 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in enzymatic and chemical deracemisation of racemic compounds
Michał
Rachwalski
*ab,
Niek
Vermue
a and
Floris P. J. T.
Rutjes
*a
aRadboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: F.Rutjes@science.ru.nl; Fax: +31 24 365 3393; Tel: +31 24 365 3202
bUniversity of Łódź, Faculty of Chemistry, Department of Organic and Applied Chemistry, Tamka 12, 91-403 Łódź, Poland. E-mail: mrach14@wp.pl; Fax: +48 042 665 5162; Tel: +48 042 635 5767
First published on 24th September 2013
Abstract
Deracemisation of racemic compounds is still the most important strategy to produce optically pure compounds despite many recent advances in asymmetric synthesis. Especially deracemisation approaches that give rise to single enantiomers are preferred, which can be achieved either by invoking organocatalysts, metal complexes or enzymes – leading to dynamic kinetic resolution – or by applying other deracemisation techniques based on stereoinversion or enantioconvergence. In this tutorial review, we will provide an overview of new, recently developed approaches that lead to the important goal of creating organic compounds of single chirality.
 Michał Rachwalski | Dr Michał Rachwalski (1979, Częstochowa, PL) received his PhD at the Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences in Łódź (PL) under the supervision of Prof. Piotr Kiełbasiński in 2008. In 2012 he did his post-doc with Prof. F. Rutjes (Radboud University Nijmegen, NL). Presently, he is assistant professor at the University of Łódź (PL). His research interests include the synthesis and application of chiral ligands in asymmetric synthesis, the use of enzymes in organic synthesis and the chemistry of aziridines. |
 Niek Vermue | Niek Vermue (1986, Oostburg, NL) studies chemistry at Radboud University Nijmegen, where he obtained his BSc in 2010. Currently studying to obtain his MSc degree, he did internships in both the synthetic organic chemistry group of Prof. Floris Rutjes, where he focussed on the synthesis of chiral heterocycles via the asymmetric Mannich reaction, and the bioorganic chemistry group of Prof. Jan van Hest, working on biohybrid nanoparticles for drug-delivery. |
 Floris P. J. T. Rutjes | Prof. Floris Rutjes (1966, Heiloo, NL) received his PhD at the University of Amsterdam (NL) under the supervision of Prof. Nico Speckamp in 1993. He then did his post-doc with Prof. K. C. Nicolaou (The Scripps Research Institute, La Jolla, USA) resulting in a total synthesis of brevetoxin B. In 1995, he became assistant professor at the University of Amsterdam and in 1999 full professor in synthetic organic chemistry at Radboud University Nijmegen. His research interests include the application of asymmetric catalysis (bio-, transition metal- and organocatalysis) in the synthesis of biologically relevant molecules and synthesis of new molecular diagnostic tools for application in life sciences and flow chemistry. |
Key learning pointsThis tutorial review will:(1) Explain the importance of deracemisation (2) Introduce various strategies for deracemisation (3) Provide clear explanations of the mechanisms of deracemisation (4) Provide an overview of recent developments and state-of-the-art practical application of newly developed techniques (5) Offer a broader view on the general importance of chirality in (synthetic organic) chemistry |
1 Introduction
The synthesis of enantiomerically pure compounds remains a challenge in modern organic chemistry due to the importance of chirality in biological processes. The majority of biomolecules existing in living organisms are chiral and consist of only one of the two possible enantiomeric forms. As a result, molecular interactions of a racemic substrate with these biomolecules will always be different for one enantiomer than the other. This plays a particularly important role in the pharmaceutical industry, but also in other industrial sectors such as the food, and flavour and fragrance industries. As a result, it is crucial for those industries to produce many of their products (e.g. drugs, food additives, vitamins) as single enantiomers.The first person being able to separate a racemic mixture into the two enantiomers was Louis Pasteur, who as early as in 1849 physically separated tartaric acid crystals of opposite handedness. Ever since, the separation of a racemate into its two enantiomers – commonly known as resolution – has been the most prominent way to separate two enantiomers in numerous applications. This resolution is often performed through addition of an enantiopure (auxiliary) reagent leading to the preferred formation of one diastereoisomer over the other, which on the basis of different physical properties can then be separated. Removal of the auxiliary compound produces the desired enantiopure product. Another commonly used technique is treatment of a racemate with an enzyme, which recognizes one enantiomer and converts it into a different product, while the other enantiomer remains untouched. At this point, the two enantiomeric forms have different physical properties and can be readily separated. These kinetic resolutions can be carried out in small scale laboratory experiments, but also in large scale industrial processes. The limitation of such types of ‘classic’ kinetic resolutions (KR), however, is that both enantiomers can only be obtained in a maximum theoretical yield of 50%. The wish of the chemical industry to reduce costs and waste in the production of chiral building blocks led chemists to develop novel resolution procedures of racemic mixtures that proceed beyond the 50% yield. This is called deracemisation, which is defined as the conversion of a racemate into one of the corresponding enantiomers in theoretically 100% chemical yield and 100% optical purity without isolation of intermediates.
The existing different deracemisation techniques have been classified by the mechanism of action as proposed by Faber (Scheme 1).1 Inspired by the fact that deracemisation strategies continue to be discovered, we herewith provide an overview of deracemisation methods, based on the classification by Faber, with a strong focus on new and innovative approaches that have appeared in recent literature.
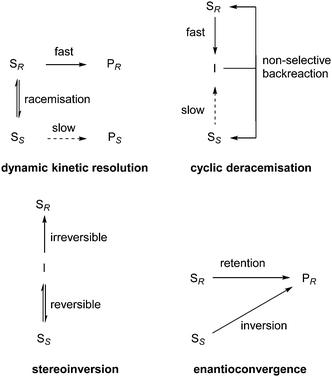 | ||
| Scheme 1 Deracemisation mechanisms proposed by Faber. | ||
2 Dynamic kinetic resolution (DKR)
Dynamic kinetic resolution (DKR) is the most widely used technique for deracemisation. It is a combination of ‘classic’ kinetic resolution, in which a racemate (SS and SR) is resolved by chemical transformation of one single enantiomer into a product (PR), with in situ racemisation of the ‘unwanted’ enantiomer (SS) of the starting material (Scheme 1). Many examples have been revealed combining metal-catalysed racemisation with enzymatic kinetic resolution. Since such chemoenzymatic DKRs have already been extensively reviewed,2,3 in this review, we will focus on DKR processes employing either organocatalysis, transition metal catalysis or enzymatic transformations. DKR of atropoisomers and so-called attrition-enhanced deracemisation will be also discussed.2.1 Organocatalysed DKR
Organocatalysis has gained much interest from organic chemists in recent years. Catalysis proceeds without metals using small organic molecules instead, thereby contributing to the sustainable (green) nature of the given conversions. Not unimportantly, the small chiral molecules that are generally employed as organocatalysts are often readily available at relatively low cost.In recent years the group of Birman reported several examples of DKR catalysed by small organic molecules. In 2010, they disclosed the use of benzotetramisole (BTM) as an enantioselective acyl transfer catalyst in the DKR of azlactones.4 In initial experiments, different amidine-based catalysts were screened in the alcoholysis of compound 1 (R = Me) using an equimolar proportion of catalyst and benzoic acid. Benzoic acid appeared to be an essential promotor in this reaction, since no conversion was observed in its absence. The best results in terms of conversion and ee were obtained using (S)-BTM 3 as the catalyst (Scheme 2). Under these slightly acidic conditions, the azlactone 1 racemizes via the corresponding enol, while the chiral acyl transfer catalyst causes the sterically congested alcohol nucleophile (4) to react faster with the (S)- than with the (R)-enantiomer. With less bulky nucleophiles, the acylation was less selective.
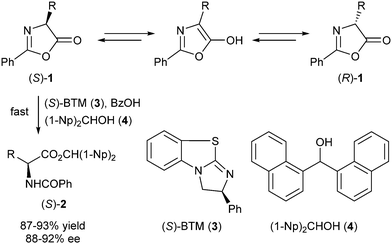 | ||
| Scheme 2 DKR of azlactones via BTM catalysis. | ||
Further variation of reaction conditions showed that catalyst loadings as low as 2 mol% were still effective though requiring longer reaction times. Lower temperatures proved to be detrimental to enantioselectivity, while elevated ones resulted in higher rates but with the same enantioselectivity. The applicability of this system to other azlactones was studied using a series of different R groups under optimal conditions. Good yields and ee's (around 90%) were obtained except for isopropyl- and 2,4-dimethoxyphenyl-substituted substrates, which proved to be resistant to alcoholysis.
One year later, the same group published DKR of azlactones using chiral Brønsted acids.5 Perhaps inspired by the necessity of a protic acid in the previous example, they reasoned that a strong chiral Brønsted acid might also be able to sufficiently activate azlactones to promote direct alcoholysis without intermediacy of a nucleophilic catalyst. Several 3,3′-disubstituted BINOL-derived phosphoric acids were screened as catalysts for the reaction of 2,5-diphenylazlactone with benzyl alcohol giving the most promising results (91% conversion, 63% ee) with 9-anthryl-substituted BINOL 6. This most likely proceeds through protonation of one of the azlactone enantiomers with the chiral phosphate acting as the counterion, followed by nucleophilic attack of the alcohol. Use of the more bulky 3,5-dimethoxyphenyl group led to substantial improvement in ee (77% with R = Bn) (Scheme 3).
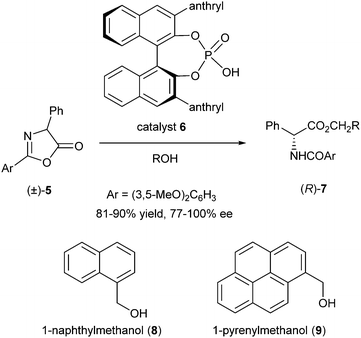 | ||
| Scheme 3 DKR of azlactones via Brønsted acid catalysis. | ||
The effect of the structure of the alcohol on enantioselectivity was also examined. After screening, 1-naphthylmethanol (8) and 1-pyrenylmethanol (9) were selected as being optimal leading to 90% conversion, 100% ee and 91% conversion, 90% ee, respectively. DKR of substituted 4-aryl azlactones under optimized conditions yielded the corresponding substituted α-arylglycine derivatives in 86–93% ee and 81–90% yield.
The most recent report of Birman's group describes the DKR of α-arylthio- and α-alkylthioalkanoic acids.6 This latest example was inspired by the serendipitous finding that α-arylthioesters are readily racemised by the previously mentioned amidine-based catalysts, thereby opening up the possibility of DKR. In this process the alkanoic acid 10 was first reacted with an activator resulting in the formation of the corresponding anhydride in the presence of base (iPr2NEt) (Scheme 4).
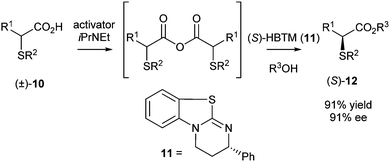 | ||
| Scheme 4 DKR of α-thioalkanoic acids. | ||
With α-phenylthiopropanoic acid 10 (R1 = Me, R2 = Ph) as a substrate, the use of DCC as an activator resulted in slow anhydride formation accompanied by the formation of side products. The use of benzoic anhydride as an activator and 1-naphthylmethanol as the nucleophile gave the ester 12 in 91% yield and 91% ee. After optimisation, (S)-homobenzotetramisole (HBTM, 11) appeared to be the best acylation catalyst. Finally, ester 12 was reduced using lithium aluminium hydride (95% yield) without any loss of optical purity.
Deracemisation by means of an enantioselective acylation catalyst was also achieved by Qu et al.7 In this case, the meso-1,2-diol 13 was non-selectively mono-acylated, after which via DKR selective acylation was performed using an enantiopure organocatalyst to selectively acylate the remaining free alcohol. A base was used to catalyse the racemisation of the starting material (Scheme 5).
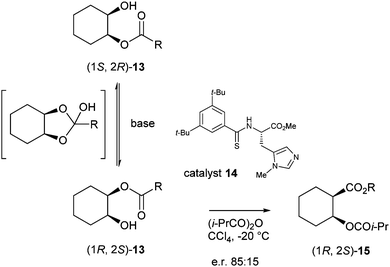 | ||
| Scheme 5 DKR of meso-1,2-diols 13. | ||
In initial experiments with monochloroacetate 13 (R = CH2Cl) and iPr2NEt as a base in CCl4 racemisation was slower than acylation resulting in decrease of ee during the reaction. Racemisation was faster with more electron-withdrawing groups on the acetate rendering it more susceptible to nucleophilic attack. However, the presence of these groups makes the acetate a worse hydrogen bond acceptor, thereby lowering the enantioselectivity of the reaction. Several halogen substituents were tested and the best results were obtained with dichloroacetylated diol 13 (R = CHCl2). Optimisation of the reaction conditions proved that DABCO was the best base (e.r. 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15) in CCl4 at −20 °C.
:15) in CCl4 at −20 °C.
Scheidt et al. reported that N-heterocyclic carbenes can also be used as organocatalysts in enantioselective acylation-based DKR to synthesize highly substituted β-lactones.8 α-Substituted β-ketoesters can undergo facile racemisation under basic conditions, which are typical for carbene generation and were therefore used as substrates in this DKR procedure (Scheme 6).
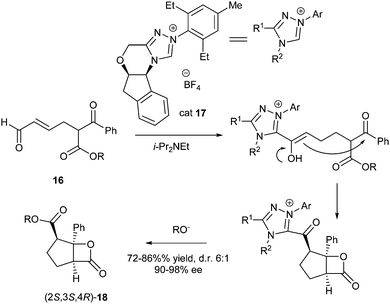 | ||
| Scheme 6 DKR of α-substituted β-ketoesters 16. | ||
With β-ketoester 16 (R = Et) as a substrate different catalysts were screened and NHC 17 was found to give the best results (72% yield, d.r. of 5:1 and 90% ee). This reaction proceeds through nucleophilic attack of the carbene onto an aldehyde, which gives rise to an umpolung so that the resulting enol, directed by the chiral NHC, diastereoselectively attacks the ketone, which then continues to form the β-lactone. The alkoxide that is released in the process liberates the NHC to give product 18. Using the same catalyst, the R-group in the substrate was varied changing also the acidity of the α-position and thus the rate of racemisation. The use of a trifluoroethyl ester led to an increase of diastereoselectivity (8:1 d.r.) with excellent ee (99%), albeit in lower yield (64%). Further optimisation of the conditions revealed that 1,2-dichloroethane and cesium carbonate were the best solvent and base, respectively. Under these conditions, substrate 16 was transformed into the desired lactone in high yield (86%), good d.r. (6:1) and excellent ee (98%).
2.2 Transition metal-catalysed DKR
Considering the asymmetric transformations that are catalyzed by transition metals, asymmetric hydrogenation stands out as a general and highly practical way to convert olefins, ketones, or imines into the corresponding enantiopure reduced products. This approach was discovered as early as in 1995, when the Japanese chemist Ryoji Noyori developed a ruthenium-based catalyst for asymmetric transfer hydrogenation of ketones and imines.9 As a result, in 2001 he was awarded the Nobel Prize for his work on asymmetric catalytic hydrogenation. Asymmetric hydrogenation of a racemate, however, will give rise to the formation of diastereoisomers. Several groups have addressed this issue, where in situ racemisation will give rise to the formation of single, enantiomerically pure products.In 2010 the group of Ratovelomanana-Vidal and Genêt reported a ruthenium-catalysed DKR in the synthesis of Taxotere®, an anti-mitotic chemotherapy drug.10 In this process, asymmetric hydrogenation was initially used for resolution of α-chloro-β-ketophosphonates 19 and esters 21. Racemisation proceeded via tautomerisation of the ketophosphonate to the corresponding enol. First, different ruthenium complexes were employed in the DKR of racemic 19 (R = Me, Scheme 7).
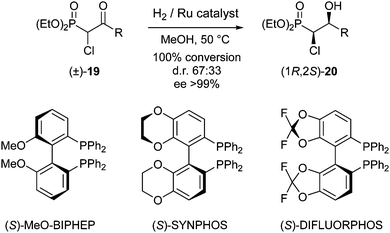 | ||
| Scheme 7 DKR of α-chloro-β-ketophosphonates. | ||
Best results were obtained using the preformed complex [RuCl((S)-SYNPHOS)2(μ-Cl)3][NH2Me2] at 50 °C, which gave 100% conversion with a d.r. of 67:33 and ee >99%. Results for substates 19 with other substituents (R = Et, iPr, Ph, p-ClC6H4) were similar, however, tert-butyl-substituted substrate 19 (R = tBu) turned out to be too sterically hindered and no formation of the desired product was observed. Finally, application of this approach to ketoester 21, the envisioned precursor in the Taxotere® synthesis, was also described (Scheme 8).
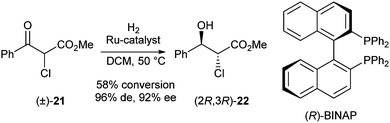 | ||
| Scheme 8 DKR of α-chloro-β-ketoesters. | ||
The best selectivity (96% de, 92% ee) was achieved using (R)-FLUOROPHOS at 50 °C, albeit in moderate conversion (58% after 48 h). In order to increase the conversion several solvents were screened with DCM giving the best compromise between conversion and selectivity.
In the same year Ratovelomanana-Vidal et al. reported a DKR of related α-alkoxy-β-ketoesters based on Noyori's catalyst.11 A number of ruthenium complexes were screened at 30 °C using 1.4 equiv. of a 5:2 formic acid–triethylamine mixture as a hydrogen source. Catalyst 24 provided the best results (>99% conversion, 95:5 d.r. and >99% ee) (Scheme 9).
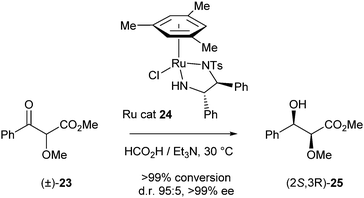 | ||
| Scheme 9 DKR of α-alkoxy-β-ketoesters. | ||
Diluting the reaction mixture from 1 to 0.2 M improved the d.r. to 97:3. Under optimised conditions DKR of a broad range of substrates was performed with very good conversions (80–99%) and excellent selectivities (d.r. 97:3, >99% ee).
In 2010, related DKR of α-alkyl-β-ketoamides via ruthenium-mediated asymmetric transfer hydrogenation was also reported.12 In initial studies compound 26 was used as a test substrate with ruthenium complex 27 as the catalyst and a mixture of formic acid and triethylamine as the hydrogen source (Scheme 10).
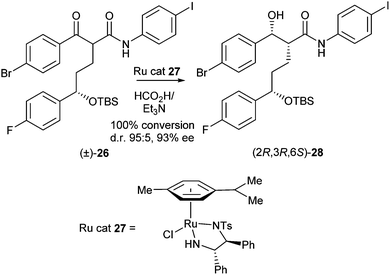 | ||
| Scheme 10 DKR of α-alkyl-β-ketoamides. | ||
The best results (100% conversion, d.r. 95:5, 93% ee) were obtained with 1 equiv. of triethylamine and 1.2 equiv. of formic acid after 8 hours. Further optimisation via modification of the catalyst using other aryl sulfonyl groups was carried out to reveal that the p-fluorobenzenesulfonyl-substituted ligand was the best catalyst (30, Scheme 11). Finally, a range of different α-alkyl-substituted aromatic β-ketoamides were successfully transformed into the corresponding syn-β-hydroxy amides in yields of 70–90%, d.r.'s of 92:8–99:1 (after crystallisation) and 92–99% ee.
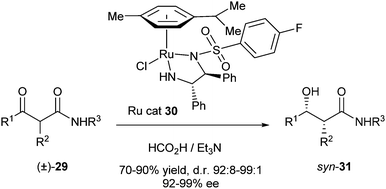 | ||
| Scheme 11 Exploration of substrate scope. | ||
In the same year, Somfai et al. reported DKR of α-amido-β-ketoesters via asymmetric transfer hydrogenation to give the corresponding anti-β-hydroxy-α-amido esters.13 The best results (95% yield, d.r. >95:5 and e.r. 97:3) were achieved using (S,S)-BnDPAE as a ligand with [RuCl2(benzene)]2 as the catalyst (Scheme 12).
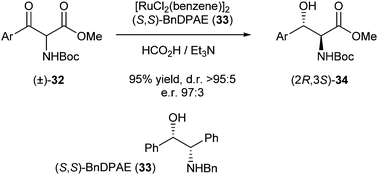 | ||
| Scheme 12 DKR of α-amido-β-ketoesters. | ||
Using differently substituted aromatic ketones in combination with methyl or ethyl ester, the products were formed in generally good yields (76–95%) and excellent enantioselectivities (96:4–99:1), except for the m-chlorophenyl-substituted ketone with the ethyl ester, which gave significantly worse results (69% yield, 66:34 e.r.).
In 2012, Johnson et al. published a DKR of α-ketoesters also via asymmetric transfer hydrogenation.14 A broad range of catalysts were screened on substrate 35 (R = Ph) in DMF at 75 °C using a 5:2 mixture of formic acid and triethylamine as the hydrogen source (Scheme 13).
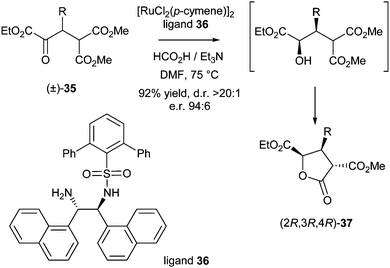 | ||
| Scheme 13 DKR of α-ketoesters. | ||
The best results were obtained using ligand 36, which gave the desired product in 92% yield, excellent d.r. (>20:1) and e.r. (94:6). Substrates incorporating electron-rich, electron-poor and heteroaryl substituents gave all products in high yields (around 90%) and enantioselectivities (typical e.r. of 95:5).
On a somewhat different note, in the same year Fu et al. reported a DKR of secondary alcohols using a combination of an enantiopure dimethylaminopyridine (DMAP) derivative and a ruthenium-based catalyst (Scheme 14).15
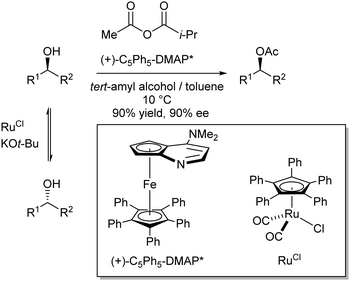 | ||
| Scheme 14 DKR of secondary alcohols. | ||
In this case, the Ru-catalyst was responsible for racemisation of the secondary alcohols (via dehydrogenation/hydrogenation through hydrogen transfer), while in a kinetic resolution only one of the enantiomeric alcohols reacts faster with the in situ formed enantiopure activated acyl-DMAP intermediate. The best results were obtained with acetyl isopropyl carbonate as the acylating agent in a 1:1 mixture of tert-amyl alcohol and toluene at 10 °C. Then, various aryl alkyl carbinols were used as substrates to give successful results (yields generally exceeding 90% and ee's around 90%).
2.3 Enzymatic DKR
In addition to enantiopure organocatalysts and transition metal complexes, enzymes can be powerful catalysts to induce enantioselective reactions due to their high substrate selectivity. In order to overcome the 50% yield barrier in enzymatic resolutions of racemic mixtures, the DKR principle has also been recently applied to a variety of enzymatic reactions.In 2011 the group of Asano reported enzyme-promoted DKR of α-aminonitriles leading to optically pure α-amino acids.16 This process consisted of three steps: (i) hydrolysis of racemic α-aminonitrile by a non-selective nitrile hydratase (NHase), (ii) stereoselective hydrolysis of the resulting α-amino acid amide by an amino acid amide hydrolase, and (iii) simultaneous racemisation of the α-amino acid amide by ACL racemase (Scheme 15).
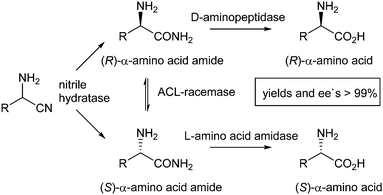 | ||
| Scheme 15 DKR of α-aminonitriles to form chiral α-amino acids. | ||
Several substrates were tested using this system and both (R)- and (S)-amino acids could be obtained in excellent yields and ee's (>99%). Such an approach, however, will always be dependent on the availability of different enzymes that are able to convert both enantiomers.
The group of Ramström used a lipase from Burkholderia cepacia (a group of Gram-negative bacteria) to catalyse the reaction between an acyl donor (phenyl acetate) and an amine to give the acetylated amine.17 In initial experiments KR of α-aminonitrile 38 was investigated (Scheme 16). The conversion into product (R)-39 was recorded at 60% with an enantiomeric purity of 88% ee. The conversion exceeding the theoretical 50% maximum of KR and the observation that the remaining starting material 38 was racemic, clearly indicated that a DKR-type process took place. Further mechanistic studies led to the conclusion that racemisation of the α-aminonitrile was catalysed by lipase via a retro-Strecker mechanism through the same active site that catalyses transacylation.
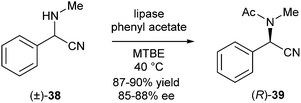 | ||
| Scheme 16 KR of α-aminonitriles promoted by lipase. | ||
Racemisation most likely occurs when a His-residue abstracts a proton from the amino group, followed by elimination of the cyanide ion (Scheme 17).
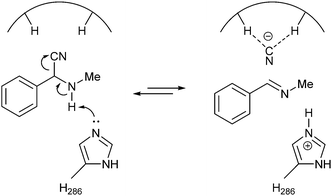 | ||
| Scheme 17 Transition state for lipase-catalysed racemisation. | ||
This method was demonstrated to give good yields (87–90%) and ee's (85–88%), but only after prolonged reaction times (7–13 days).
In 2010 Kroutil et al. published a DKR of racemic mandelic acid 40 to yield the corresponding enantiopure α-amino acid L-phenylglycine (42).18 The α-hydroxy acid was first oxidised to the α-ketoacid 41 using D-mandelate dehydrogenase (D-MDH), after which it was converted into the α-amino acid via asymmetric reductive amination with L-amino acid dehydrogenase (L-AADH) (Scheme 18).
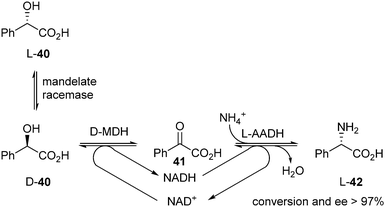 | ||
| Scheme 18 DKR of mandelic acid 40. | ||
Because oxidation and reduction took place simultaneously, the NADH/NAD+ cofactor was recycled in this system. In the screening process, 10 out of 24 tested AADHs gave excellent results (>97% conversion, >97% ee), all favouring L-42.
D'Arrigo et al. reported in 2011 an enzymatic DKR of N-Boc-amino acid thioesters.19 In this process, the authors take advantage of the fact that thioesters, as opposed to the resulting amides, are prone to racemisation under basic conditions. The formation of the amide bond can be catalysed in an enantioselective manner by proteases. Subtilisin, commercially available under the name alcalase, is known to be stable in organic solvents (an aqueous reaction will lead to hydrolysis of the thioester) and in the presence of bases. In this system, ammonia or amines can be used as nucleophiles (Scheme 19).
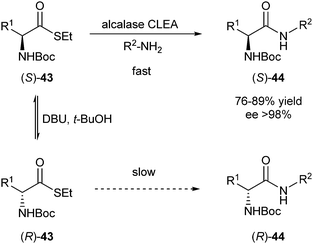 | ||
| Scheme 19 DKR of N-Boc-amino acid thioesters. | ||
Using this system, yields of 76–89% were obtained with ee's of >98% with 43 (R1 = Bn) as a substrate and three different nucleophiles (R2 = H, nPr, Bn). Other substrates (R1 = PhCH2CH2, nBu) were transformed with lower selectivity.
In 2010, Gotor et al. published two reports about DKR using Baeyer–Villiger monooxygenases (BVMOs). In one of them, a DKR of 2-alkyl-1-indanones to form the corresponding dihydroisocoumarins was described.20 An enzymatic DKR of racemic 2-methyl-1-indanone 45a was performed in a biphasic system of Tris-HCl buffer with 5% hexane, in which racemisation most likely proceeds via the corresponding enol. The ring expansion was carried out using a single mutant of thermostable phenylacetone monooxygenase (M446G PAMO) from Thermobifida fusca in the presence of glucose-6-phosphate dehydrogenase (G6PDH) to recycle NADP+ (Scheme 20).
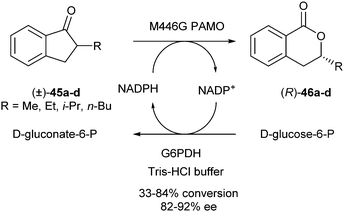 | ||
| Scheme 20 DKR of 2-alkyl-1-indanones. | ||
It was found that longer reaction times (7 days) are beneficial for conversions (84%, compared to 33% after 3 days), but ee's decreased (82% in comparison with 92% after 3 days) due to partial racemisation of the lactone under the reaction conditions.
A second report of the Gotor group described DKR of benzyl ketones catalysed by BVMOs.21 Racemic substrate 47 (R1 = R2 = Me) was subjected to DKR catalysed by 4-hydroxyacetophenone monooxygenase (HAPMO) from Pseudomonas fluorescens in the presence of G6PDH to regenerate NADPH from NADP+. Racemisation of the substrate occurred spontaneously under basic conditions, and was enhanced by the presence of an anionic exchange resin (Scheme 21). Several resins were tested and the best results (86% conversion, 84% ee after 116 hours) were obtained for (R)-48 using Dowex MWA-1, a weak anionic exchange resin.
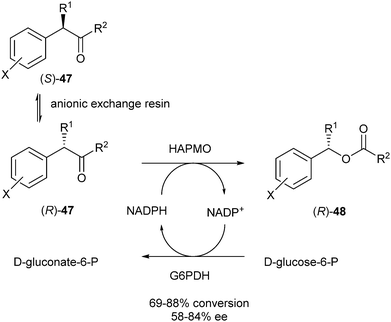 | ||
| Scheme 21 DKR of benzyl ketones. | ||
With differently substituted aromatic rings (X = m-Me, m-CF3, p-Cl) conversions varied from 69 to 88% with ee's of the products varying from 58 to 81%. Clearly, the results using this system are strongly dependent on the substrate structure.
In 2011, Yang et al. reported a DKR of secondary aromatic alcohols using an acidic resin known as CD8604, which is known to racemise benzylic alcohols, presumably via formation of the benzylic cation and attack again from water. In addition, Novozyme 435 (a commercially available immobilised preparation of lipase B from Candida antarctica) was used for the kinetic resolution.22 Different acyl donors were investigated using this system (Scheme 22).
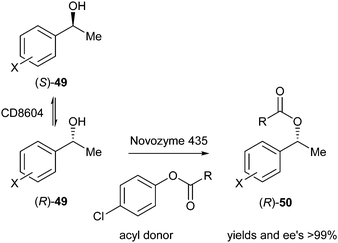 | ||
| Scheme 22 DKR of aromatic alcohols using different acyl donors. | ||
It was found that longer chain acyl donors (R = Bu, Pent, etc.) strongly inhibited non-selective transesterification catalysed by the resin due to steric effects of the substrates, as well as to formation of micromicelles in the pores of the resin. After screening of acyl donors with varying chain length (R = Me to octyl), 4-chlorophenyl valerate (R = Bu) was found to be the most appropriate in DKR of phenylethanol (yield >99%, ee >99% after 6 hours). Employing this acyl donor in DKR of 1-phenylethanols with various substitution patterns in the phenyl ring resulted in yields and ee's up to 99% in almost all cases. Finally, the reusability of this system was examined up to 20 times without any loss in yield or ee.
DKR of the racemic Boc-protected vicinal diamine 51 catalysed by lipase B from Candida antarctica (CAL-B) was described by Rebolledo et al.23 First, KR of (±)-51 was performed to find that the use of 1-phenylethyl acetate as the acyl donor and methyl tert-butyl ether as solvent at 28 °C gave the best results (Scheme 23).
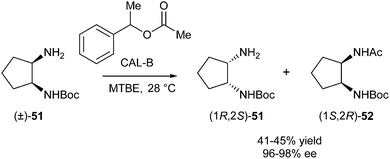 | ||
| Scheme 23 KR of vicinal diamine 51. | ||
Then, other monocarbamates were subjected to the same conditions. Surprisingly, yields and ee's pointed towards racemisation taking place, thus leading to DKR as opposed to KR. This racemisation is most likely due to intramolecular migration of the alkoxycarbonyl group between both vicinal amines. Reaction conditions for DKR of 53 (R = Bn) were optimised, and using MTBE–Et3N 10:1 as a solvent and increasing the temperature to 50 °C resulted in high yield and ee (94 and 96%, respectively) (Scheme 24).
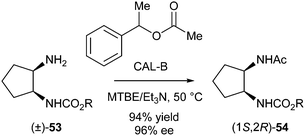 | ||
| Scheme 24 DKR of a vicinal diamine. | ||
In 2010, Nelson et al. reported DKR via a vinylogous Payne rearrangement.24 Epoxides 55 and 56 were intermediates in the synthesis of the natural product scyphostatin. Both diastereoisomers could be separated, but appeared to reequilibrate to the mixture of 55 and 56 upon heating after 3 days. Thus, this equilibrating mixture was used in DKR. The use of lipase Amano PS (commercially available lipase PS from Burkholderia cepacia) allowed for selective acetylation of 56 using vinyl acetate as the acyl donor in hexane at ambient temperature (Scheme 25). It is important to remark that since the starting compound is not a racemic mixture, but a mixture of enantiopure diastereoisomers, this formally is not a resolution. Nevertheless, after a 14-day reaction, starting from a 1:1 mixture of 55 and 56, diastereomerically pure acetate 57 was produced in yield of 46%.
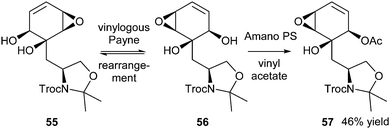 | ||
| Scheme 25 DKR of epoxides via vinylogous Payne rearrangement. | ||
The group of Giacomini described a DKR via reduction of arylpropionic aldehydes to (2S)-2-arylpropanols promoted by alcohol dehydrogenases (ADHs).25 Several racemic substituted 2-arylpropanals 58 (readily racemisation at higher pH values via enolisation) were tested and after optimisation of reaction conditions (pH, cosolvent) for every substrate, good yields and enantiomeric ratios were obtained (Scheme 26). A noteworthy result was obtained for ketoprofenal 60 which bears two carbonyl groups. HLADH exhibited a high degree of selectivity, since no traces of possible ketone reduction by-products were detected.
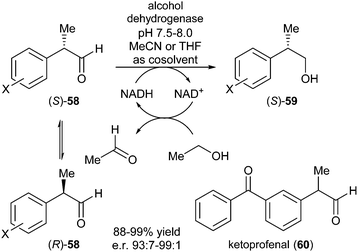 | ||
| Scheme 26 DKR of 2-arylpropanals. | ||
2.4 Other DKRs
Two special cases of DKR are discussed in this section, namely, (1) DKR of atropisomers, compounds being chiral due to hindered rotation around a single bond, and (2) so-called attrition-enhanced deracemisation, a process employing the property of some compounds to crystallise as a conglomerate in DKR processes.In 2010, an example of DKR of atropisomers was reported by Miller et al.26 Compound 61 was triply brominated in reaction with NBS in the presence of a catalytic amount of N-methylpyrrolidine (Scheme 27). This reaction gave rise to compound 62, which due to the increased steric bulk at one of the aromatic rings, showed the occurrence of two atropisomers.
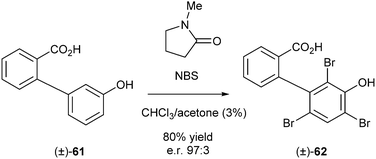 | ||
| Scheme 27 Triple bromination using NBS. | ||
The enantiopure tripeptide 63, which catalyses aromatic electrophilic substitution, was selected as the best catalyst for further study with NBP as a bromine source (Scheme 28). In this case, the catalyst kinetically favours bromination of one biaryl enantiomer over the other, which prior to the bromination are in equilibrium, but are after the bromination are not. In this way the racemic mixture is resolved.
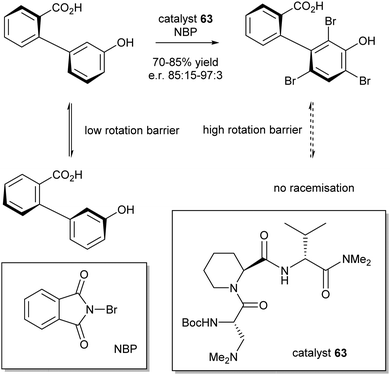 | ||
| Scheme 28 DKR of atropisomers via triple bromination with NBP. | ||
A range of differently substituted biaryl compounds were tested and all the desired products could be obtained in good yields (70–85%) and moderate to good e.r.'s (85:15–97:3).
A different mechanochemical approach to DKR of atropisomers was reported in 2012 by Wiggins and Bielawski.27 Enantiopure atropisomers were racemised by applying mechanical force using poly(methyl acrylate) (PMA) chains on (R)- and (S)-BINOL in combination with ultrasound. Thus, by treatment of a racemic mixture of polymer-functionalised BINOL with deoxycholic acid and cholesterol esterase, esters of (S)-BINOL are selectively hydrolysed (Scheme 29). Upon subjecting the remaining enantiopure starting material to ultrasound, the atropisomer racemises, ultimately producing only (S)-BINOL, which could be isolated in 90% yield and >98% ee.
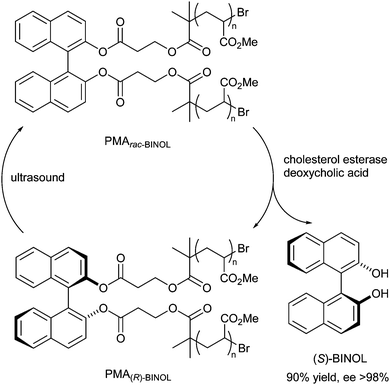 | ||
| Scheme 29 DKR via mechanochemical racemisation. | ||
An entirely different, recently discovered resolution process is so-called attrition-enhanced deracemisation. This is a process that can only be applied to compounds that crystallise as a conglomerate, i.e. the enantiomers form separate solid phases. Furthermore, compounds must be prone to deracemisation, e.g. by deprotonation under basis conditions. The driving force for deracemisation is a process called Ostwald ripening, meaning that dissolution of smaller crystals is favoured over larger ones. The resolution is not based on kinetics, as in a ‘classical’ DKR, but on thermodynamics, and thus it can be called dynamic thermodynamic resolution (DTR).
Noorduin et al. reported in 2008 a deracemisation of phenylglycine derivative 64 using this technique.28 The compound readily racemised under basic conditions making it a potentially good substrate for attrition-enhanced deracemisation. Nearly racemic mixtures of 64 were suspended in MeOH and ground by magnetic stirring in the presence of glass beads. Once the equilibrium between the solution and solid states had been established, DBU was added as a base to initiate racemisation (Scheme 30).
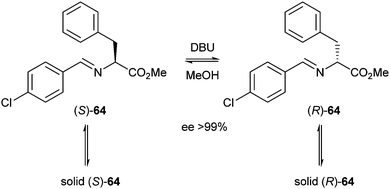 | ||
| Scheme 30 Equilibria during attrition-enhanced deracemisation. | ||
Because of the slight excess of one enantiomer, this enantiomer will have a slightly larger surface area for crystallisation and will therefore crystallise faster, thus shifting all equilibria until the other enantiomer is depleted in the solid state. The ee increases exponentially during the reaction.
The same group reported in 2009 the use of this technique in the deracemisation of naproxen.29 While naproxen itself crystallises as a racemic compound, its ethyl and methyl esters 65 and 66 both crystallise as conglomerates. Interestingly, the solubility of 66 in methanol is lower than that of 65. This difference can be used to generate a supersaturated solution of 66 starting from a saturated solution of 65. To introduce a gradual feed of racemic 66, a mixture of rac-65 (92 mol%) and (S)-66 (8 mol%) was partially dissolved in MeOH–NaOMe and ground at 700 rpm (Scheme 31).
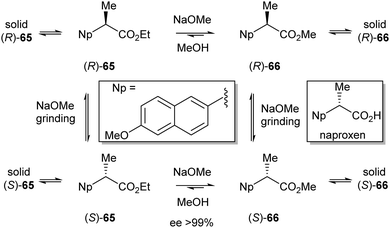 | ||
| Scheme 31 Attrition-enhanced deracemisation of naproxen. | ||
During the reaction all solid material 65 was converted into 66. Simultaneously, crystalline 66 was deracemised via attrition-enhanced deracemisation, in favour of the (S)-enantiomer. It also appeared that the use of a gradual feed decreased the deracemisation time to 15 hours.
3 Cyclic deracemisation and stereoinversion
Cyclic deracemisation and stereoinversion are techniques involving chemically stable, achiral intermediates (Scheme 1). In cyclic deracemisation, the achiral intermediate (I) is non-selectively converted into a racemic mixture of the substrate (back reaction). The rates of conversion of the two enantiomers SR and SS into the intermediate I differ significantly, thus leading to accumulation of the slow-reacting enantiomer SS. In a stereoinversion process, a single enantiomer of the substrate (SS) is transformed into an achiral intermediate I, followed by irreversible conversion to the desired (opposite) enantiomer (SR).Märkle and Lütz reported deracemisation of amino acids via a cyclic process in 2008, combining stereoselective enzymatic oxidation with non-selective electrochemical reduction.30 In a test reaction, the electrochemical reduction of 4-methyloxovalerate (69) in ammonia buffer at pH 10 (to establish an equilibrium between keto acid 69 and imino acid 68) was pursued. The electrolysis potential was chosen to reduce only the imino acid 68. So far, the keto acid could be converted into the racemate of leucine 67. A D-amino acid oxidase (D-AAO) was applied to selectively oxidise the D-amino acid (Scheme 32).
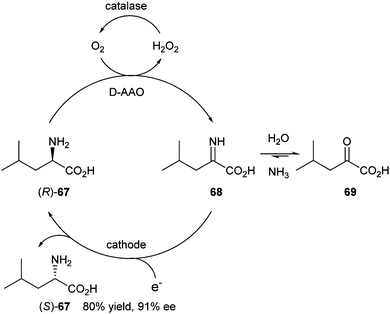 | ||
| Scheme 32 Deracemisation of amino acid via cyclic deracemisation. | ||
Starting from racemic leucine, a yield of 80% of L-leucine (S)-67 could be obtained with 91% ee. In addition, starting from enantiopure D-leucine a yield of 60% of (S)-67 with 77% ee was obtained.
Kroutil et al. reported deracemisation of α-chiral primary amines via stereoinversion.31 ω-Transaminases (ω-ATAs) were used to catalyse stereoselective oxidation of one amine enantiomer to the corresponding ketone (kinetic resolution). This ketone was then selectively reconverted to a single enantiomer of the desired amine (Scheme 33).
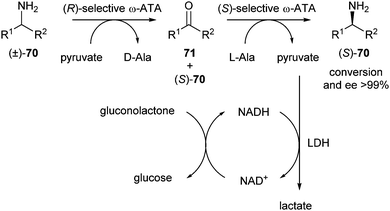 | ||
| Scheme 33 Deracemisation of primary amines via stereoinversion. | ||
In the second step, the equilibrium lies towards the substrates (ketone, alanine) rather than towards the products (amine, pyruvate). To shift the equilibrium towards the products, the pyruvate formed was removed by reduction using lactate dehydrogenase (LDH). With this system, enantiomers of a range of primary amines were accessible, depending on the order of addition of ω-ATAs and were obtained in generally good conversions and excellent ee's (>99% in most cases).
In 2011, the group of Patel reported three enzymatic routes to (S)-amino acid 72 from racemic mixtures, two of which proceeded via a stereoinversion mechanism, while the third one was a cyclic deracemisation.32 The basic strategy was to employ an (R)-amino acid oxidase to convert the (R)-enantiomer of the amino acid into an achiral keto acid intermediate, in combination with another enzyme, to reduce the keto acid selectively to the (S)-amino acid (Scheme 34). The oxidase was inhibited under the reductive amination conditions, so a two-step procedure was required. Using this process, a 54% yield of (S)-72 was obtained in >99% ee.
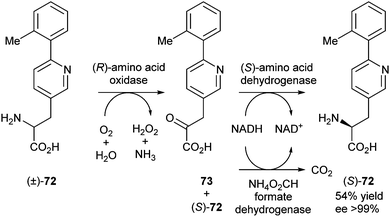 | ||
| Scheme 34 Deracemisation of amino acids via stereoinversion. | ||
In a similar procedure (R)-amino acid oxidase was combined with (S)-aminotransferase from Burkholderia sp. After 6 hours, oxidation was complete as indicated by the ee of the (S)-amino acid of >99%. The reaction was continued for an additional 16 hours to complete the conversion of 73 into (S)-72. The yield was 85% with an ee of 99.5% (Scheme 35).
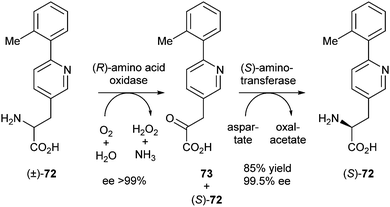 | ||
| Scheme 35 Deracemisation of an amino acid via stereoinversion. | ||
The third strategy described by this group was a cyclic deracemisation. The use of a Celite-immobilised (R)-amino acid oxidase was combined with chemical reduction of the imine with a borane–ammonia complex. This reaction occurred before the enzyme-bound imine was hydrolysed to the keto acid (Scheme 36).
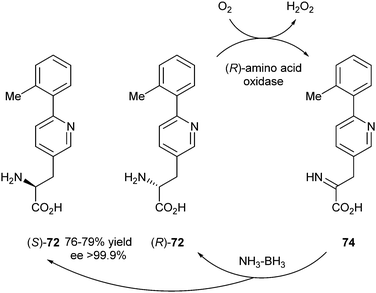 | ||
| Scheme 36 Cyclic deracemisation of an amino acid. | ||
The reaction was performed in a phosphate buffer solution with ammonium formate at different pH values. The best yields (76–79%) were obtained at pH 6.0–7.0 in >99.9% ee.
In 2012, Yun et al. described deracemisation of the unnatural amino acid homoalanine using D-amino acid oxidase (DAAO) and ω-transaminase (ω-TA).33 DAAO stereospecifically catalyses oxidation of D-amino acids into the corresponding imino acids, which are spontaneously hydrolysed to the corresponding keto acids and ammonia. The ω-TA from Vibrio fluvialis was used to catalyse a transfer of the amino group from the amino donor onto the carbonyl moiety of the amino acceptor, in this case keto acid 76 (Scheme 37). After shaking at 37 °C for 8 hours, a 99% conversion was reached with >99% ee.
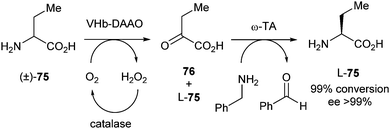 | ||
| Scheme 37 Deracemisation of homoalanine via stereoinversion. | ||
4 Whole cell-mediated deracemisation
The use of whole cells as biocatalysts in enzymatic processes has the advantage that time-consuming and thus expensive cell lysis and/or protein purification procedures are not necessary. This means that whole cell preparations are often used in production scale enzymatic processes. A disadvantage, however, is that because of the large amounts of different enzymes present in living cells, the exact mechanism of whole-cell-mediated deracemisation cannot always be completely elucidated. For that reason, the whole cell examples are not shared under the categories given in Scheme 1.In 2011, Mehmetoğlu et al. described deracemisation of benzoin acetate to form benzoin by whole cells of the fungus Rhizopus oryzae.34 In previous studies it had become apparent that R. oryzae possesses both lipase and racemase activity. Additionally, this preparation exhibits alcohol dehydrogenase (ADH) activity, which catalyses the undesired oxidation of benzoin 78 to benzil 79 (Scheme 38).
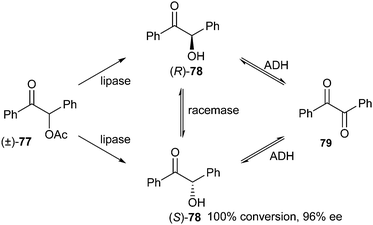 | ||
| Scheme 38 Deracemisation of benzoin acetate by R. oryzae. | ||
The best results were obtained using cells treated with 20 kHz ultrasound. A 100% conversion of racemic acetate 77 in favour of (S)-78 with 45% ee was achieved in one day. After 7 days, the ee had increased to 89%. The effect of pH on the medium was also investigated and a pH of 6.0 in combination with ultrasonic homogenisation at 20 kHz gave the best ee (96%) and conversion (100%).
Alcántara and Molinari also described a whole cell-mediated deracemisation of benzoin to the (S)-enantiomer employing whole cells from the non-conventional yeast Pichia glucozyma.35 The reaction showed successful deracemisation with (S)-78 being the only enantiomer present in 85% yield. Benzil 79 was detected (10–15%) in the organic phase during the reaction, indicating an oxidation–reduction cascade taking place (Scheme 39). The reactions performed in highly hydrophobic solvents such as hexane and isooctane took place with excellent ee's (>98%) and yields (91% for isooctane).
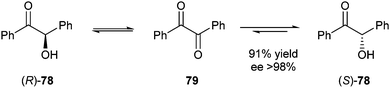 | ||
| Scheme 39 Proposed mechanism of deracemisation of benzoins. | ||
In the same year, Solís et al. reported deracemisation of ibuprofen nitrile 80 mediated by cells from Nocardia corallina B-276 to ibuprofen amide 81 and its subsequent hydrolysis to ibuprofen 82 (Scheme 40).36
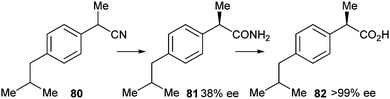 | ||
| Scheme 40 Hydrolysis of ibuprofen nitrile. | ||
Using a suspension of harvested cells in phosphate buffer yielded 12% of unreacted 80, while products 81 and 82 were obtained in 15 and 73% yield, respectively, after 72 hours. The presence of amide 81 indicates a two-step process rather than a direct nitrilase-catalysed transformation of the nitrile 80 into the acid 82. While the remaining amide 81 showed an ee of 38%, the resulting acid 82 was enantiomerically pure (>99% ee in favour of the (R)-enantiomer).
Kurina-Sanz and Orden et al. described deracemisation of secondary alcohols by a chemoenzymatic sequence of oxidation/reduction using plant cells.37 The reactivity of six cell lines towards the oxidation of racemic 1-phenylethanol (83a) was screened. Only whole cells from Gardenia jasminoides were able to resolve the racemic mixture providing the (R)-isomer with 90–92% ee after 6 days. In order to investigate the substrate scope of this whole cell catalyst, whole cell-mediated kinetic resolution of various 1-phenylethanol derivatives was performed. Substrates 83c and d gave the best results and were therefore selected for a chemoenzymatic deracemisation using NaBH4 as the non-selective reducing agent (Scheme 41).
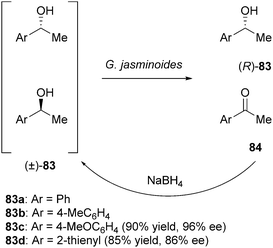 | ||
| Scheme 41 Deracemisation of secondary alcohols using G. jasminoides cells. | ||
For those substrates the (R)-alcohols 83c and d were obtained in yields of 90 and 85%, respectively, with good ees (96 and 86%).
Parvulescu et al. described a combination of enantioselective deoxygenation of racemic sulfoxides using E. coli bacteria with heterogeneous-catalytic oxidation of the resulting sulfite by a Ta2O5–SiO2 catalyst (Scheme 42).38
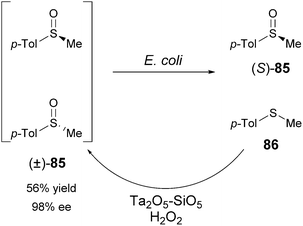 | ||
| Scheme 42 Deracemisation of aromatic sulfoxides using E. coli cells. | ||
The enantioselective reduction of racemic methyl p-tolyl sulfoxide (MTSO, 85b) was repeated three times with fresh E. coli cells leading to 49.3% conversion and 97.3% ee of (S)-MTSO. The resulting thioether was subjected to chemical oxidation leading to 22% conversion. The resulting racemic sulfoxide was subjected to a new deracemisation cycle. After three cycles, (S)-MTSO (85) was obtained in 56% yield and 98% ee.
5 Enantioconvergence
The last category mentioned in Scheme 1 to achieve deracemisation is enantioconvergence. In this approach deracemisation is accomplished by the use of enantioconvergent methods which transform the two enantiomers of the substrate (SS and SR) into a single enantiomer of the product (PR). The key feature of this strategy is that each enantiomer of the substrate follows a different reaction pathway towards a single enantiomer of the product.In 2009, Matsumoto et al. reported deracemisation of monotosylated diols combining enzymatic hydrolysis with a Mitsunobu esterification.39 Racemic 2-acetoxyhexyl tosylate 87 (R = Bu) was chosen as a representative target compound. Its hydrolysis using lipase PS via a KR-type process had already been described. The Mitsunobu reaction of alcohol 88 into (S)-87 was achieved with 100% yield and 99% ee using solid-supported PPh3, DEAD and acetic acid in toluene at room temperature. Using these conditions, the two-step deracemisation of a series of four racemic 2-acetoxyalkyl tosylates was performed. Deracemisation of 87 (R = Bu, Me, CH2OPMB, CH2Cl) was successful in all cases with good to excellent yields (73–91%) and ee's (93–98%) (Scheme 43).
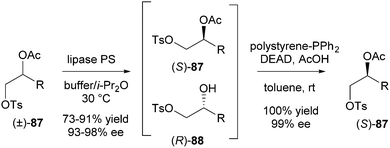 | ||
| Scheme 43 Two-step deracemisation using an enantioconvergent strategy. | ||
Sawamura et al. reported in 2010 a direct enantioconvergent transformation of racemic allylic ethers.40 These compounds have a ‘robust’ chiral center that cannot be efficiently racemised and therefore are not suitable substrates for DKR. The racemic substrate 89a was reacted with bis(pinacolato)diboron (94) in the presence of Cu(OtBu) and chiral phosphine ligand (R,R)-QuinoxP* (91, Scheme 44).
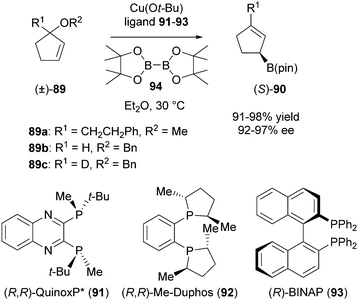 | ||
| Scheme 44 Deracemisation of allylic ethers via enantioconvergence. | ||
Complete conversion was reached within 24 hours to give the corresponding allylboronate (S)-90 in high yield (98%) and excellent ee (97%). Substrates 89b and c were also employed to investigate the details of this process. Both were successfully converted into the corresponding (S)-allylboronates with good yields (92 and 91%, respectively) and 92% ee. The enantioconvergence has been rationalised by showing that the achiral in situ formed Cu–boron nucleophile reacts with the allylic ether in an anti-type SN2′ substitution with one substrate enantiomer, while the chiral phosphine-coordinated Cu–boron nucleophile reacts in a syn-fashion with the other substrate enantiomer.
In 2011, the group of Faber reported a deracemisation of secondary alcohols employing a stereoselective alkylsulfatase which catalyses the hydrolysis of a sulfate ester with inversion of configuration at the stereocentre.41 The alkylsulfatase from Pseudomonas sp. DSM 6611, termed Pisa1, showed excellent stereoselectivity in the hydrolysis of linear, branched and cyclic sec-alkyl sulfates. To show the feasibility of an enantioconvergent deracemisation procedure employing this newly discovered enzyme, enzymatic hydrolysis was combined with chemical hydrolysis under acidic conditions (Scheme 45).
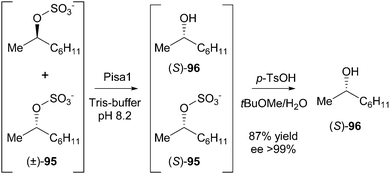 | ||
| Scheme 45 Enantioconvergent deracemisation of sec-alkyl sulfates. | ||
Racemic 2-octyl sulfate (95) was treated with Pisa1 to yield, via hydrolysis and concomitant inversion of the stereocentre, equimolar amounts of (S)-95 and (S)-96 in a kinetic resolution. The remaining alkyl sulfate could be chemically hydrolysed with retention of configuration, thus leading to a successful deracemisation affording enantiopure (S)-96 (>99% ee) in 87% isolated yield from the racemate.
6 Conclusions
Among the various techniques applied to obtain enantiopure products, deracemisation of racemates remains an utmost important topic in scientific and applied chemical research. While dynamic kinetic resolution is still the most predominant deracemisation method to produce single enantiomers, other elegant strategies such as cyclic deracemisation, stereoinversion and enantioconvergence have also been increasingly reported in recent years. The number of approaches on deracemisation strategies is still expanding, thereby making a diversity of chiral products accessible for a wide range of applications. Considering the fact that in the past few years entirely new approaches for deracemisation have been discovered, we feel that novel strategies in this exciting field will continue to emerge in the years to come.Notes and references
- K. Faber, Chem.–Eur. J., 2001, 7, 5004 CrossRef CAS.
- I. Hussain and J.-E. Bäckvall, Enzyme Catalysis in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 1777–1806 Search PubMed.
- J. H. Lee, K. Han, M.-J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999 CrossRef CAS.
- X. Yang, G. Lu and V. B. Birman, Org. Lett., 2010, 12, 892 CrossRef CAS PubMed.
- G. Lu and V. B. Birman, Org. Lett., 2011, 13, 356 CrossRef CAS PubMed.
- X. Yang and V. B. Birman, Angew. Chem., Int. Ed., 2011, 50, 5553 CrossRef CAS PubMed.
- J.-L. Cao and J. Qu, J. Org. Chem., 2010, 75, 3663 CrossRef CAS PubMed.
- D. T. Cohen, C. C. Eichman, E. M. Phillips, E. R. Zarefsky and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 7309 CrossRef CAS PubMed.
- For a review, see: T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393 CAS.
- S. Prévost, S. Gauthier, M. C. C. De Andrade, C. Mordant, A. R. Touati, P. Lesot, P. Savignac, T. Ayad, P. Phansavath, V. Ratovelomanana-Vidal and J.-P. Genêt, Tetrahedron: Asymmetry, 2010, 21, 1436 CrossRef.
- D. Cartigny, K. Puntener, T. Ayad, M. Scalone and V. Ratovelomanana-Vidal, Org. Lett., 2010, 12, 3788 CrossRef CAS PubMed.
- J. Limanto, S. W. Krska, B. T. Dorner, E. Vazquez, N. Yoshikawa and L. Tan, Org. Lett., 2010, 12, 512 CrossRef CAS PubMed.
- B. Seashore-Ludlow, P. Villo, C. Häcker and P. Somfai, Org. Lett., 2010, 12, 5274 CrossRef CAS PubMed.
- K. M. Steward, E. C. Gentry and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 7329 CrossRef CAS PubMed.
- S. Y. Lee, J. M. Murphy, A. Ukai and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 15149 CrossRef CAS PubMed.
- K. Yasukawa, R. Hasemi and Y. Asano, Adv. Synth. Catal., 2011, 353, 2328 CrossRef CAS.
- P. Vongvilai, M. Linder, M. Sakulsombat, M. Svedendahl Humble, P. Berglund, T. Brinck and O. Ramström, Angew. Chem., Int. Ed., 2011, 50, 6592 CrossRef CAS PubMed.
- V. Resch, W. M. F. Fabian and W. Kroutil, Adv. Synth. Catal., 2010, 352, 993 CrossRef CAS.
- D. Tessaro, L. Cerioli, S. Servi, F. Viani and P. D'Arrigo, Adv. Synth. Catal., 2011, 353, 2333 CrossRef CAS.
- A. Rioz-Martinez, G. de Gonzalo, D. E. Torres Pazmiño, M. W. Fraaije and V. Gotor, J. Org. Chem., 2010, 75, 2073 CrossRef CAS PubMed.
- C. Rodríguez, G. de Gonzalo, A. Rioz-Martinez, D. E. Torres Pazmiño, M. W. Fraaije and V. Gotor, Org. Biomol. Chem., 2010, 8, 1121 Search PubMed.
- G. Xu, Y. Chen. J. Wu, Y. Cheng and L. Yang, Tetrahedron: Asymmetry, 2011, 22, 1373 CrossRef CAS.
- F. J. Quijada, V. Gotor and F. Rebolledo, Org. Lett., 2010, 12, 3602 CrossRef CAS PubMed.
- T. R. Hoye, C. S. Jeffrey and D. P. Nelson, Org. Lett., 2010, 12, 52 CrossRef CAS PubMed.
- P. Galletti, E. Emer, G. Gucciardo, A. Quintavalla, M. Pori and D. Giacomini, Org. Biomol. Chem., 2010, 8, 4117 CAS.
- J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS PubMed.
- K. M. Wiggins and C. W. Bielawski, Angew. Chem., Int. Ed., 2012, 51, 1640 CrossRef CAS PubMed.
- B. Kaptein, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 7226 CrossRef CAS PubMed.
- W. L. Noorduin, B. Kaptein, H. Meekes, W. J. P. van Enckevort, R. M. Kellog and E. Vlieg, Angew. Chem., Int. Ed., 2009, 48, 4581 CrossRef CAS PubMed.
- W. Märkle and S. Lütz, Electrochim. Acta, 2008, 53, 3175 CrossRef.
- D. Koszelewski, D. Clay, D. Rozzel and W. Kroutil, Eur. J. Org. Chem., 2009, 2289 CrossRef CAS.
- Y. Chen, S. L. Goldberg, R. L. Hanson, W. L. Parker, I. Gill, T. P. Tully, M. A. Montana, A. Goswami and R. N. Patel, Org. Process Res. Dev., 2011, 2, 241 CrossRef.
- Y.-M. Seo, S. Mathew, H.-S. Bea, Y.-H. Khang, S.-H. Lee, B.-G. Kim and H. Yun, Org. Biomol. Chem., 2012, 10, 2482 CAS.
- R. Songür, B. Lurçi, E. Bayraktar, U. Mehmetoğlu and A. S. Demir, Artif. Cells, Artif. Cells, Blood Substitutes, Immobilization Biotechnol., 2011, 39, 162 CrossRef PubMed.
- M. C. Fragnelli, P. Hoyos, D. Romano, R. Gandolfi, A. R. Alcántara and F. Molinari, Tetrahedron, 2012, 68, 523 CrossRef CAS.
- R. Lievano, H. I. Pérez, N. Manjarrez, A. Solís and M. Solís-Oba, Molecules, 2012, 17, 3148 CrossRef CAS PubMed.
- C. Magallanes-Noguera, M. M. Ferrari, M. Kurina-Sanz and A. A. Orden, J. Biotechnol., 2012, 160, 189 CrossRef CAS PubMed.
- M. Tudorache, S. Nica, E. Bartha, I. Lupan and V. I. Parvulescu, Appl. Catal. A, 2012, 441–442, 42 CrossRef CAS.
- Y. Shimada, K. Usuda, H. Okabe, T. Suzuki and K. Matsumoto, Tetrahedron: Asymmetry, 2009, 20, 2802 CrossRef CAS.
- H. Ito, S. Kunii and M. Sawamura, Nat. Chem., 2010, 2, 972 CrossRef CAS PubMed.
- M. Schober, P. Gadler, T. Knaus, H. Kayer, R. Birner-Grunberger, G. Christian, P. Macheroux, U. Wagner and K. Faber, Org. Lett., 2011, 13, 4296 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2013 |