Chiral salen Mn(III) complexes immobilized onto crystalline aluminium oligo-styrenyl phosphonate-hydrogen phosphate (AlSPP) for heterogeneous asymmetric epoxidation†
Xuemei
Huang
,
Xiangkai
Fu
*,
Ziyong
Jia
,
Qiang
Miao
and
Guoming
Wang
College of Chemistry and Chemical Engineering Southwest University, Research Institute of Applied Chemistry Southwest University, The Key Laboratory of Applied Chemistry of Chongqing Municipality, The Key Laboratory of Eco-environments in Three Gorges Reservoir Region Ministry of Education, No. 2 Tiansheng Road, BeiBei District, Chongqing, 400715, China. E-mail: fxk@swu.edu.cn; Fax: +86 2368254000; Tel: +86 2368253704
First published on 14th September 2012
Abstract
A novel type of organic polymer–inorganic hybrid material layered crystalline aluminium oligo-styrenyl phosphonate-hydrogen phosphate (AlSPP) was designed and prepared in the absence of any template. The chiral salen Mn(III) complex immobilized onto alkoxy-modified AlSPP was synthesized and characterized by FT-IR, diffusion reflection UV-vis, AAS, N2 volumetric adsorption, SEM, TEM, XRD, TG and AFM. The supported catalysts displayed superior catalytic activities in the asymmetric epoxidation of α-methylstyrene and indene with m-CPBA as an oxidant. Delightfully, the heterogeneous catalysts afforded a remarkable increase in conversion and ee values without the expensive O-coordinating axial bases for the asymmetric epoxidation of olefins. The catalysts can be easily recovered and reused with a slight decrease in activity and enantioselectivity after ten cycles. Furthermore, they could also be efficiently used in large-scale reactions with the enantioselectivity being maintained at the same level, which provided the potentiality for application in industry.
1. Introduction
Due to the great importance of the chiral compounds in the manufacture of drugs, vitamins, fragrances, and optical materials, the synthesis of chiral building blocks has attracted special attention. Chiral epoxides are among the most important intermediates that can be readily converted into various chiral compounds via regioselective ring-opening or functional transfer reactions. Chiral Mn(salen)Cl complexes1,2 exhibit excellent activity and enantioselectivity for the homogeneous asymmetric epoxidation of various unfunctionalized olefins.3–6 However, reusability is a major problem due to the decomposition of the catalyst and the formation of inactive dimeric μ-oxo-Mn(VI)7 in the homogeneous phase, which limits the practical applications of chiral Mn(III) salen catalysts in both synthetic chemistry and industrial processes. The heterogeneous catalytic systems can potentially solve these problems. In addition to these merits, the solid surface or the nanopores of the supports may provide an unexpected effect on asymmetric catalytic performance for heterogeneous asymmetric epoxidations. Therefore, immobilizing the chiral salen Mn(III) complex on various supports becomes the most effective method, such as immobilizing onto organic polymers;8 inorganic supports—clays,9 MCM-41,10 SBA-15,11 activated silica,12 zeolites;13 and organic polymer–inorganic hybrid materials.14 One of the most outstanding advantages is that the immobilized catalyst can realize the separation and reusability by simple filtration and can enhance the catalyst stability since the generation of the dimeric μ-oxo-Mn(VI) could be avoided.Our research has, for many years, been concerned with metal phosphonate chemistry for catalysts and catalyst supports.14 Recently, we have reported a series of organic–inorganic hybrid phosphonate-phosphates M(HPO4)2−x(O3P-G)x·nH2O (M is Zr, Zn, G is organic groups) as various kinds of catalysts or catalyst supports to immobilize the chiral salen Mn(III) complex. The immobilized chiral salen Mn(III) complex showed higher enantioselectivity than that of the corresponding homogeneous catalyst and could be reused several times in asymmetric epoxidation of unfunctionalized olefins under the same conditions.
To the best of our knowledge, it is reported that aluminium phosphates and aluminium phosphonates are types of layered multi-functional materials with high thermal and chemical stability.15,16 In the past few decades, a series of aluminium phosphates and aluminium phosphonates with various microporous, mesoporous holes were synthesised by hydrothermal methods and played an important role in adsorption separation, catalyst and catalyst carrier, soil science and sewage treatment, especially in the catalytic field due to the characteristic structure with different molecular size, uniform pores, continuous pores and big crystal surface.17 In order to make the material possess both the advantages of organic polymers (such as low mass, flexibility) and inorganic materials (such as high strength, heat stability and ordered structural), therefore, in this work, a new type of layered crystalline inorganic–organic polymer hybrid material AlSPP was prepared by the reaction of oligo-styrenyl phosphonate with aluminium acetate dihydrate and sodium dihydrogen phosphate under mild conditions without any organic template. Alkoxy-modified AlSPP were applied to immobilize the chiral salen Mn(III) complexes through axial coordination for the first time. The immobilized catalysts (4a–4f) effectively catalyzed epoxidation of α-methylstyrene and indene with m-CPBA as the oxidant. The results were obviously better than those achieved with the corresponding homogeneous catalyst. Notably, the immobilized catalysts could be reused up to ten times without significant loss of activity and enantioselectivity, and also could be applied in large-scale reactions with the conversion and enantioselectivity being maintained at the same level. Furthermore, the immobilized catalysts showed high reactivity in the absence of NMO, which was entirely different from most results reported.
2. Experimental
2.1 Materials and methods
(1R,2R)-(−)-1,2-Diaminocyclohexane, acetate aluminium, chloromethyl methyl ether (toxic compound), indene, α-methylstyrene, n-nonane, N-methylmorpholine N-oxide (NMO) and m-chloroperbenzoic acid (m-CPBA) were supplied by Alfa Aesar. Other commercially available chemicals were laboratory-grade reagents from local suppliers. The chiral salen ligand and the chiral homogeneous catalyst salen Mn(III) were synthesized according to the standard literature procedures, and further identified by analysis and comparison of IR spectra with the literature.18,19FT-IR spectra were recorded from KBr pellets using a Bruker RFS100/S spectrophotometer (USA) and diffuse reflectance UV-vis spectra of the solid samples were recorded in a spectrophotometer with an integrating sphere using BaSO4 as the standard. TG analyses were performed on a SBTQ600 thermal Analyzer (USA) with a heating rate of 20 °C min−1 from 25 to 1000 °C under flowing N2 (100 mL min−1). The Mn content of the catalysts was determined by TAS-986G (Pgeneral, China) atomic absorption spectroscopy. SEM were performed on a KYKY-EM3200 (KYKY, China) microscope. TEM were obtained on a TECNAI10 (PHILIPS, Holand) apparatus. The interlayer spacings were obtained on a DX-1000 automated X-ray power diffractometer, using Cu Kα radiation and an internal silicon powder standard with all samples. The patterns were generally measured between 3.00° and 80.00° with a step size of 0.02° min−1 and X-ray tube settings of 36 kV and 20 mA. Nitrogen adsorption isotherms were measured at 77 K on a 3H-2000I (Huihaihong, China) volumetric adsorption analyzer using the BET method. The conversions (with n-nonane as an internal standard) and the ee values were analyzed by gas chromatography (GC) with a Shimadzu GC2010 (Japan) instrument equipped with a chiral column (HP19091 G-B213, 30 m × 30 m × 0.32 mm × 0.25 μm) and an FID detector, injector 200 °C, detector 300 °C. The column temperature was in the range of 80–150 °C.
2.2 Synthesis of layered crystalline aluminium oligo-styrenyl phosphonate-hydrogen phosphate (AlSPP)
The synthesis and characterization of OSPNA have been reported earlier by our group.20OSPNA (14.74 g, 36.5 mmol) was dissolved in 100 mL THF at room temperature for 30 min. The solution of sodium dihydrogen phosphate (14.84 g, 77.56 mmol) and aluminium acetate (14.86 g, 75.02 mmol) was added dropwise and stirred continuously. The reaction mixture was kept at 66 °C for 72 h and at room temperature overnight. The pH value was adjusted to 7.0 by adding Et3N and the solvent was evaporated under reduced pressure. The white solid AlSPP was filtered off, washed with deionized water and dried at 60 °C for 24 h in the vacuum with 90% yield. IR (KBr): νmax/cm−1 3100 (CH), 1631, 1590, 1349 (C6H5), 1154 (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1074, 1033 (PO32−).
O), 1074, 1033 (PO32−).
2.3 Synthesis of the supported catalyst (Scheme 1)
P–OH), 1630, 1395, 1353 (C6H5), 1155 (PO), 701 (C–Cl) cm−1.
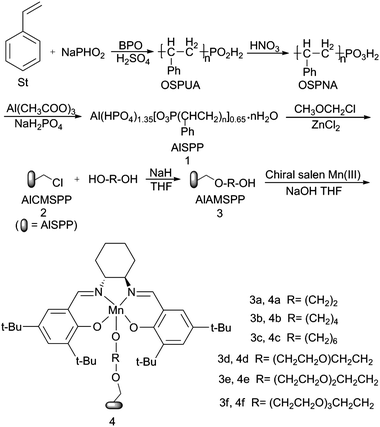 | ||
| Scheme 1 Synthetic route of the heterogeneous catalysts. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and the mixture was stirred and kept at 80 °C for 24 h. Subsequently, the mixture was neutralized and the product was filtered off, washed and dried in the vacuum to obtain AlAMSPP with the reaction yields always exceeded 90%. The products were abbreviated as 3a–3f in turn.
:1) and the mixture was stirred and kept at 80 °C for 24 h. Subsequently, the mixture was neutralized and the product was filtered off, washed and dried in the vacuum to obtain AlAMSPP with the reaction yields always exceeded 90%. The products were abbreviated as 3a–3f in turn.
2.4 Asymmetric epoxidation
The activity of the prepared catalysts was tested for the epoxidation of unfunctionalized olefins in CH2Cl2 at 0 °C for 6 h using m-CPBA/NMO as an oxidant and with alkene (1 mmol), n-nonane (internal standard, 1 mmol), NMO (5 mmol, if necessary), the immobilized catalysts 4a–4f (0.05 mmol, based on the Mn amount in the catalyst) or homogeneous (0.05 mmol). Oxidant m-CPBA (2 mmol) was added in portions over 5 minutes. After reaction, Na2CO3 (2 mL, 1.0 M) was added to quench the reaction. And the organic layer was dried over sodium sulfate, and the catalyst was precipitated out from the solution by adding hexane and kept for subsequent use without further purification. The conversion and ee values of the epoxide were determined by GC.2.5 The reusability of the catalyst
In a typical recirculation, an equal volume of hexane was added to the reaction mixture after the reactions. Thereafter, the organic phase was separated, and the catalyst was washed with hexane and deionized water, and dried over vacuum at 60 °C. The recovered dried solid catalyst was weighed and reused in the next run. In every run the same proportion of the substrate-to-catalyst and solvent-to-catalyst was retained.2.6 Large-scale asymmetric epoxidation
In order to detect the potential of the catalyst's industrial applications, we performed large-scale asymmetric epoxidation reactions. Catalyst 4b (1.25 mmol), n-nonane (25 mmol) and α-methylstyrene (25 mmol) in CH2Cl2 (100 mL) at 0 °C were stirred for 30 min. Then, m-CPBA (50 mmol) was added to the solution step by step. After 6 h, Na2CO3 (100 mL, 1.0 M) was added to quench the reaction. And the organic layer was dried over sodium sulfate, and the catalyst was precipitated out from the solution by adding hexane and kept for subsequent use without further purification. The conversion and ee values of the epoxide were determined by GC.3. Results and discussions
3.1 Spectral analysis
The most informative evidence, which confirmed the anchoring of the chiral salen Mn(III) complex to the alkoxy-modified AlSPP, was obtained by FT-IR spectra (Fig. 1). Comparing the IR spectra of the heterogeneous salen Mn(III) complexes with the neat chiral salen Mn(III), all the samples (4a–4f) and the chiral salen Mn(III) complex showed similar bands at 1635–1645 cm−1, attributed to the vibration of the imine group. The IR band at 1400 cm−1 was due to C–H vibration of the alkyl groups. The stretching vibration at 1074–1085 cm−1 was assigned to the characteristic flexible vibrations of the C–O band. Moreover, catalysts 4a–4f showed strong absorption peaks at 542–558 cm−1, assigned to the Mn–N bond of the salen ligand.14c Hence, it was suggested that the chiral salen Mn(III) had been successfully immobilized onto the alkoxy-modified AlSPP support.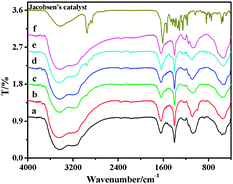 | ||
| Fig. 1 FT-IR spectra of 4a–4f (a–f) and Jacobsen's catalyst. | ||
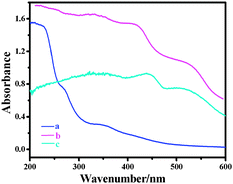
UV-vis spectra gave more obvious evidence for the successful immobilization (Fig. 2). The spectra of 4b showed similar characteristic absorption peaks to the chiral salen Mn(III). The band at 335 nm could be attributed to the charge transfer transition of the salen ligand. The band at 433 nm was due to the ligand-to-metal charge transfer transition, and the band at 510 nm was assigned to the d–d transition of the salen Mn(III) complex.21 For the immobilized salen Mn(III) catalysts, all the characteristic bands appeared in their spectra and exhibited a blue shift from 335, 433 and 510 nm to 330, 420 and 504 nm, which indicated that an interaction existed between the salen Mn(III) complex and the alkoxy-modified AlSPP support.
3.2 The X-ray diffraction analysis
The powder XRD patterns of the modified supports and the heterogeneous catalyst are shown in Fig. 3. As can be seen from Fig. 3, the XRD pattern of AlSPP (a) obviously displayed strong and sharp peaks which revealed that AlSPP was in the crystal state. XRD data also showed that the crystalline AlSPP (a) was a layered complex whose interlayer distance calculated according to the Bragg equation (nλ = 2dsinθ) was about 5.4 Å. The powder XRD patterns of AlAMSPP and salen Mn(III)/AlAMSPP showed similar periodic structures to AlSPP. Compared with AlSPP, the characteristic peaks of AlAMSPP and salen Mn(III)/AlAMSPP showed a small shift toward larger 2θ values (from 16.6 to 18.3, 21.2, respectively). The same phenomenon was found by Li22 that the sulfonic sodium modified mesoporous MCM-41(PhSO3Na-2-MCM(1.6)) and the immobilized catalyst (PhSO3Mn(salen)-2-MCM(1.6)) showed a small shift toward larger 2θ values compared with MCM-41. These observations indicated that the interactions existed between the salen Mn(III) complex and the alkoxy-modified AlSPP support.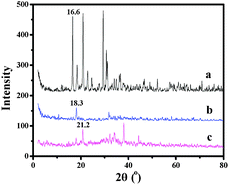 | ||
| Fig. 3 XRD patterns of AlSPP (a), AlAMSPP (b) and catalyst 4b (c). | ||
3.3 Thermal gravimetric analysis
As described in the TG curves (Fig. 4a), it could be inferred that AlSPP lost 5.4 molar equivalents of surface-bound or intercalated water below 100 °C. The decomposition temperature of AlSPP was over 100 °C. The sharp weight loss of 20.5% in the temperature range of 100–200 °C was attributed to the decomposition of the appended organic fragments. The weight loss of 8.6% in the temperature range of 200–600 °C was due to the conversion of aluminium phosphonates to aluminium phosphate. Simultaneously, according to catalyst 4b (Fig. 4b), the initial weight loss of 4.3% below 100 °C was ascribed to the loss of the surface-bound or intercalated water in this stage. In the temperature range of 100–350 °C, the organic moieties decomposed with a total of 15.7% weight loss. The weight loss in the temperature range of 350–600 °C was owing to the phase changes from aluminium phosphonates to aluminium phosphate and the decomposition of the chiral salen Mn(III) complex.14a–d Obviously, catalyst 4b still had high stability over 150 °C. Generally, heterogeneous catalysis reactions were carried out below 150 °C. Therefore, catalyst 4b had adequate thermal stability to be applied in heterogeneous catalytic reactions.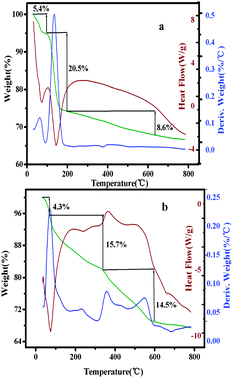
3.4 X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) is a powerful technique to investigate the electronic properties of the species formed on the surface. The XPS spectra of the heterogeneous catalyst 4b are described in Fig. 5. The neat chiral salen Mn(III) complex exhibited the Mn 2p3/2 core level peak at a binding energy of 642.1 eV. The immobilized salen Mn(III) complex showed a binding energy at 641.7 eV. Compared with the neat salen Mn(III) complex, the immobilized catalyst showed a chemical shift of 0.4 eV which was consistent with earlier literature data.23 The electron density of the manganese atom decreased, might be because the metal Mn was in a different coordination environment in AlAMSPP compared with the neat chiral salen Mn(III) complex. After reused ten times, catalyst 4b showed a binding energy at 641.2 eV which elaborated that the catalyst was relatively stable.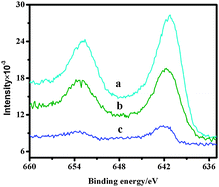 | ||
| Fig. 5 XPS spectra of recycled catalyst 4b (a); catalyst 4b (b) and (c) neat salen Mn(III). | ||
3.5 Nitrogen adsorption–desorption isotherms and Mn content of the immobilized catalysts
The corresponding textural parameters calculated by N2 adsorption–desorption isotherms are presented in Table 1. BJH analysis (Fig. 6) showed that the distribution of the pores was broad and non-uniform, and the average pore size was about 19.6 nm which was extremely larger than many inorganic supports (MCM-41, MCM-48)24 and also larger than the reported organic–inorganic hybrid phosphonate-phosphates M(HPO4)2−x(O3P-G)x·nH2O (M is Zr, Zn, G is organic groups) by our group.14 The surface area reached 25 m2 g−1 with a pore volume of 0.25 cm3 g−1. The local environment inside the mesopores and the pore size of the support did affect the enantioselectivity of the epoxidation reaction. In general, the pore size, pore volume and surface area presented regular changes with the regular changes of linkage groups R in salen Mn(III)/AlAMSPP.| Sample | S e/m2 g−1 | V p/cm3 g−1 | D p/nm | Mn(salen)/mmol g−1 |
|---|---|---|---|---|
| AlSPP | 25.4 | 0.25 | 19.6 | — |
| 4a | 48.5 | 0.17 | 6.82 | 0.38 |
| 4b | 37.6 | 0.14 | 7.04 | 0.42 |
| 4c | 27.8 | 0.12 | 7.18 | 0.45 |
| 4d | 142.7 | 0.28 | 5.02 | 0.53 |
| 4e | 124.5 | 0.34 | 5.35 | 0.62 |
| 4f | 117.2 | 0.53 | 9.12 | 0.65 |
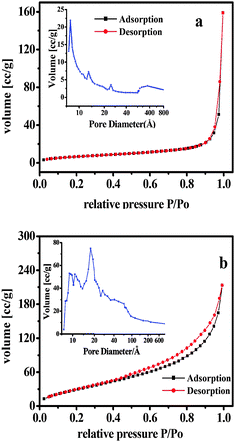 | ||
| Fig. 6 Isotherms and distribution of pore diameter of AlSPP (a), catalyst 4b (b). | ||
Meanwhile, compared with the parent support, a large increase in BET surface area (25.4 to 142.7), but a large decrease in pore size (19.6 to 6.82) were observed for catalysts 4d–4f, inversely, the immobilized catalyst 4a–4c only got a slight increase in BET surface area (25.4 to 27.8). It could be due to the fact that linkage groups polyol in catalysts 4d–4f had more oxygen atom to coordinate the chiral salen Mn(III) than the diol groups in catalysts 4a–4c, so that there were more chiral salen Mn(III) in the nanopores of polyol-modified AlSPP. The peculiar appearance was entirely different to most of the results that the surface area and pore volume decreased sharply after incorporation of the chiral salen Mn(III) complex into the support, attributed to the fact that the pores of the support were filled with the chiral salen Mn(III) complex.25 On the basis of this, it could be deduced that the chiral salen Mn(III) complexes were mainly immobilized on the external surface of AlSPP for catalysts 4d–4f and mainly presented inside the nanopores for catalysts 4a–4c. In other words, there were two forms of immobilization of the ligand: inner type and outer type.26 The amount of chiral salen Mn(III) anchored onto AlSPP was in the range of 0.38–0.65 mmol g−1 (Table 1) ascertained by AAS based on the Mn element. The Mn content also further comfirmed that chiral salen Mn(III) in the nanopores of polyol-modified AlSPP was more than that of the diol-modified AlSPP. In view of these facts, it could be inferred that the chiral salen Mn had been successfully attached.
3.6 The hypothesized layered structure of AlSPP
The layered crystalline structure of AlSPP has been indicated by XRD (Fig. 3). Fig. 7a demonstrates the forming process of AlSPP. When an aqueous solution of hydrated aluminium acetate was added, the phosphate and the phosphonic acid in the polymer reacted with Al3+ immediately to form a white colloidal precipitate of AlSPP. During the process, almost all phosphate participated in forming the white colloidal precipitate of AlSPP, while most of the organic phosphoric acid in the polymer participated in forming or entering into the white colloidal precipitate and little free phosphoric acid in the polymer covered on the surface of white colloidal precipitate of AlSPP, that is to say, hybrid aluminium oligo-styrenyl phosphonate-hydrogen phosphate Al(HPO4)1.5−x(O3PG)x·nH2O (x = 0–1.5) was generated by partial displacement of phosphate residue (P)–OH by organic groups (P)–G. Simultaneously, hydrophobic segments of polystyrene in the polymer, which exist in half or part of holes and cavums with different volumes and shapes, also covered on the surface of the white colloidal precipitate of AlSPP. Therefore, AlSPP possesses dual properties due to the hydrophobic nature of polystyrene parts and the hydrophilic nature of phosphate parts. Meanwhile, AlSPP could be reacted with other complexes either in aqueous solution or in organic solvent even in mixed solvent which was the character that many other materials do not possess.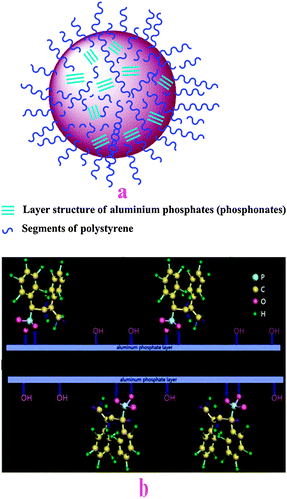 | ||
| Fig. 7 Deduced structure models of AlSPP. | ||
From the formation process of AlSPP, we know that the hybrid aluminium oligo-styrenyl phosphonate-hydrogen phosphate Al(HPO4)1.5−x(O3PG)x·nH2O (x = 0–1.5) was generated by partial displacement of phosphate residue (P)–OH with organic groups (P)–G. Al(HPO4)1.5−x(O3PG)x·nH2O was different from the pure aluminium phosphonate Al2(O3PG)3·H2O (G is organic groups) in which the phosphate residue (P)–OH on lamellar surfaces of Al2(HPO4)3·H2O was completely displaced by the organic groups (P)–G in many cases. The ideal structure of AlSPP might be considered to be that the phosphate residue (P)–OH on its lamellar surfaces was regularly and alternately displaced by organic groups (P)–G, as AABAABAAB distribution, here A(P)–OH (the phosphate residue) and B(P)–G (the organic groups of phosphonate). Fig. 7b is presented here as two-dimensional surface ideal forms of AlSPP.
3.7 Surface morphology
Scanning electron microscopy (SEM), transmission electron microscopy (TEM) and atomic force microscopy (AFM) were performed to understand the surface morphology of the support and the catalyst. The SEM image of AlSPP (Fig. 8a) indicated that the support AlSPP had a layered crystalline structure as confirmed by XRD (Fig. 3). And Fig. 8a also indicates that AlSPP consisted irregularly of many small and big layered particles and the particles were aggregates of lots of minor crystalline grains. And the particle diameters of the support were in the scope of microns. Meanwhile, the support with various caves, holes, pores and channels with different shapes and sizes existed in every particle. Some micropores and secondary channels would increase the surface area of the catalyst26 (comfirmed by nitrogen adsorption–desorption). Catalyst 4b (Fig. 8b) took on the same structure with AlSPP, and various caves, holes, pores and channels with different shapes and sizes existed in every particle, but the structure was more loose. | ||
| Fig. 8 SEM photograph of AlSPP (a), catalyst 4b (b) and TEM photograph of AlSPP (c), the catalyst 4b (d). | ||
The TEM photography of AlSPP (Fig. 8c) manifested that the structure of the support was spheroid, its channels, holes and cavums could be discerned clearly, and their sizes were about 20–40 nm. Based on the interlayer spacings of AlSPP, 0.54 nm, determined by XRD, it could be inferred that each primary small particle consisted of stacks of 10–15 layers of AlSPP. While for the heterogeneous catalyst 4b (Fig. 8d), the configuration was loose, and its channels, holes and cavums also existed in it.
AFM images of the catalyst 4b are shown in Fig. 9. The morphology and the roughness of the catalyst 4b nanoparticles were visualized by AFM. Fig. 9 shows the 2D and 3D AFM images of catalyst 4b, respectively. The images of AFM showed that the small particles of catalyst 4b were in the nanometre range which possessed irregular or random packed granulation and the morphology of its surfaces was rough (also confirmed by N2 adsorption–desorption). It was important to emphasize that the average diameters of these pores and secondary channels among the layers of the catalyst were far greater than substrates. According to this, it could be deduced that substrates would have more chance to transfer to the internal catalytic active sites in solution.
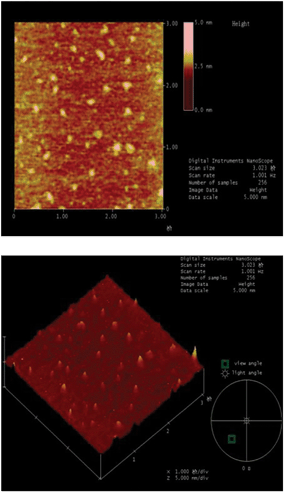 | ||
| Fig. 9 AFM images of catalyst 4b. | ||
3.8 Catalytic performance of the heterogeneous catalysts
The results in Table 2 show that the homogeneous chiral salen Mn(III) complex was active and enantioselective for the epoxidation of α-methylstyrene and indene in CH2Cl2 with m-CPBA as an oxidant, but the enantioselectivity was not very high (entries 1, 9). Delightfully, the ee values were improved by using the heterogeneous catalyst, especially, the conversion could reach 97% and ee value could be up to 95% (entry 13) for α-methylstyrene which was remarkably improved compared with the homogeneous catalyst (ee: 58%, entry 9). Similar results were obtained by Kim and Shin25d and Xiang et al.10a Kim and Shin reported that, for the asymmetric epoxidation of α-methylstyrene, the ee value increased from 51 to 59% after immobilization of chiral Mn(III) salen on the siliceous MCM-41 by multi-step grafting. It was also reported that the ee value was increased from 52% to 81% for asymmetric epoxidation of α-methylstyrene after immobilizing chiral salen Mn(III) on phenoxy-modified zirconium poly(styrene-phenylvinylphosphonate)phosphate (ZPS-PVPA).14f It had been reported that the alkoxy-modified MCM-41, ITQ-2 and ITQ-6 immobilized the salen Mn(III) catalyst in the asymmetric epoxidation of indene with NaClO as an oxidant, the ee value only got 56% for 70 h.27 Delightfully, in this paper, the ee values reached 91%–97% for 6 h with the alkoxy-modified AlSPP immobilized salen Mn(III) as the catalyst in the asymmetric epoxidation of indene. Li et al.10a reported that chiral salen Mn(III) axially immobilized on MCM-41 via phenoxyl groups, presented a 72% ee value in the NaClO aqueous/organic biphasic system whereas the same complex immobilized by the phenyl sulfonic group could give a 78% ee value for epoxidation of α-methylstyrene. Hutchings28 also found that the confinement effect originated from the zeolite cage could improve the chiral induction for the asymmetric aziridination of styrene in the zeolite cages. Therefore, we could conclude that the spatial effect including the surface effect originated from the supports as well as the immobilization modes are considered to be the main reasons for the increase in ee values. Here, in this paper, the spatial effect was attributed to the microenvironment effect and confinement effect20 which were provided by the layered and porous structure of AlSPP and the balance adjustment between the hydrophobic nature of polystyrene parts and the hydrophilic nature of phosphate parts. And these features were different from either pure polystyrene or pure phosphates. The flexibility of the alkoxy linker between the salen Mn(III) and the support was capable of tuning the transitional and rotational freedoms and potentially leading to enhanced catalytic site interactions. However, the conversions were lower than the homogeneous catalyst, which was mainly due to the slow diffusion of the reactant and the oxidant into the mesopores of the AlSPP in the multiphase reaction system.
| Entry | Substrateb | Catalyst | Oxidant system | Time/h | T/°C | Conv.c (yield%) | eed (%) | TONe |
|---|---|---|---|---|---|---|---|---|
| a Reactions were carried out at 0 °C in CH2Cl2 (2 mL) with α-methylstyrene or indene (1 mmol), n-nonane (internal standard, 1 mmol), m-CPBA (2 mmol), NMO (5 mmol, if necessary), heterogeneous catalysts (0.05 mmol, based on the Mn element). The conversion and the ee values were determined by GC with chiral capillary columns HP19091G-B 213, 30 m × 30 m × 0.32 mm × 0.25 μm. b A = indene, B = α-methylstyrene. c Isolated epoxide. d (S)-form. e Total TON = mmol epoxide/mmol Mn. | ||||||||
| 1 | A | Jacobsen | m-CPBA | 6 | 0 | 99(98) | 72 | 20 |
| 2 | 4a | m-CPBA | 6 | 0 | 92(90) | 94 | 18 | |
| 3 | 4a | m-CPBA/NMO | 6 | 0 | 42(38) | 35 | 8 | |
| 4 | 4b | m-CPBA | 6 | 0 | 93(92) | 95 | 19 | |
| 5 | 4c | m-CPBA | 6 | 0 | 95(94) | 97 | 19 | |
| 6 | 4d | m-CPBA | 6 | 0 | 95(89) | 91 | 19 | |
| 7 | 4e | m-CPBA | 6 | 0 | 97(83) | 92 | 19 | |
| 8 | 4f | m-CPBA | 6 | 0 | 98(85) | 94 | 20 | |
| 9 | B | Jacobsen | m-CPBA | 6 | 0 | 96(95) | 58 | 19 |
| 10 | 4a | m-CPBA | 6 | 0 | 93(82) | 92 | 19 | |
| 11 | 4a | m-CPBA/NMO | 6 | 0 | 54(43) | 38 | 11 | |
| 12 | 4b | m-CPBA | 6 | 0 | 96(95) | 93 | 19 | |
| 13 | 4c | m-CPBA | 6 | 0 | 97(81) | 95 | 19 | |
| 14 | 4d | m-CPBA | 6 | 0 | 97(92) | 82 | 19 | |
| 15 | 4e | m-CPBA | 6 | 0 | 98(87) | 89 | 20 | |
| 16 | 4f | m-CPBA | 6 | 0 | 99(79) | 93 | 20 | |
It was also found that the catalytic reaction performance (including conversions and ee values) increased with the increasing linkage lengths. Immobilized catalysts gave increased ee values from 91% to 94% and the conversions increased from 95% to 98% for the asymmetric epoxidation of indene accompanied with the increase of the linkage lengths (entries 6–8). The results might be attributed to the fact that active intermediates Mn(V) could attack the substrate more expediently with increasing linkage length. Similar results were obtained by Li.22 Compared with catalysts 4a–4c, the conversions of catalysts 4d–4f were much higher, but the ee values were much lower. It might be due to the fact that the linkage groups polyol in catalysts 4d–4f had more oxygen atom to coordinate the chiral salen Mn(III) than the diol groups in catalysts 4a–4c. Therefore, there were more chiral salen Mn(III) in the nanopores of polyol-modified AlSPP (confirmed by N2 adsorption–desorption), namely, there were more active centers in catalysts 4d–4f compared with catalysts 4a–4c. So the catalytic reactivities of catalysts 4d–4f were much higher than catalysts 4a–4c. Simultaneously, the increased active centers in catalysts 4d–4f made the steric hindrance between the chiral salen ligand and the olefins increased, so that the olefins could not attack the active centers with the best directions.
It is well known that the axial base generally could facilitate faster reaction rates, higher epoxide yields, and improved enantioselectivity. However, it was interesting to find out here that a remarkable increase in conversion and ee values was obtained in the absence of expensive O-coordinating axial base with the heterogeneous catalysts, especially for the epoxidation of indene (entry 2 vs. 3, con. 92% vs. 42%; ee 94% vs. 35%). Similar results were also obtained for α-methylstyrene (entry 10 vs. 11, con. 93% vs. 54%, ee 92% vs. 38). These observations were not in agreement with other literature reported earlier for both homogeneous and heterogeneous systems, where O-coordinating axial bases were rather essential for improving catalyst stability and enantioselectivity.29 The exceptional phenomenon, originated in the chiral salen Mn(III) complex immobilized on phenoxy-modified ZPS-PVPA, has been reported by our group recently.14f It was not in accordance with the catalysts axially coordinatively immobilized on diamine or polyamine modified organic-inorganic hybrid support ZPS-PVPA.14c The unusual phenomenon, in this text, induced that the alkoxy linker group had the similar properties in electronic structure and coordination performance with O-coordinating axial bases such as NMO (N-methylmorpholine N-oxide). Addition of O-coordinating axial additive NMO to the asymmetric epoxidation might cause competition with alkoxy linker groups, resulting in the formation of a well-defined transition-state geometry and conformation of catalysts 4a–4f being suppressed, thus decreasing the conversion and chirality recognization. Further studies concerning the mechanism of this novel behaviour of these immobilized catalysts are currently in progress.

| Cycle | Time/h | Conv.b (yield%) | eec (%) | TONd | Initial recovered catalyst (g) |
|---|---|---|---|---|---|
| a Reactions were carried out at 0 °C in CH2Cl2 and the same proportion of the substrate-to-catalyst and solvent-to-catalyst was retained. The conversion and the ee values were determined by GC with chiral capillary columns HP19091G-B 213, 30 m × 30 m × 0.32 mm × 0.25 μm. b Isolated epoxide. c (S)-form. d Total TON = mmol epoxide/mmol Mn. | |||||
| Fresh | 6 | 96(95) | 93 | 19 | 0.120 |
| 0.120 | |||||
| 1 | 6 | 96(95) | 93 | 19 | 0.120 |
| 0.119 | |||||
| 2 | 6 | 95(94) | 93 | 18 | 0.119 |
| 0.118 | |||||
| 3 | 6 | 95(93) | 93 | 18 | 0.118 |
| 0.117 | |||||
| 4 | 6 | 93(90) | 92 | 16 | 0.117 |
| 0.116 | |||||
| 5 | 7 | 91(90) | 92 | 15 | 0.116 |
| 0.115 | |||||
| 6 | 7 | 90(89) | 91 | 14 | 0.115 |
| 0.112 | |||||
| 7 | 8 | 90(89) | 91 | 14 | 0.112 |
| 0.110 | |||||
| 8 | 8 | 87(85) | 89 | 12 | 0.110 |
| 0.109 | |||||
| 9 | 10 | 85(83) | 89 | 10 | 0.109 |
| 0.106 | |||||
| 10 | 12 | 84(82) | 86 | 8 | 0.106 |
| 0.103 | |||||
The loss of Mn element could be due to the leaching of the salen Mn(III) complex from the support and the loss of the hyperfine granules of the heterogeneous salen Mn(III) catalyst during the stirring and filtrating process. This deduced that the heterogeneous chiral salen Mn(III) catalysts were relatively stable. One reason was attributed to the effective isolation of the Mn sites on the solid support AlSPP, whereas the Mn(salen)Cl catalysts were prone to oxidative degradation30 and dimerization to inactive μ-oxo-Mn(IV) species31 under homogeneous catalytic conditions. Another reason was due to the novel structure of the AlSPP support. AlSPP consisted of hydrophobic oligostyrenyl moieties and hydrophilic aluminium phosphonate-phosphate parts and possessed self-assembled layered structure on the nanometre scale. Generally, layered aluminium phosphonate was relatively stable, while the layered structure maybe destroyed under some extreme conditions such as base solutions. However, the virgin layered structure and channels, holes and caves could be roughly recovered under aqueous phase conditions, which was helpful to carry out self-assembly for layered aluminium phosphonate. Hence, these kinds of heterogeneous catalysts have the singular advantage of recyclability.
| Entry | Time(h) | Conv.d (yield%) | eee (%) | TONf |
|---|---|---|---|---|
| a The same as in Table 2. b The usage amounts of reagents were α-methylstyrene (25 mmol), n-nonane (25 mmol), heterogeneous catalyst 4b (1.25 mmol, based on the Mn element), m-CPBA (50 mmol), respectively. c The usage amounts of reagents were α-methylstyrene (50 mmol), n-nonane (50 mmol), heterogeneous catalyst 4b (2.5 mmol, based on the Mn element), m-CPBA (100 mmol), respectively. d Isolated epoxide. e (S)-form. f Total TON = mmol epoxide/mmol Mn. | ||||
| 1b | 6 | 95(94) | 93 | 19 |
| 2c | 6 | 93(91) | 92 | 19 |
4. Conclusions
In summary, novel types of immobilized chiral salen Mn(III) complexes onto AlAMSPP were synthesized and applied as catalysts in the asymmetric epoxidation of unfunctionalized olefins. The new heterogeneous catalysts indicated the comparable or even higher conversions and enantioselectivities than the corresponding homogeneous catalyst under the same conditions, which was mainly attributed to the spatial constraint effects of the special structure of AlSPP. Especially, it was found here that the heterogeneous catalysts afforded a remarkably increase in conversion and ee values in the absence of expensive O-coordinating axial bases for the asymmetric epoxidation of olefins. Moreover, the heterogeneous catalyst could be conveniently separated from the reaction system by simple precipitation in hexane and reused at least ten times with insignificant loss of ee and activity. Remarkably, this organocatalyzed asymmetric epoxidation reaction can be performed on a large-scale with the catalytic ability being maintained at the same level, which offers a great possibility for applications in industry.Acknowledgements
This work was financially supported by the National Ministry of Science and Technology Innovation Fund for High-tech Small and Medium Enterprise Technology (NO.09C26215112399) and the National Ministry of Human Resources and Social Security Start-up Support Projects for Students Returned to Business, Office of Human Resources and Social Security Issued 2009(143).References
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801 CrossRef CAS.
- R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Tetrahedron Lett., 1990, 31, 7345 CrossRef CAS.
- M. Palucki, P. J. Pospisil, W. Zhang and E. N. Jacobsen, J. Am. Chem. Soc., 1994, 116, 9333 CrossRef CAS.
- E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063 CrossRef CAS.
- B. D. Brandes and E. N. Jacobsen, J. Org. Chem., 1994, 59, 4378 CrossRef CAS.
- B. D. Brandes and E. N. Jacobsen, Tetrohydron Lett., 1995, 36, 5123 CAS.
- K. P. Bryliakov, O. A. Kholdeeva and M. P. Vanina, J. Mol. Catal. A: Chem., 2002, 47, 178 Search PubMed.
- (a) L. Canali, E. Cowan, H. Deleuze, C. L. Gibsona and D. C. Sherrington, Chem. Commun., 1998, 2561 RSC; (b) C. E. Song, E. J. Roh, B. M. Yu, D. Y. Chi, S. C. Kim and K. J. Lee, Chem. Commun., 2000, 615 RSC; (c) K. Smith and C. H. Liu, Chem. Commun., 2002, 886 RSC.
- (a) P. Das, I. Kuzniarska-Biernacka, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mol. Catal. A: Chem., 2006, 248, 135 CrossRef CAS; (b) I. Kuzniarska-Biernacka, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mol. Catal. A: Chem., 2007, 278, 82 CrossRef CAS; (c) I. Kuzniarska-Biernacka, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, Catal. Lett., 2010, 134, 63 CrossRef CAS; (d) I. Kuźniarska-Biernacka, C. Pereira, A. P. Carvalho, J. Piresb and C. Freire, Appl. Clay. Sci., 2011, 53, 195 CrossRef.
- (a) S. Xiang, Y. L. Zhang, Q. Xin and C. Li, Chem. Commun., 2002, 2696 RSC; (b) F. Bigi, L. Moroni, R. Maggi and G. Sartori, Chem. Commun., 2002, 716 RSC; (c) H. D. Zhang, S. Xiang and C. Li, Chem. Commun., 2005, 1209 RSC; (d) R. I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, S. Singh, P. H. Pandia and R. V. Jasra, J. Catal., 2005, 235, 28 CrossRef CAS; (e) D. M. Zhao, J. Q. Zhao, S. S. Zhao and W. Y. Wang, J. Inorg. Organomet. Polym. Mater., 2007, 17, 653 CrossRef CAS; (f) K. Yu, Z. C. Gu, R. N. Ji, L. L. Lou and S. X. Liu, Tetrahedron, 2009, 65, 305 CrossRef CAS.
- (a) L. Saikia, D. Srinivas and P. Ratnasamy, Appl. Catal., A, 2006, 309, 144 CrossRef CAS; (b) L. Saikia, D. Srinivas and P. Ratnasamy, Microporous. Mesoporous Mater., 2007, 104, 225 CrossRef CAS; (c) D. P. Serrano, J. Aguado and C. Vargas, Appl. Catal., A, 2008, 335, 172 CrossRef CAS; (d) S. Sahoo, P. Kumar, F. Lefebvre and S. B. Halligudi, Appl. Catal., A, 2009, 354, 17 CrossRef CAS.
- (a) A. R. Silva, J. L. Figueiredo, C. Freire and B. D. Castro, Microporous. Mesoporous Mater., 2004, 68, 83 CrossRef CAS; (b) M. Cardoso, A. R. Silva, B. D. Castro and C. Freire, Appl. Catal., A, 2005, 285, 110 CrossRef CAS; (c) S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork and M. S. Saeedi, Appl. Catal., A, 2010, 381, 233 CrossRef CAS.
- V. Mirkhani, M. Moghadam, S. Tangestaninejad, B. Bahramian and A. Mallekpoor-Shalamzari, Appl. Catal., A, 2007, 321, 49 CrossRef CAS.
- (a) R. F. Bai, X. K. Fu, H. B. Bao and W. S. Ren, Catal. Commun., 2008, 9, 1588 CrossRef CAS; (b) W. S. Ren and X. K. Fu, J. Mol. Catal. A: Chem., 2009, 312, 40 CrossRef CAS; (c) B. W. Gong, X. K. Fu and J. X. Chen, J. Catal., 2009, 262, 9 CrossRef CAS; (d) W. S. Ren, X. K. Fu, H. B. Bao, R. F. Bai, P. P. Ding and B. L. Sui, Catal. Commun., 2009, 10, 788 CrossRef CAS; (e) X. B. Tu, X. K. Fu, X. Y. Hu and Y. D. Li, Inorg. Chem. Commun., 2010, 13, 404 CrossRef CAS; (f) X. C. Zou, X. K. Fu, Y. D. Li, X. D. Tu, S. D. Fu, Y. F. Luo and X. J. Wu, Adv. Synth. Catal., 2010, 352, 163 CrossRef CAS; (g) J. Huang, X. K. Fu, G. Wang, C. Li and X. Y. Hu, Dalton. Trans., 2011, 40, 3631 RSC; (h) J. Huang, X. K. Fu and Q. Miao, Appl. Catal., A, 2011, 407, 163 CrossRef CAS; (i) J. Huang, X. K. Fu, G. Wang and Q. Miao, J. Solid State Chem., 2011, 184, 2605 CrossRef CAS; (j) Z. K. Hu, X. K. Fu, Y. D. Li and X. B. Tu, Appl. Organomet. Chem., 2011, 25, 128 CrossRef CAS; (k) X. Y. Hu, X. K. Fu, J. W. Xu and C. W. Wang, J. Organomet. Chem., 2011, 696, 2797 CrossRef CAS; (l) J. Huang, X. K. Fu, G. Wang, Y. Q. Ge and Q. Miao, J. Mol. Catal. A: Chem., 2012, 357, 162 CrossRef CAS.
- A. Clearfield and D. S. Thakur, Appl. Catal., A, 1986, 26, 1 CrossRef CAS.
- S. Nakayama and K. J. Itoh, J. Eur. Ceram. Soc., 2003, 23, 1047 CrossRef CAS.
- J. T. Zhao, L. Yan and X. Q. Xin, Eur. J. Inorg. Chem., 2000, 5, 801 Search PubMed.
- A. Razouk, M. Sahlaoui and M. Sajieddine, Appl. Surf. Sci., 2009, 255, 8695 CrossRef CAS.
- X. B. Ma, J. Liu, L. S. Xiao, R. Cheng, J. Q. Zhou and X. K. Fu, J. Mater. Chem., 2009, 19, 1098 RSC.
- Y. Sui, X. K. Fu, X. B. Ma, J. R. Chen and R. Q. Zeng, React. Funct. Polymers, 2005, 64, 55 CrossRef.
- R. Tan, D. H. Yin, N. Y. Yu, Y. Jin, H. H. Zhao and D. I. Yin, J. Catal., 2008, 255, 287 CrossRef CAS.
- H. D. Zhang, Y. M. Zhang and C. Li, J. Catal., 2006, 238, 369 CrossRef CAS.
- A. R. Silva, J. L. Figueiredo, C. Freire and B. D. Castro, Microporous Mesoporous Mater., 2004, 68, 83 CrossRef CAS.
- R. N. Ji, K. Yu, L. L. Lou, Z. C. Gu and S. X. Liu, J. Inorg. Organomet. Polym. Mater., 2010, 20, 675 CrossRef CAS.
- (a) D. M. Zhao, J. Q. Zhao, S. S. Zhao and W. Y. Wang, J. Inorg. Organomet. Polym. Mater., 2007, 17, 653 CrossRef CAS; (b) K. Yu, Z. C. Gu, R. N. Ji, L. L. Lou, F. Ding, C. Zhang and S. X. Liu, J. Catal., 2007, 252, 312 CrossRef CAS; (c) R. I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, K. Pathak and R. V. Jasra, J. Catal., 2006, 238, 134 CrossRef CAS; (d) G. J. Kim and J. H. Shin, Tetrahedron Lett., 1999, 40, 6827 CrossRef CAS.
- J. Huang, X. K. Fu and Q. Miao, Catal. Sci. Technol., 2011, 1, 1472 CAS.
- I. Domínguez, V. Fornés and M. J. Sabater, J. Catal., 2004, 228, 92 CrossRef.
- S. Taylor, J. Gullick, P. McMorn, D. Bethell, P. C. Bulman Page, F. E. Hancock, F. King and G. J. Hutchings, J. Chem. Soc., Perkin Trans., 2001, 2, 1714 Search PubMed.
- R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. T. Patel, P. K. Lyer and R. V. Jasra, J. Catal., 2002, 209, 99 CrossRef CAS.
- S. H. Zhao, P. R. Ortiz, B. A. Keys and K. G. Davenport, Tetrahedron Lett., 1996, 37, 2725 CrossRef CAS.
- B. B. De, B. B. Lohray, S. Sivaram and P. K. Dhal, Tetrahedron: Asymmetry, 1995, 6, 2105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20394d |
| This journal is © The Royal Society of Chemistry 2013 |