Ni2O3–Au+ hybrid active sites on NiOx@Au ensembles for low-temperature gas-phase oxidation of alcohols†
Guofeng
Zhao
a,
Huanyun
Hu
a,
Wei
Chen
a,
Zheng
Jiang
b,
Shuo
Zhang
b,
Jun
Huang
*c and
Yong
Lu
*a
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, Shanghai 200062, China. E-mail: ylu@chem.ecnu.edu.cn; Fax: +86 21-62233424
bShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
cSchool of Chemical and Biomolecular Engineering, University of Sydney, NSW 2006, Australia. E-mail: jun.huang@sydney.edu.au
First published on 22nd October 2012
Abstract
An interesting ensemble of NiOx@Au (i.e. 20–30 nm gold particles partially covered with very small NiOx segments) were clearly identified to be highly active for the low-temperature gas-phase oxidation of alcohols. On such active NiOx@Au ensembles, large amounts of Ni2O3–Au+ hybrid active sites were defined, taking a step closer to identifying the low-temperature activity. By their nature, Ni2O3 specimens not only promote the formation of Au+ ions and stabilize them but also act as an oxygen supplier to transfer oxygen species onto the Au+ sites to react with alcohol.
Bulk gold is extremely inert for catalysis.1a However, supported gold nanoparticles (Au NPs) have recently gained tremendous attention as an excellent catalyst and have blossomed in many fields.1 Up to now, multiple factors, including Au particle size and chemical state,2,3 the support properties4 (such as type,4a morphology (shape)4b–d and size4e–g) and Au–support interactions,2a,c,d have been found to contribute to the distinct catalytic activity of supported Au NPs. Among them, the small size of the Au NPs (<5 nm) has been considered as one dominant factor which contributes to higher catalytic activity, due to the size-related low coordination, unsaturated surface atoms which often act as active sites.2a–d
However, some recent contributions reported just the opposite behavior in that large gold-based particles were also active in many reactions, such as Au–Pd core–shell particles (∼10 nm) for the liquid-phase selective oxidation of alcohols,5a Au/SiO25b with large Au particles (∼15 nm) and Au–Cu/SiO25c with large AuCu alloy particles (∼25 nm) for the gas-phase selective oxidation of alcohols, Au/Al2O3 with Au particles (50–100 nm) for the dehydrogenation of amines to imines5d and monolithic nanoporous gold for the gas-phase selective oxidative coupling of methanol to methyl formate.5e These results are astonishing but unfortunately, the origin of this adverse behavior is not deeply discussed therein. Moreover, the catalytic activity of the large-sized Au particles could be further enhanced by depositing transition-metal oxides with low coverage onto a single gold crystal, such as CeO2/Au(111)6a and TiO2/Au(111).6a,b The oxide–Au interface has been proposed to explain the adverse behavior of CeO2(TiO2)/Au(111),6a,b but the structure and nature of the active site at the oxide–Au interface is still unclear.
More recently, we have reported a microfibrous-structured gold catalyst (Au/Ni-fiber) which exhibited an extremely high activity and selectivity in the low-temperature gas-phase oxidation of alcohols,7 within which the formation of active NiOx@Au ensembles is critical to promote the high catalytic activity. Despite the above advance, the origin of the catalytic performance over such ensembles, the finer structure and better understood nature of the active sites over the ensembles and the mechanism of alcohol oxidation over the active sites are still lacking or ambiguous. A clear understanding is of great importance for optimizing the current catalytic processes and rational design of novel catalysts. To this end, three issues will be addressed in the present research: (1) penetrate the indispensability of the nano-architecture of the NiOx@Au ensembles contributing to higher catalytic activity; (2) reveal the activity originally from the specific hybrid active sites hidden on such NiOx@Au ensembles; (3) describe how the hybrid active sites promote alcohol oxidation.
Generally, the activity of catalysts relies on the physical structure of their active ensembles. The base NiOx@Au/Ni-fiber catalyst obtained via a gold galvanic deposition method (see the catalyst preparation in the ESI† for details) was checked by HRTEM, whereby small NiOx segments were found to partially cover the large gold particle to form interesting NiOx@Au ensembles (Fig. S1, ESI†). With these ensembles, the NiOx@Au/Ni-fiber catalyst shows much higher low-temperature activity and TOF for the gas-phase oxidation of benzyl alcohol (conversion of 95% at 250 °C, Entry 1 in Table 1), compared with the Au/SiO2 catalyst (conversion of 6% at 250 °C, Entry 2 in Table 1) and maintained an optimum equilibrium state for a long time in the reaction environment (Fig. S2, ESI†).
| Entry | Catalysta,b | DAud (nm) | DNiOxd (nm) | Conv. (%) | Sel. (%) | TOFe (h−1) |
|---|---|---|---|---|---|---|
| The mass balance of 99.9%, with an experimental error of 0.2%, was achievable (see Note in Fig. S2 in the ESI for details).a For all the catalysts, the gold loading is 4.0 wt% and NiOx loading is 3.2 wt% if not otherwise specified. The catalysts were pre-activated by undergoing the gas-phase oxidation of benzyl alcohol at 380 °C for 1 h using a molar ratio of O2 to alcoholic hydroxyl (O2/hydroxyl) = 0.6 and weight hourly space velocity (WHSV) = 20 h−1, and were then examined at 250 °C using O2/hydroxyl = 0.6 and WHSV = 20 h−1.b The catalyst preparation is described in ESI.c The Au and NiOx loadings for the NiOx@Au/Ni-fiber catalyst were 3.8 wt% and 2.4 wt% (see characterization in the ESI).d The particle size was estimated from the XRD patterns using Scherrer's equation.e TOF (turnover frequency) was calculated based on the amount of surface Au atoms (see Table S1 in the ESI for details). | ||||||
| 1 | NiOx@Au/Ni-fiberc | 25 | 5 | 95 | 99 | 17097 |
| 2 | Au/SiO2 | 27 | — | 6 | 99 | 1156 |
| 3 | Au/Ni-fiber-W | 25 | — | 31 | 99 | 5591 |
| 4 | NiOx/Ni-fiber | — | 5 | 6 | 99 | — |
| 5 | NiOx@Au/Ni-fiber-W | 25 | 6 | 93 | 99 | 16720 |
| 6 | Au/Ti-fiber | 30 | — | 4 | 99 | 839 |
| 7 | NiOx/Ti-fiber | — | 5 | 5 | 99 | — |
| 8 | NiOx@Au/Ti-fiber | 27 | 5 | 92 | 99 | 17907 |
| 9 | Au/NiO–Ni-fiber | 24 | — | 30 | 99 | 5155 |
| 10 | NiOx@Au/NiO–Ni-fiber | 24 | — | 93 | 99 | 16031 |
To penetrate the indispensability of such active NiOx@Au ensembles for the low-temperature gas-phase oxidation of alcohol, a series of model catalysts were designed, prepared, (see the catalyst preparation in the ESI† for details), and then compared by testing the gas-phase oxidation of benzyl alcohol. For comparison, a Au/Ni-fiber-W catalyst with only Au particles was prepared by removing NiOx segments from the NiOx@Au ensembles (Fig. S3 and S4, ESI†), over which, not surprisingly, the benzyl alcohol conversion and TOF was markedly reduced (Entries 1 and 3 in Table 1). In addition, just NiOx supported on Ni-fibers also delivered a very low benzyl alcohol conversion of 6% (Entry 4 in Table 1). We wondered what would happen with post-loading NiOx onto the Au particles over the Au/Ni-fiber-W catalyst. After that, the regeneration of the active NiOx@Au ensembles was observed clearly by TEM (Fig. S5A and S5B, ESI†), which showed that the large Au particles were partially covered with small NiOx segments. As a result, benzyl alcohol conversion increased to 93% over the NiOx@Au/Ni-fiber-W catalyst (Entry 5 in Table 1), almost as high as that over the NiOx@Au/Ni-fiber catalyst (Entry 1 in Table 1).
Subsequently, the Ni-fibers were replaced by Ti-fibers to avoid in situ formation of the special NiOx@Au ensembles, since a galvanic reaction cannot proceed between Ti-fibers and HAuCl4. Analogously, only Au particles or NiOx supported on the Ti-fibers induced a very low benzyl alcohol conversion of <5% (Entries 6 and 7 in Table 1). Excitingly, after post-introducing NiOx artificially onto the Au/Ti-fiber catalyst, the conversion and TOF were remarkably promoted (Entry 8 in Table 1) along with the formation of the NiOx@Au ensembles (Fig. S5C and S5D, ESI†). This again indicated that the NiOx@Au ensembles are essential for the low-temperature gas-phase oxidation of alcohols.
Are Au@NiO ensembles (i.e. a NiO plate partially covered with 20–30 nm Au particles) still able to deliver a high catalytic activity? To answer this question, Au particles were supported onto NiO–Ni-fiber (obtained by calcining pure sintered Ni-fibers at 600 °C in air for 2 h) to form Au@NiO ensembles (see Discussion 1 in the ESI† for details). Unfortunately, over such ensembles, a benzyl alcohol conversion of only 30% was obtained with 99% selectivity (Entry 9 in Table 1, similar to the result for the Au/Ni-fiber-W catalyst with NiOx removal (Entry 3 in Table 1). Likewise, by again post-adding NiOx onto the Au/NiO–Ni-fiber catalyst to form NiOx@Au ensembles on the NiO–Ni-fiber support, the benzyl alcohol conversion was sharply promoted to 93% and the TOF restored to a high level, similar to that over the NiOx@Au/Ni-fiber catalyst (Entries 1 and 10 in Table 1).
Therefore, all the results undoubtedly demonstrate that, upon building the nano-architecture of the NiOx@Au ensembles, the catalysts with such NiOx@Au ensembles always show promising reactivity in the oxidation reaction, even when changing the support fibers or preparation methods. This has firmly proven the indispensability and popularity of the special active NiOx@Au ensemble structure for the low-temperature gas-phase oxidation of alcohols by ruling out Au@NiO ensembles as well as only Au particles or NiOx segments. For the metal–oxide catalysts, the metal–oxide interaction is a very important factor for their catalytic performance.8 Indeed, many studies have reported that Au–oxides interations also exist at their interface.2a,c,d,9 However, the active sites over such NiOx@Au ensembles and their corresponding catalytic nature are not yet clear.
To define the structure of the active sites over such ensembles, surface-sensitive X-ray photoelectron spectroscopy (XPS) was used to study the NiOx@Au/Ni-fiber catalyst (i.e., Entry 1 catalyst in Table 1). Fig. 1 shows the XPS spectra in the O 1s, Au 4f and Ni 2p regions for several NiOx@Au/Ni-fiber samples undergoing different reaction conditions. On the surface of the working catalyst sample (obtained after undergoing a reaction with benzyl alcohol in the presence of O2 (O2/hydroxyl = 0.6) at 250 °C for 0.5 h), apart from Au0 (Au 4f (7/2): 84.0 eV) and NiO (Ni 2p: 854.4 eV; O1s: 529.7 eV), not only were large amounts of Ni2O3 specimens (Ni 2p: 856.2 eV; O 1s: 531.8 eV) formed but considerable amounts of Au+ specimens were also clearly detected, with a Au 4f (7/2) peak at 84.8 eV (Fig. 1a–c). Note that O atoms adsorbed onto the Au+ sites generally exist in the form of OH groups that also provide an O 1s peak at approximately 532 eV (Fig. S6, ESI†).10 CeO2 (or Fe2O3, TiO2)–Au interfaces have been previously reported and proposed to generate cationic gold and activate molecular oxygen for oxidation reactions.4e,f,6d,11
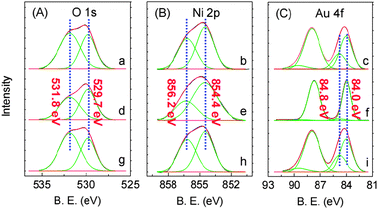 | ||
| Fig. 1 The XPS spectra in the O 1s (A), Ni 2p (B) and Au 4f (C) regions of the surface of several catalysts. (a–c) The working NiOx@Au/Ni-fiber catalyst, the same as the catalyst in Entry 1 in Table 1; (d–f) The spent catalyst, obtained by placing the working catalyst in a benzyl alcohol stream in the absence of O2 for 0.5 h; (g–i) The spent catalyst, catalyzing benzyl alcohol oxidation by re-feeding O2 again under the same conditions as in (A-a). | ||
On the surface of the working catalyst, the Au+ surface content to the total amount of surface gold atoms is 31%, which was obtained by calculating the area of the corresponding XPS peaks (AAu+/(AAu0 + AAu+) = 0.31), and the Ni2O3 surface content to the total amount of surface nickel oxides is 43% (ANi2O3/(ANiO + ANi2O3) = 0.43) with a surface Au+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni3+ peak area ratio of 1:45 (Fig. 1a–c, Table S2, ESI†). As the working catalyst underwent the reaction with benzyl alcohol in the absence of O2 for 0.5 h (the resulting catalyst was called the spent catalyst), the benzyl alcohol conversion was decreased from 95% to only 0.7% (Table S3, Fig. S7, ESI†) while the Au+ surface content disappeared completely (Fig. 1f, Table S2, ESI†). Interestingly, the Ni2O3 surface content was decreased from 43% to 33% with the stoichiometric reduction of the O 1s peak for Ni2O3, while the NiO surface content was increased from 57% to 67%. Combined with the fact that the whole peak area for Ni remained unchanged, this observation definitely indicates the transformation of Ni2O3 to NiO when reacting without O2 (Fig. 1d and e, Table S2, and the more detailed Discussion 2 in the ESI†). Subsequently, after the spent catalyst was reacted with benzyl alcohol by re-feeding O2 at 250 °C, the Au+ and Ni2O3 surface content both recovered to previous levels, with a Au+ content of 33% and a Ni2O3 content of 42% (Fig. 1g–i, Table S2, ESI†), and the benzyl alcohol conversion returned to 93%.
:Ni3+ peak area ratio of 1:45 (Fig. 1a–c, Table S2, ESI†). As the working catalyst underwent the reaction with benzyl alcohol in the absence of O2 for 0.5 h (the resulting catalyst was called the spent catalyst), the benzyl alcohol conversion was decreased from 95% to only 0.7% (Table S3, Fig. S7, ESI†) while the Au+ surface content disappeared completely (Fig. 1f, Table S2, ESI†). Interestingly, the Ni2O3 surface content was decreased from 43% to 33% with the stoichiometric reduction of the O 1s peak for Ni2O3, while the NiO surface content was increased from 57% to 67%. Combined with the fact that the whole peak area for Ni remained unchanged, this observation definitely indicates the transformation of Ni2O3 to NiO when reacting without O2 (Fig. 1d and e, Table S2, and the more detailed Discussion 2 in the ESI†). Subsequently, after the spent catalyst was reacted with benzyl alcohol by re-feeding O2 at 250 °C, the Au+ and Ni2O3 surface content both recovered to previous levels, with a Au+ content of 33% and a Ni2O3 content of 42% (Fig. 1g–i, Table S2, ESI†), and the benzyl alcohol conversion returned to 93%.
X-ray absorption near-edge structure (XANES) is sensitive to the oxidation state of atoms on the surface of metallic particles,12a–d because there is an intense whiteline in the compounds containing a metallic cation.12a To further verify the high-low-high associate-changes of Au+ and Ni2O3, the above three catalysts were also investigated by XANES at the Au L3 and Ni K edge, with the results shown in Fig. 2A. The working catalyst sample shows a similar XANES pattern with the reported mixture of Au+ complexes and Au0 particles (Fig. 2A-a),12e definitely indicating the existence of Au+ ions. Interestingly, as the working catalyst underwent the oxidation reaction with only benzyl alcohol for 0.5 h in the absence of O2, the intensity of the whiteline decreases and the resulting XANES pattern is similar to that of Au foil, indicating that the Au+ content decreases to almost zero (Fig. 2A-b and c). After re-feeding O2 to the spent catalyst, the whiteline intensity increases to the previous level of the working catalyst, indicating that the Au+ ions are regenerated with a content close to the working catalyst level (Fig. 2A-d). Indeed, the same trend is also observed in the changes of the Au–O coordination (Fig. 2B). The working catalyst shows a Au–O coordination at a distance of 1.7 Å (Fig. 2B-a), a similar distance to that of other reported catalysts.12f Over the spent catalyst, the Au–O coordination almost disappears (Fig. 2B-b and c), while after re-feeding O2 into the reaction stream, the Au–O coordination likewise increases to the working catalyst level (Fig. 2B-d). Regarding Ni2O3, the existence of the Ni-fiber support leads to a very low Ni2O3/Ni0 molar ratio. This means the XANES technique is not as sensitive as the XPS method in tracking the changes of the Ni2O3 content in the above cases. Even so, the high-low-high changes of the whiteline intensity of the XANES pattern at the Ni K edge could still be clearly distinguished (Fig. S8, ESI†). Likewise, the absorption intensity at 8350 eV for the spent catalyst was slightly lower than that for the working catalyst and subsequently, could be revived to the previous level of the working catalyst by re-feeding O2 into the reaction stream. As expected, similar evolution behaviors of XANES for both Au and Ni were also observed for the NiOx@Au/Ti-fiber catalyst in the reaction processes with-without-with O2 (Fig. S9, ESI†). Combined with the XPS results, it is reasonable to infer that the high-low-high change of the absorption intensity at 8350 eV can be assigned to the high-low-high change of the Ni2O3 content (see Discussion 3 in the ESI† for details).
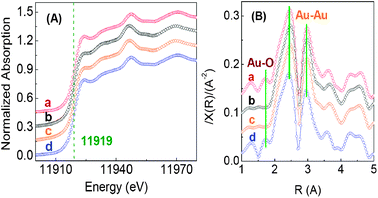 | ||
| Fig. 2 XANES of the Au L3 edge (A) and the modulus of the fourier transform Au L3-edge signal (B) of several catalysts. (a) The same was used sample as in Fig. 1A-a; (b) The same sample was used as in Fig. 1A-d; (c) Au foil; (d) The same sample was used as in Fig. 1A-g. | ||
Along with the high-low-high surface content of Au+, the benzyl alcohol conversion clearly shows a high-low-high evolution behavior as well. Over the spent catalyst, the benzyl alcohol conversion decreased from 95% to 0.7%, with a significant decrease of the Au+ surface content from 31% to 1.7%. In this case, an associated reduction of Ni2O3 from 43% to 33% was observed. These results clearly indicate that the combination of Ni2O3 and NiO with the absence of Au+ (i.e., the spent catalyst) was inactive for the low-temperature gas-phase oxidation of alcohols by the direct-oxidation dehydrogenation route.13
On the surface of the NiOx@Au ensembles, it is worth noting that the content change of Au+ in the reaction processes with-without-with O2 was solely associated with a parallel change of Ni2O3 but not NiO (Fig. 1). This unique relationship between Ni2O3 and Au+ indicates a synergistic interaction between the Au particles and Ni2O3 specimens rather than NiO. By their nature, the Ni2O3 specimens are proposed to play a key role not only in promoting the formation of Au+ ions and stabilizing them, but also in acting as a surface-active-oxygen species supplier.
When looking at Table 1, the Au/Ni-fiber-W and Au/NiO–Ni-fiber catalysts (Entries 3 and 9 in Table 1) both delivered much lower benzyl alcohol conversions (∼30%) and TOFs (5100–5600 h−1) than the NiOx@Au/Ni-fiber catalyst (conversion of 95%, TOF of ∼17000 h−1, Entry 1 in Table 1), but much higher values than the Au/Ti-fiber catalyst (conversion of 4%, TOF of 839 h−1, Entry 6 in Table 1). Although the NiOx segments were not presented on the Au particles for these two samples, the formation of a NiOx circumference at the interface between the Au particles and fibrous support (Scheme S1, ESI†) was unavoidable during their preparation. This likely induced a similar synergistic effect as in the NiOx@Au ensembles in order to form some Au+ with Ni2O3 specimens, as evidenced by XPS peaks at Au 4f (7/2) = 84.7 eV and Ni 2p = 856.2 eV (Fig. S10, ESI†). The amount of Au+–Ni2O3 active sites over these two samples, with a surface Au+:Ni3+ peak area ratio of 1:43 (close to that (1:45) for NiOx@Au/Ni-fiber), is only 35–37% of that over the NiOx@Au/Ni-fiber catalyst (Tables S2 and S4). This might be the reason for the higher reactivity over both of them compared to the Au/Ti-fiber catalyst but a much lower reactivity than the NiOx@Au/Ni-fiber catalyst (Table 1).
On the basis of the above investigation, a Ni2O3–Au+ hybrid active site was defined to be the genesis of the high low-temperature activity and the NiOx@Au ensembles facilitated the generation of high density Ni2O3–Au+ sites (Au+ content of 31–33% and Ni2O3 content of 42–43%, Fig. 1) on the catalyst surface during oxidation. As shown in Fig. 3, the oxygen molecules can be abstracted by the oxygen vacancy of the Ni2O3 specimens to be stored as precursors to active oxygen species. Such abstracted oxygen atoms (O 1s B.E.: ca. 532 eV, Fig. 1) can be transferred onto the Au+ sites to become active oxygen species which react with alcohol while releasing an oxygen vacancy for the next catalytic cycle.
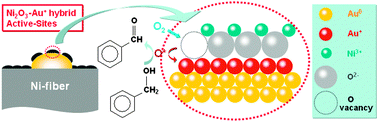 | ||
| Fig. 3 The structure of the Ni2O3–Au+ hybrid active sites on the NiOx@Au ensembles for the working catalyst and the reaction mechanism of benzyl alcohol oxidation over the active sites. | ||
We thank the NSF of China (20973063, 21076083, 21273075), the MOST of China (2011CB201403), the Fundamental Research Funds for the Central Universities, the Shanghai Rising-Star Program (10QH1400800), the Shanghai Leading Academic Discipline Project (B409) and the Electron Spectroscope Center of the East China Normal University for the TEM measurements.
Notes and references
- (a) M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405 CrossRef CAS; (b) M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS; (c) G. J. Hutchings, J. Catal., 1985, 96, 292 CrossRef CAS.
- (a) G. C. Bond and D. T. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319 CrossRef CAS; (b) M. S. Chen and D. G. Goodman, Science, 2004, 306, 252 CrossRef CAS; (c) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef; (d) B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709 CrossRef CAS; (e) G. J. Hutchings, Dalton Trans., 2008, 5523 RSC.
- J. C. Fierro-Gonzalez and B. C. Gates, Chem. Soc. Rev., 2008, 37, 2127 RSC.
- (a) M. Comotti, W. C. Li, B. Spliethoff and F. Schüth, J. Am. Chem. Soc., 2006, 128, 917 CrossRef CAS; (b) P. X. Huang, F. Wu, B. L. Zhu, X. P. Gao, H. Y. Zhu, T. Y. Yan, W. P. Huang, S. H. Wu and D. Y. Song, J. Phys. Chem. B, 2005, 109, 19169 CrossRef CAS; (c) J. Li, N. Ta, Y. Li and W. J. Shen, Chin. J. Catal., 2008, 29, 823 CAS; (d) R. Si and M. Flytzani-Stephanopoulos, Angew. Chem., Int. Ed., 2008, 47, 2884 CrossRef CAS; (e) J. Guzman, S. Carrettin and A. Corma, J. Am. Chem. Soc., 2005, 127, 3286 CrossRef CAS; (f) A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS; (g) A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331 CrossRef CAS; (h) J. K. Edwards, B. Solsona, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS.
- (a) D. I. Enache, J. K. Edwards, P. Landon, B. Solsna-Espriu, A. F. Carely, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS; (b) S. Biella and M. Rossi, Chem. Commun., 2003, 378 RSC; (c) C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384 CrossRef CAS; (d) B. Zhu, M. Lazar, B. G. Trewyna and R. J. Angelici, J. Catal., 2008, 260, 1 CrossRef CAS; (e) A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319 CrossRef CAS.
- (a) J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757 CrossRef CAS; (b) T. Fujitani, I. Nakamura, T. Akita, M. Okumura and M. Haruta, Angew. Chem., Int. Ed., 2009, 48, 9515 CrossRef CAS; (c) S. Shaikhutdinov, Y. N. Sun, Z. H. Qin, M. Lewandowski, E. Carrasco, M. Sterrer and H. J. Freund, J. Catal., 2009, 266, 359 CrossRef; (d) Q. Fu, W. X. Li, Y. Yao, H. Liu, H. Y. Su, D. Ma, X. K. Gu, L. Chen, Z. Wang, B. Wang and X. H. Bao, Science, 2010, 328, 1141 CrossRef CAS.
- G. F. Zhao, H. Y. Hu, M. M. Deng and Y. Lu, Chem. Commun., 2011, 47, 9642 RSC.
- (a) S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170 CrossRef CAS; (b) S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121 CAS.
- (a) M. Baron, O. Bondarchuk, D. Stacchiola, S. Shaikhutdinov and H. J. Freund, J. Phys. Chem. C, 2009, 113, 6042 CrossRef CAS; (b) T. Risse, S. Shaikhutdinov, N. Nilius, M. Sterrer and H. J. Freund, Acc. Chem. Res., 2008, 41, 949 CrossRef CAS; (c) L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 311, 195 CrossRef.
- L. C. Wang, L. He, Q. Liu, Y. M. Liu, M. Chen, Y. Cao, H. Y. He and K. N. Fan, Appl. Catal., A, 2008, 344, 150 CrossRef CAS.
- (a) G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41 CrossRef CAS; (b) S. Y. Yu, H. Huang, H. B. Liu, Z. N. Chen, R. Zhang and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 686 CrossRef CAS; (c) S. Carrettin, P. Concepcion, A. Corma, J. M. Lopez Mieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538 CrossRef CAS; (d) D. Matthey, J. G. Wang, S. Wendt, J. Matthiesen, R. Schaub, E. Lægsgaard, B. Hammer and F. Besenbacher, Science, 2007, 315, 1692 CrossRef CAS; (e) N. Weiher, A. M. Beesley, N. Tsapatsaris, L. Delannoy, C. Louis, J. A. van Bokhoven and S. L. M. Schroeder, J. Am. Chem. Soc., 2007, 129, 2240 CrossRef CAS; (f) Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935 CrossRef CAS; (g) A. Trovarelli, Catal. Rev. Sci. Eng., 1996, 38, 439 CrossRef CAS.
- (a) J. Guzman and B. C. Gates, J. Am. Chem. Soc., 2004, 126, 2672 CrossRef CAS; (b) M. K. Oudenhuijzen, J. A. van Bokhoven, J. T. Miller, D. E. Ramaker and D. C. Koningsberger, J. Am. Chem. Soc., 2005, 127, 1530 CrossRef CAS; (c) D. E. Ramaker, M. Teliska, Y. Zhang, A. Stakheev and D. C. Koningsberger, Phys. Chem. Chem. Phys., 2003, 5, 4492 RSC; (d) D. E. Ramaker and D. C. Koningsberger, Phys. Rev. Lett., 2002, 89, 139701 CrossRef CAS; (e) J. A. van Bokhoven, C. Louis, J. T. Miller, M. Tromp, O. V. Safonova and P. Glatzel, Angew. Chem., Int. Ed., 2006, 45, 4651 CrossRef CAS; (f) J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240, 222 CrossRef CAS.
- (a) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS; (b) R. A. Sheldon, I. Arends, G. J. Ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst preparation, testing and characterization details; additional XRD patterns, TEM images, XPS spectra, XANES spectra; data analysis and discussion; Fig. S1–S10; Tables S1–S4; Scheme S1; supplementary discussion 1 to 3. See DOI: 10.1039/c2cy20579c |
| This journal is © The Royal Society of Chemistry 2013 |