BisP*)3(μ3-Cl)2][BF4], (right); ORTEP 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances [Å] and angles [°] for [Rh3((R,R)-Me-DuPHOS)3(μ3-Cl)2][BF4]/[Rh3((R,R)-t-Bu-BisP*)3(μ3-Cl)2][BF4]: Rh–Rh 3.175–3.275 (2)/3.230–3.267 (2), Rh–P 2.172–2.179 (2)/2.181–2.187 (2), Rh–Cl 2.460–2.478 (4)/2.442–2.475 (4), P–Rh–P 85.4–85.7 (1)/84.9–85.7 (1), Cl–Rh–Cl 81.5–81.6 (1)/80.2–80.7 (1). | ||
Because of the increased steric demand of chloride in comparison to the methoxy or hydroxy bridge present in analogous trinuclear complexes, the trinuclear μ3-chloro-bridged complex possesses longer Rh–Rh and Rh–X (X = Cl or OMe/OH) bonds and wider X–Rh–X angles. Furthermore, in comparison to the diolefin complexes [Rh(Me-DuPHOS)(COD)][BF4]13 the Rh–P bond is considerably shortened.
The second species present in the isolated solid is the neutral dinuclear complex [Rh2(Me-DuPHOS)2(μ2-Cl)2]. The identity of this species, initially postulated based on NMR data and as a possible intermediate in the formation of the trinuclear complex according to Saito's hypothesis (Scheme 1), was confirmed by X-ray analysis.14
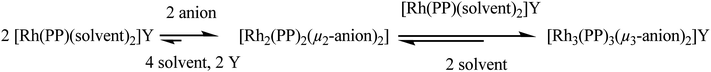 | ||
| Scheme 1 General reaction sequence for the formation of trinuclear μ3-halide bridged complexes in agreement with Saito et al.5 | ||
Hence, the addition of NaCl to [Rh(Me-DuPHOS)(MeOH)2][BF4] results in the formation of two complexes, the cationic trinuclear complex [Rh3(Me-DuPHOS)3(μ3-Cl)2][BF4] (31P{1H}-NMR, δ 96.8 ppm, JP–Rh = 203.1 Hz) and the neutral dinuclear complex [Rh2(Me-DuPHOS)2(μ2-Cl)2] (31P{1H}-NMR δ 96.5 ppm, JP–Rh = 199.6 Hz). While the first type of complex is described here for the first time, neutral dinuclear complexes have been known for some time.
To assess whether the formation of both complexes takes place through a reversible consecutive reaction, the neutral complex [Rh2(Me-DuPHOS)2(μ2-Cl)2] (intermediate in Scheme 1) was treated with the cationic solvate complex [Rh(Me-DuPHOS)(MeOH)2][BF4] in a THF–MeOH mixture.15 The 31P-NMR spectrum (Fig. S2, ESI†) proves that indeed the trinuclear complex is formed, in agreement with the sequence of equilibria described in Scheme 1. Similar results are detailed in ref. 4b.
It is evident that the formation of such trinuclear species must be taken into account if suitable anions which can act as bridging ligands are present in solution.
The reaction of the DIPAMP solvate complex [Rh(DIPAMP)(MeOH-d4)2][BF4]16 with a methanolic NaCl solution also leads to a 31P-NMR spectrum which shows two doublets, Fig. S3a (ESI†). Unlike the experimental findings observed when the Me-DuPHOS ligand was used, the resulting precipitate can be redissolved at elevated temperature. Upon slow cooling, yellow needles precipitated, which were isolated and characterized by X-ray analysis, Fig. S4 (ESI†). Hence, for DIPAMP too the trinuclear μ3-chloro-bridged complex is formed: [Rh3(DIPAMP)3(μ3-Cl)2][BF4] (31P{1H}-NMR δ 72.5 ppm, JP–Rh = 203.5 Hz). Comparison of the 31P-NMR data of the second complex with literature values confirms the presence of the neutral dinuclear complex [Rh2(DIPAMP)2(μ2-Cl)2] (31P{1H}-NMR δ 74.7 ppm, JP–Rh = 199.6 Hz) analogous to the one formed with Me-DuPhos.14103Rh-NMR measurements indicate similar electronic environments in both species as well: the corresponding chemical shifts are −144 ppm for the trinuclear and −77 ppm for the dinuclear complex. In the case of the Me-DuPHOS complexes the corresponding values are −306 ppm (trinuclear complex) and −280 ppm (dinuclear complex).
An interesting aspect is the dependence of the ratio of trinuclear to dinuclear complex on NaCl concentration. Formally, for the formation of the trinuclear complex 2 equivalents of Cl are necessary per 3 Rh i.e. 2/3 Cl per 1 Rh, in the case of the dinuclear complex it is 2 Cl per 2 Rh. Indeed, the experiment shows that the share of the dinuclear complex [Rh2(DIPAMP)2(μ2-Cl)2] increases with increasing NaCl concentration, which can be explained with Scheme 1 in the absence of the solvate complex (MeOH-d4: δ 80.6 ppm, JP–Rh = 208.1 Hz), compare Fig. S3a/b (ESI†).
Transferring the solvate complex [Rh(t-Bu-BisP*)(MeOH-d4)2][BF4]4a into a Young-NMR tube containing solid NaCl leads to a homogeneous solution and to the 31P-NMR spectrum shown in Fig. S5 (ESI†). Also in this case the trinuclear complex [Rh3(t-Bu-BisP*)3(μ3-Cl)2][BF4] (86.8 ppm, JP–Rh = 204.5 Hz) is formed, as well as another so far unknown species (84.6 ppm, JP–Rh = 199.3 Hz) which is assumedly the dinuclear complex [Rh2(t-Bu-BisP*)2(μ2-Cl)2]. The molecular structure of [Rh3(t-Bu-BisP*)3(μ3-Cl)2][BF4] is shown in Fig. 3, right.17
Using Tangphos as the ligand, two species are observed in the 31P-NMR spectrum as well if a solution of NaCl dissolved in MeOH is added to the solvate complex, Fig. S6 (ESI†). One species is again the trinuclear complex [Rh3(Tangphos)3(μ3-Cl)2][BF4] (120.5 ppm, JP–Rh = 200.9 Hz), its detailed characterization has been discussed in ref. 18. The hitherto unknown second species (120.2 ppm, JP–Rh = 198 Hz) is most probably the corresponding dinuclear complex [Rh2(Tangphos)2(μ2-Cl)2].
In contrast to what was observed with Me-DuPHOS, DIPAMP, t-Bu-BisP* and Tangphos, the addition of a methanolic solution of NaCl to the BINAP solvate complex [Rh(BINAP)(MeOH-d4)2][BF4]6 leads to the formation of only one species, as the 31P-NMR spectrum of the dry precipitate redissolved in CH2Cl2-d2 proves. Comparison with published data confirms that the dinuclear complex [Rh2(BINAP)2(μ2-Cl)2] (δ 49.6 ppm, JP–Rh = 195.5 Hz, in CD2Cl2) was formed.14 However, the trinuclear complex might have formed, but rapidly depleted from solution because of a shift in equilibrium due to the crystallization of the dinuclear complex. To verify that indeed only one species had been formed, the experiment was repeated in a MeOH–CH2Cl2 mixture as the dinuclear complex [Rh2(BINAP)2(μ2-Cl)2] is well soluble in CH2Cl2. The 31P-NMR spectrum (Fig. S7a, ESI†) now shows two doublets. The newly formed species is most probably the trinuclear cation [Rh3((R)-BINAP)3(μ2-Cl)2]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 which is in good agreement with the dependence of its relative concentration from that of NaCl, vide supra; if the NaCl concentration increases more dinuclear complex is detected by 31P-NMR, Fig. S7a/b (ESI†).
19 which is in good agreement with the dependence of its relative concentration from that of NaCl, vide supra; if the NaCl concentration increases more dinuclear complex is detected by 31P-NMR, Fig. S7a/b (ESI†).
Also in the case of Et-DuPHOS – in analogy to BINAP – only one species is formed upon the addition of NaCl to the solvate complex [Rh(Et-DuPHOS)(MeOH)2][BF4].12 The initially formed voluminous precipitate crystallized overnight yielding yellow crystals of the neutral dinuclear complex [Rh2((R,R)-Et-DuPHOS)2(μ2-Cl)2] (δ 91.1 ppm, JP–Rh = 199.7 Hz, in THF-d8); the molecular structure is shown in Fig. 4.
![Molecular structure of [Rh2((R,R)-Et-DuPHOS)2(μ2-Cl)2]; ORTEP, 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances [Å] and angles [°]: Rh–Rh 3.308 (1), Rh–P 2.158–2.161 (1), Rh–Cl 2.409–2.435 (1), P–Rh–P 85.4–86.2 (1), Cl–Rh–Cl 83.7–84.7 (1).](/image/article/2013/CY/c2cy20591b/c2cy20591b-f4.gif) | ||
| Fig. 4 Molecular structure of [Rh2((R,R)-Et-DuPHOS)2(μ2-Cl)2]; ORTEP, 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances [Å] and angles [°]: Rh–Rh 3.308 (1), Rh–P 2.158–2.161 (1), Rh–Cl 2.409–2.435 (1), P–Rh–P 85.4–86.2 (1), Cl–Rh–Cl 83.7–84.7 (1). | ||
With the ligands Et-DuPHOS and BINAP it is hence shown for the first time that under certain conditions (MeOH as the solvent and high chloride concentration) it is possible to form selectively the neutral μ2-chloro-bridged complex by addition of chloride ions to the corresponding cationic solvate complex [Rh(PP)(MeOH)2][BF4]. Usually these neutral μ2-anion-bridged dinuclear complexes are formed via ligand exchange, e.g. from [Rh2(COD)2(μ2-Cl)2] and a chelating diphosphine.14
μ-Bromo-bridged multinuclear complexes
The synthesis of μ-bromo-bridged multinuclear complexes was investigated with Me-DuPhos and DIPAMP as examples. The addition of a methanolic solution of NaBr11 to the solvate complex [Rh(Me-DuPHOS)(MeOH)2][BF4] resulted in a yellow precipitate. The precipitate was redissolved in CH2Cl2 (31P-NMR spectrum: Fig. S9, ESI†) and layered with diethyl ether. Single crystals of both the neutral dinuclear and the cationic trinuclear complex could be isolated and analyzed by X-ray analysis, Fig. 5.20![Molecular structure of [Rh2((S,S)-Me-DuPHOS)2(μ2-Br2)] (left) and of the cation in [Rh3((R,R)-Me-DuPHOS)3(μ3-Br)0,8(μ3-Cl)1,2][BF4] (right); ORTEP, 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances [Å] and angles [°] for [Rh2((S,S)-Me-DuPHOS)2(μ2-Br2)]/[Rh3((R,R)-Me-DuPHOS)3(μ3-Br)0,8(μ3-Cl)1,2][BF4]: Rh–Rh 3.263 (1)/3.221–3.274 (1), Rh–P 2.164–2.172 (1)/2.168–2.176 (1), Rh–Br 2.526–2.572 (1)/2.538–2.571 (8), P–Rh–P 86.0–86.1 (1)/85.4–85.7 (1), Br–Rh–Br 85.1–85.2 (1)/85.2–85.9 (2).](/image/article/2013/CY/c2cy20591b/c2cy20591b-f5.gif) | ||
| Fig. 5 Molecular structure of [Rh2((S,S)-Me-DuPHOS)2(μ2-Br2)] (left) and of the cation in [Rh3((R,R)-Me-DuPHOS)3(μ3-Br)0,8(μ3-Cl)1,2][BF4] (right); ORTEP, 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances [Å] and angles [°] for [Rh2((S,S)-Me-DuPHOS)2(μ2-Br2)]/[Rh3((R,R)-Me-DuPHOS)3(μ3-Br)0,8(μ3-Cl)1,2][BF4]: Rh–Rh 3.263 (1)/3.221–3.274 (1), Rh–P 2.164–2.172 (1)/2.168–2.176 (1), Rh–Br 2.526–2.572 (1)/2.538–2.571 (8), P–Rh–P 86.0–86.1 (1)/85.4–85.7 (1), Br–Rh–Br 85.1–85.2 (1)/85.2–85.9 (2). | ||
In the case of DIPAMP, after the addition of NaBr to the solvate complex [Rh(DIPAMP)(MeOH)2][BF4] a red brown precipitate was instantly formed. Single crystals suitable for X-ray analysis, however, could not be isolated. Still, NMR measurements showed that under the described experimental conditions only one species was formed. From the NMR spectroscopic data (1H-NMR, Fig. S10, 31P-NMR, Fig. S11, ESI†) we can infer that only the neutral dinuclear complex [Rh2(DIPAMP)2(μ2-Br)2] was formed. The coupling constant in the 31P-NMR spectrum JP–Rh = 198.3 Hz is very similar to the ones of the μ2-chloro-bridged dinuclear complexes of DIPAMP and Me-DuPHOS (for both 199.6 Hz).
μ-Halide-bridged multinuclear complexes: deactivating species
As already mentioned in the introduction, μ3-methoxy- and μ3-hydroxy-bridged trinuclear complexes affect the efficiency of asymmetric hydrogenations as they are not active in hydrogenation.3To investigate the influence of μ-halide-bridged multinuclear complexes on the catalytic activity, the hydrogenation of methyl-(Z)-α-acetamidocinnamate (mac) and dimethyl itaconate with the solvate complex [Rh(DIPAMP)(MeOH)2][BF4] was monitored in the presence of sodium salts (Cl, Br, I).
At a first glimpse hydrogen consumption curves for the hydrogenation of mac with Rh(DIPAMP)(MeOH)2][BF4]3,21 in the presence of NaCl show no effect. A closer look at higher conversions, however, unequivocally shows that – if compared to the reference curve – the deviations towards the end of hydrogenations become more apparent with increasing NaCl concentration, Fig. S12 (ESI†). In other words, as the concentration of the chiral olefin drops towards the end of the hydrogenation the halide successfully competes for the solvate complex.
Fig. 6 illustrates the influence of different halides on the test hydrogenation of mac with the DIPAMP system in methanol.
![Hydrogen consumption curves for the hydrogenations of 1.0 mmol methyl (Z)-α-acetamidocinnamate with 0.01 mmol [Rh(DIPAMP)(MeOH)2][BF4] under addition of sodium halides: red: without additive; green: 0.1 mmol NaCl, blue: 0.1 mmol NaBr, gray 0.1 mmol NaI (in each case the sodium halide was put into a glass ampoule together with the substrate); conditions: 15.0 mL MeOH at 25.0 °C and 1.01 bar overall pressure. For clarity the green, blue and gray curve are each displayed with a 1 min shift compared with the previous curve.](/image/article/2013/CY/c2cy20591b/c2cy20591b-f6.gif) | ||
| Fig. 6 Hydrogen consumption curves for the hydrogenations of 1.0 mmol methyl (Z)-α-acetamidocinnamate with 0.01 mmol [Rh(DIPAMP)(MeOH)2][BF4] under addition of sodium halides: red: without additive; green: 0.1 mmol NaCl, blue: 0.1 mmol NaBr, gray 0.1 mmol NaI (in each case the sodium halide was put into a glass ampoule together with the substrate); conditions: 15.0 mL MeOH at 25.0 °C and 1.01 bar overall pressure. For clarity the green, blue and gray curve are each displayed with a 1 min shift compared with the previous curve. | ||
The enantioselectivity (97%) of all experiments was identical, in the range of reproducibility. Only with NaI, where a substantial deactivation was observed, the enantioselectivity dropped to 93%.
With prochiral olefins such as dimethyl itaconate which form much less stable substrate complexes, a much more pronounced interference during the hydrogenation is expected.22 In Fig. 7 the hydrogen consumption curves for the hydrogenation of dimethyl itaconate with [Rh(DIPAMP)(MeOH)2][BF4] after addition of NaCl and NaBr (0.1 mmol each) are shown.
![Hydrogen consumption for the hydrogenation of 1.0 mmol dimethyl itaconate with 0.01 mmol [Rh(DIPAMP)(MeOH)2][BF4] under addition of different sodium halides: red: no additive (88% ee); green: 0.1 mmol NaCl, (82% ee); blue: 0.1 mmol NaBr, (80% ee); in each case the halide was added to the substrate in a glass ampoule); conditions: each 15.0 mL MeOH at 20.0 °C (due to the fact that at 25.0 °C without additive an influence of diffusion cannot be excluded) and 1.01 bar overall pressure.](/image/article/2013/CY/c2cy20591b/c2cy20591b-f7.gif) | ||
| Fig. 7 Hydrogen consumption for the hydrogenation of 1.0 mmol dimethyl itaconate with 0.01 mmol [Rh(DIPAMP)(MeOH)2][BF4] under addition of different sodium halides: red: no additive (88% ee); green: 0.1 mmol NaCl, (82% ee); blue: 0.1 mmol NaBr, (80% ee); in each case the halide was added to the substrate in a glass ampoule); conditions: each 15.0 mL MeOH at 20.0 °C (due to the fact that at 25.0 °C without additive an influence of diffusion cannot be excluded) and 1.01 bar overall pressure. | ||
The graph clearly indicates that indeed small amounts of halide are enough to induce significant deactivations. In the case of NaI, after the addition of 0.1 mmol no consumption of hydrogen was observed.
The above investigations show that with DIPAMP both the trinuclear and dinuclear complexes are formed with NaCl and selectively the dinuclear one with NaBr in MeOH. Instead no conclusion can be drawn about the exact nature of the multinuclear species formed in the presence of NaI. Only few examples of μ2-iodo bridged dinuclear complexes have been described in the literature.23
The following example proves how important the formation of the catalytically inactive trinuclear μ3-halide-bridged complex can be. Treatment of the solvate complex [Rh(DIPAMP)(MeOH-d4)2][BF4] with the prochiral olefin ((Z)-3-[1-(dimethylamino)-2-methylpent-2-en-3-yl]phenol),24 Fig. S13 (ESI†) (and also with its hydrogenation product (3-[(2R,3S)-1-(dimethylamino)-2-methylpentan-3-yl]phenol, Fig. S14 (ESI†)) leads to a 31P-NMR spectrum which shows that almost 34% of the signal intensity corresponds to the catalytically inactive trinuclear μ3-chloro-bridged complex [Rh3(DIPAMP)3(μ3-Cl)2][BF4]. The complex results from traces of chloride left in the substrate after its synthesis.
Conclusions
We have shown for the first time that the addition of halides to the cationic solvate complexes of the type [Rh(PP)(solvent)2][anion] leads to the formation of trinuclear μ3-halide-bridged complexes. The corresponding complexes [Rh3(PP)3(μ3-X)2][BF4] with X = Cl or Br and diphosphines PP Me-DuPHOS, Et-DuPHOS, DIPAMP, t-Bu-BisP* and Tangphos were characterized – in most cases – also by X-ray analysis.Such complexes are formed together with the neutral μ2-halide bridged dinuclear ones. Depending on the steric bulk of the diphosphine, the μ-bridging anion and their solubility, the dinuclear species is formed selectively and this represents a new synthesis route to this type of complexes, which are known from the literature.
The formation of the trinuclear species evidently takes place through a reversible consecutive reaction from the neutral dinuclear complexes as the intermediate.
By reducing the concentration of the active catalyst, the in situ formation of these μ-halide-bridged multinuclear complexes in catalytic reactions leads to a decrease in activity or even to a total inactivity. This was proven experimentally in the case of asymmetric hydrogenation. The required halides – in most cases chloride – are usually present as impurities in the substrates (also when produced industrially). The extent of deactivation, known from enzyme catalysis as competitive inhibition, depends on several factors: the type of halide, the ratio of stability constants of multinuclear halide complexes and of substrate complexes, and the concentration of halide and substrate in solution.
Experimental section
General information
All manipulations were carried out under oxygen- and moisture-free conditions under argon using standard Schlenk or drybox techniques.Diethyl ether was distilled from sodium benzophenone ketyl immediately prior to use. MeOH was freshly distilled from Magnesia turnings prior to use, MeOH-d4 from LiAlH4 while CH2Cl2-d2 were distilled from CaH2. Subsequent removal of traces of oxygen for both deuterated solvents was carried out of application of six freeze–thaw cycles. NaCl (99.5%, Merck), NaBr (> 99% Sigma Aldrich) and NaI (> 99% Fluka) were used as received.
NMR
31P{1H}, 13C{1H}, 13C DEPT, and 1H NMR spectra were obtained on a Bruker ARX-300 or ARX-400 spectrometer at 297–298 K and were referenced internally to the deuterated solvent (13C, CD2Cl2: δreference = 54 ppm, CD3OD: δreference = 49.2 ppm) or to protic impurities in the deuterated solvent (1H, CDHCl2: δreference = 5.31 ppm, CD3OD: δreference = 3.32 ppm). For chemical shifts in 31P{1H} NMR spectra, 85% H3PO4 was used as an external standard. The chemical shifts are given in ppm.X-ray structure determination
Diffraction data were collected on a STOE-IPDS II diffractometer using graphite monochromated Mo-Kα radiation. The structure was solved by direct methods (SHELXS-97)25 and refined by full matrix least square techniques against F2 (SHELXL-97).25 XP (Siemens Analytical X-ray Instruments, Inc.) was used for structure representations.[Rh3(PP*)3(μ3-Cl)2][BF4]
These complexes were obtained by hydrogenation of the [Rh(PP*)(diolefin)]BF4 complex in MeOH (1 mL) for the, respectively, prehydrogenation time and subsequent removal of hydrogen gas from the yellow solution by application of three freeze–thaw cycles. A saturated solution of NaCl in MeOH was added to the frozen out solvate complex and an orange (in general) precipitate could be isolated.[Rh2(PP*)2(μ2-Cl)2]
Ligand (PP*) and [Rh2(COD)2(μ2-X)2] were dissolved in THF. At −78 °C the ligand was slowly added to [Rh2(COD)2(μ2-X)2] and the mixture was stirred at room temperature for 2 h.After adding hexane orange or red crystals were formed.
Acknowledgements
We thank Prof. X. Zhang (Chiral Quest (Suzhou) Inc.) for generously supplying us with the Tangphos ligand. We are much obliged to Cornelia Pribbenow, who carried out the hydrogenations.Notes and references
-
(a) W. S. Knowles and M. J. Sabacky, J. Chem. Soc., Chem. Commun., 1968, 1445–1446 RSC
; (b) H. B. Kagan and T. P. Dang, J. Am. Chem. Soc., 1972, 94, 6429–6433 CrossRef CAS
- J. Halpern, D. P. Riley, A. S. C. Chan and J. J. Pluth, J. Am. Chem. Soc., 1977, 99, 8055–8057 CrossRef CAS
- A. Preetz, W. Baumann, H.-J. Drexler, C. Fischer, J. Sun, A. Spannenberg, O. Zimmer, W. Hell and D. Heller, Chem.–Asian J., 2008, 3, 1979–1982 CrossRef CAS
-
(a) C. Fischer, C. Kohrt, H.-J. Drexler, W. Baumann and D. Heller, Dalton Trans., 2011, 40, 4162–4166 RSC
- T. Yamagata, K. Tani, Y. Tatsuno and T. Saito, J. Chem. Soc., Chem. Commun., 1988, 466–468 RSC
- A. Preetz, C. Fischer, C. Kohrt, H.-J. Drexler, W. Baumann and D. Heller, Organometallics, 2011, 30, 5155–5159 CrossRef CAS
- From the hydrogenation product the THP ether of the 4-amino-2(R)-methylbutan-1-ol, an important intermediate towards the receptor antagonist TAK-637, can be synthesized.
- C. J. Cobley, I. C. Lennon, C. Praquin and A. Zanotti-Gerosa, Org. Process Res. Dev., 2003, 7, 407–411 CrossRef CAS
-
(a) D. J. Ager, A. H. M. de Vries and J. G. de Vries, Chem. Soc. Rev., 2012, 41, 3340–3380 RSC
- [2-(3-Methoxy-phenyl)-cyclohex-1-enylmethyl]-dimethylamine is a member of a class of 1,2-disubstituded cyclohexene derivatives which are important intermediates in the synthesis of pharmaceutically active compounds acting on the central nervous system. The challenge is the establishment of the desired relative and absolute stereochemistry of two new stereogenic centers responsible for the pharmacological effect. [2-(3-Methoxy-phenyl)-cyclohex-1-enylmethyl]-dimethylamine can be transformed by a stereoselective hydrogenation to Tramadol, an important analgesic drug used world-wide.
- Elemental analysis of a saturated NaCl solution in methanol showed that in 1.0 mL MeOH ca. 7 mg of NaCl can be dissolved, which corresponds to 0.12 mmol. 105 mg NaBr (1.0 mmol) and 496 mg NaI (3.3 mmol) can be dissolved in 1.0 mL MeOH.
- A. Preetz, H.-J. Drexler, C. Fischer, Z. Dai, A. Börner, W. Baumann, A. Spannenberg, R. Thede and D. Heller, Chem.–Eur. J., 2008, 14, 1445–1451 CrossRef CAS
- H.-J. Drexler, S. Zhang, A. Sun, A. Spannenberg, A. Arrieta, A. Preetz and D. Heller, Tetrahedron: Asymmetry, 2004, 15, 2139–2150 CrossRef CAS
- A. Meißner, A. Preetz, H.-J. Drexler, W. Baumann, A. Spannenberg, A. König and D. Heller, in preparation.
- The dinuclear complex is very soluble in THF, however scarcely soluble in MeOH. Vice versa, the trinuclear complex is more soluble in MeOH than in THF.
- A. Preetz, W. Baumann, C. Fischer, H.-J. Drexler, T. Schmidt, R. Thede and D. Heller, Organometallics, 2009, 28, 3673–3677 CrossRef CAS
- All attempts to isolate [Rh3(t-Bu-BisP*)3(μ3-Cl)2][BF4] from the reaction of the solvate complex with NaCl failed. However, we succeeded while trying to isolate the product complex [Rh(t-Bu-BisP*)(macH2)][BF4] from CH2Cl2: the hydrogenation product of mac was added to the solvate MeOH complex. The solvent was removed in vacuo and the remaining solid was redissolved in CH2Cl2 and layered with diethyl ether. After a few days single crystals of [Rh3(t-Bu-BisP*)3(μ3-Cl)2][BF4] suitable for X-ray analysis crystallized.
- C. Kohrt, S. Hansen, H.-J. Drexler, U. Rosenthal, A. Schulz and D. Heller, Inorg. Chem., 2012, 51, 7377–7383 CrossRef CAS
- The molecular structure of the trinuclear complex obtained in a different way is shown in Fig. S8, ESI†.
- The μ3-bridges of the trinuclear complex are disordered (population: 0.6 Cl, 0.4 Br). The only Cl source can be the solvent CH2Cl2 from which the complex was recrystallized. Only one of the four possible populations is shown.
- C. R. Landis and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1746–1754 CrossRef CAS
- The asymmetric hydrogenation of dimethyl itaconate as well as the structure and stability of the catalyst–substrate complex with the DIPAMP system are described in T. Schmidt, Z. Dai, H.-J. Drexler, W. Baumann, C. Jäger, D. Pfeifer and D. Heller, Chem.–Eur. J., 2008, 14, 4469–4471 CrossRef CAS
-
(a) J. M. Buriak, J. C. Klein, D. G. Herrington and J. A. Osborn, Chem.–Eur. J., 2000, 6, 139–150 CrossRef CAS
- The prochiral olefin (Z)-3-[1-(dimethylamino)-2-methylpent-2-en-3-yl]phenol is also an important precursor of the new analgesic drug Tapentadol which is as an open-chain analogue of Tramadol. The chirality of the new stereogenic centers is very important for the pharmacological effect. The (R,R) stereoisomer of Tapentadol is a novel, centrally acting analgesic with a dual mode of action: μ-opioid receptor (MOR) agonism and norepinephrine reuptake inhibition. Tapentadol is the first oral centrally acting analgesic to be approved in 2008 in the United States for more than a decade. The corresponding (S,S) enantiomer is a very strong opioid. The (R,S) or (S,R) stereoisomers have no analgesic effect.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1997, 64, 112–122 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: X-Ray data and experimental procedures. CCDC 883479 for [Rh3(t-Bu-BisP*)3(μ3-OH)2][BF4], CCDC 883473 for [Rh3((R,R)-Me-DuPHOS)3(μ3-Cl)2][BF4], CCDC 883480 for [Rh3((R,R)-t-Bu-BisP*)3(μ3-Cl)2][BF4], CCDC 883475 for [Rh2((R,R)-Et-DuPHOS)2(μ2-Cl)2], CCDC 883478 for [Rh2((S,S)-Me-DuPHOS)2(μ2-Br2)], CCDC 883477 for [Rh3((R,R)-Me-DuPHOS)3(μ3-Br)0,8(μ3-Cl)1,2][BF4], CCDC 883474 for [Rh3((R,R)-DIPAMP)3(μ3-Cl)2][BF4] and CCDC 883476 for [Rh3((R)-BINAP)3(μ3-Cl)2][BF4]. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c2cy20591b |
| This journal is © The Royal Society of Chemistry 2013 |