 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Perspective on the Martini model
Siewert J. Marrink*a and D. Peter Tielemanb
aGroningen Biomolecular Sciences and Biotechnology Institute and Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 7, 9747AG Groningen, The Netherlands. E-mail: s.j.marrink@rug.nl; Fax: +31-50-3634398; Tel: +31-50-3634457
bDepartment of Biological Sciences and Institute for Biocomplexity and Informatics, University of Calgary, 2500 University Dr. NW, Calgary, AB T3L 1L1, Canada. E-mail: tieleman@ucalgary.ca; Fax: +1-403-2899311; Tel: +1-403-2202966
First published on 24th May 2013
Abstract
The Martini model, a coarse-grained force field for biomolecular simulations, has found a broad range of applications since its release a decade ago. Based on a building block principle, the model combines speed and versatility while maintaining chemical specificity. Here we review the current state of the model. We describe recent highlights as well as shortcomings, and our ideas on the further development of the model.
Introduction
The use of coarse-grained (CG) models in a variety of simulation techniques has proven to be a powerful tool to probe the spatial and temporal evolution of systems on the microscale, beyond what is feasible with traditional all-atom (AA) models. A large diversity of coarse-graining approaches is available, ranging from qualitative, often solvent-free, models to models including chemical specificity. Models within the latter category are typically parameterized based on comparison to atomistic simulations, a so-called bottom-up or systematic multi-scale approach. They are designed to match a set of specific target distributions from the atomistic simulations, using iterative Boltzmann,1,2 force matching,3,4 minimization of relative entropy,5 or conditional reversible work6 approaches. For an extended overview of these methods, the reader is referred to recent reviews.7–11The Martini force field,12–14 developed by the groups of Marrink and Tieleman, has also been derived in close connection with atomistic models, especially for bonded interactions. However, the philosophy of our coarse-graining approach is different. Instead of focusing on an accurate reproduction of structural details at a particular state point for a specific system, we aim for a broader range of applications without the need to reparameterize the model each time. We follow a top-down approach by extensive calibration of the non-bonded interactions of the chemical building blocks against experimental data, in particular thermodynamic data such as oil/water partitioning coefficients. Processes such as lipid self-assembly, peptide–membrane binding, and protein–protein recognition depend critically on the degree to which the constituents partition between polar and non-polar environments.
The overall aim of our coarse-graining approach is to provide a simple model that is computationally fast and easy to use, yet flexible enough to be applicable to a large range of biomolecular systems. Example input files for many systems can be downloaded from http://cgmartini.nl. The first version of the CG force field, with parameters for lipids only, was published by the Marrink group in 2004. The name ‘Martini’ for the force field was coined in 2007 with the release of version 2.0 for lipids.13 The subsequent extension to peptides and proteins14 was released as version 2.1, with recent improvements to version 2.2, the current version.15
Although the Martini model was originally developed for use with the GROMACS software suite,16 the general form of the potential energy functions has allowed other groups to implement the Martini model into other major simulation packages such as NAMD,17 GROMOS18 and Desmond.19 Note that the groups of Schulten17 and Sansom20 have developed CG protein force fields compatible with the Martini lipid force field, but different from the Martini protein force field.
In this perspective we describe the current state of the Martini model. We highlight its applications, carefully consider its shortcomings, and sketch the road ahead. The rest of this perspective is organized as follows. Below we describe the design principles of the Martini model, and provide a comprehensive overview of the field of applications. Subsequently, the limitations of the model are described. An extensive outlook section ends this paper.
Design principles of the Martini model
Mapping
The Martini model is based on a four-to-one mapping, i.e., on average four heavy atoms plus associated hydrogens are represented by a single interaction centre. The four-to-one mapping was chosen as an optimum between computational efficiency on the one hand and chemical representability on the other hand. Mapping of water is consistent with this choice, as four real water molecules are mapped to a CG water bead. Ions are represented by a single CG bead, which represents both the ion and its first hydration shell. To represent the geometry of small ring-like fragments or molecules (e.g., benzene, cholesterol, and several of the amino acids), the general four-to-one mapping approach is too coarse. Ring-like molecules are therefore mapped with a higher resolution of up to two non-hydrogen atoms to one Martini particle. The Martini mapping of major classes of (bio)molecules is shown in Fig. 1.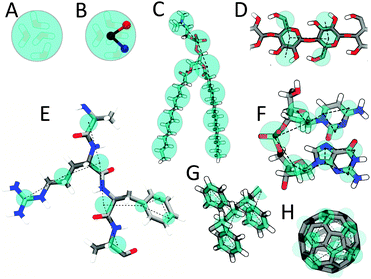 | ||
| Fig. 1 Martini mapping examples of selected molecules. (A) Standard water particle representing four water molecules. (B) Polarizable water molecule with embedded charges. (C) DMPC lipid. (D) Polysaccharide fragment. (E) Peptide. (F) DNA fragment. (G) Polystyrene fragment. (H) Fullerene molecule. In all cases Martini CG beads are shown as cyan transparent beads overlaying the atomistic structure. | ||
Based on the chemical nature of the underlying structure, the CG beads are assigned a specific particle type with more or less polar character. The Martini model has four main types of particle: polar (P), non-polar (N), apolar (C), and charged (Q). Within each type, subtypes are distinguished either by a letter denoting the hydrogen-bonding capabilities (d = donor, a = acceptor, da = both, 0 = none) or by a number indicating the degree of polarity (from 1 = low polarity to 5 = high polarity), giving a total of 18 particle types or ‘building blocks’.
Non-bonded interactions
Non-bonded interactions are described by a Lennard-Jones (LJ) 12-6 potential. The strength of the interaction, determined by the value of the LJ well-depth εij, depends on the interacting particle types i,j. The value of ε ranges from εij = 5.6 kJ mol−1 for interactions between strongly polar groups to εij = 2.0 kJ mol−1 for interactions between polar and apolar groups, mimicking the hydrophobic effect. The effective size of the particles is governed by the LJ parameter σ = 0.47 nm for all normal particle types. For the special class of particles in ring-like molecules, slightly reduced parameters are defined to model ring–ring interactions: σ = 0.43 nm, and εij is scaled to 75% of the standard value. The full interaction matrix can be found in the original publication.13 In addition to the LJ interaction, charged groups (type Q) bear a charge ±e and interact via a Coulombic energy function. Coulombic interactions are screened implicitly with a relative dielectric constant εrel = 15 to account for the reduced set of partial charges and resulting dipoles that occur in an atomistic force field. Note that the non-bonded potential energy functions are used in a shifted form. The non-bonded interactions are cut off at a distance rcut = 1.2 nm. The LJ potential is shifted from rshift = 0.9 nm to rcut. The electrostatic potential is shifted from rshift = 0.0 nm to rcut. Shifting of the electrostatic potential in this manner mimics the effect of a distance-dependent screening. To alleviate some of the limitations of the implicit screening of electrostatic interactions caused primarily by a water model without charges or dipole, polarizable water models were also recently introduced for use with Martini.21,22 With these models a much lower dielectric constant is used because of the increased explicit screening (εrel = 2.5 for the Martini polarizable water model,21εrel = 1.3 for the Big Multipole Water (BMW) model22).The non-bonded interactions of the Martini model have been parameterized based on a systematic comparison to experimental thermodynamic data. Specifically, the free energy of hydration, the free energy of vaporization, and the partitioning free energies between water and a number of organic phases were calculated for each of the 18 different CG particle types. Martini reproduces the correct trend in free energies of hydration and vaporization. However, the actual values are systematically too high, implying that the CG condensed phase is not as stable with respect to the vapour phase as it should be. The same is true with respect to the solid phase. This is a known consequence of using a LJ 12-6 interaction potential, which has a limited fluid range (see ‘Limitation’ section below). As long as its applications are aimed at studying the condensed phase and not at reproducing gas/fluid or solid/fluid coexistence regions, the most important thermodynamic property is the partitioning free energy. Importantly, the water/oil partitioning behaviour of a wide variety of compounds can be accurately reproduced with the current version of Martini.
Bonded interactions
Bonded interactions are described by a standard set of potential energy functions common in classical force fields, including harmonic bond and angle potentials, and multimodal dihedral potentials. Proper dihedrals are primarily used to impose secondary structure on the peptide backbone. Improper dihedrals are mainly used to prevent out-of-plane distortions of planar groups. LJ interactions between nearest neighbours are excluded.To parameterize the bonded interactions, we use structural data either directly derived from the underlying atomistic geometry (such as bond lengths of rigid structures) or obtained from comparison to atomistic simulations. In the latter procedure, the higher resolution simulations are first converted into a “mapped” CG (MCG) simulation by identifying the centre of mass of the corresponding atoms as the MCG bead. Second, the distribution functions are calculated for the mapped simulation and compared to those obtained from a true CG simulation. Subsequently the CG parameters are systematically changed in an iterative way until satisfactory overlap of the distribution functions is obtained.
For proteins, the PDB databank has been used as an additional source of atomistic reference geometries. Based on MCG distributions from the PDB, bonds, angles, and dihedral interactions were optimized. Importantly, the bonded parameters depend on the sequence, and are used to stabilize the secondary structure elements of the protein; the lack of directional hydrogen bonds prevents realistic folding at the Martini level of coarse-graining. On top of this, an elastic network can be used to further constrain the protein close to a particular, e.g. native, state. The elastic network approach, named ElNeDyn,23 has been optimized with respect to atomistic reference simulations.
Simulation parameters
The force field terms are coupled to the simulation algorithms Martini was parameterized for, although there is some flexibility in this regard. Key parameters include the timestep, neighbour list update frequency, cut-off radii for the nonbonded potentials, and the exact form of the switch function. Martini simulations generally are stable with timesteps of up to 40 fs or 20 fs in the presence of rings such as in cholesterol or proteins. Lower values than 20 fs we consider an inefficient use of computer time as errors in the model will strongly dominate errors due to numerical integration time steps.24 Energy conservation also depends strongly on the use of neighbour lists, but the implementation of these lists differs in different software and for GROMACS in different versions. More frequent updates or the use of large buffer zones removes errors associated with neighbour lists but come at a computational cost as well. Martini is parameterized with a switch function that reduces both LJ and Coulomb interactions to zero at 1.2 nm. The form of this function together with the accurate calculation of temperature turned out to be important in implementing Martini in other software as errors may lead to more serious temperature artefacts.25Validation
Currently, the Martini force field provides parameters for a variety of biomolecules, including many different lipids,12,26–28 sterols,13,29 peptides and proteins,14,20,30 sugars,31,32 polymers,33–36 nanoparticles,37–39 dendrimers,40,41 and more. This family of Martini molecules is constructed using the building block principle, i.e. mapping the underlying chemical structure to the corresponding flavor of CG particle types, and stringing them together to reproduce the overall topology of the target molecule. The basic assumption underlying Martini is that the carefully parameterized properties of the individual beads are transferable to the molecule as a whole. This basic assumption requires validation, which may come either from comparing to more detailed atomistic simulations or to experimental data. Recent validation examples of the first category are: partitioning of amino acid side chain analogues (SCAs) across the membrane, which shows profiles matched to within 1–2 kT for most SCAs;42 dimerization of SCAs in solvents of different polarities, reproducing most atomistic dimerization free energies to within 1 kT;43 and the potential of mean force (PMF) between a pair of fullerenes in different solvents, reproducing the atomistic profiles.39 Examples of validation with respect to experimental data are: the area per lipid, typically reproduced to within 0.1–0.2 nm2 or experimental accuracy for many types of lipid membranes;12,13 the ternary phase behaviour of lipid mixtures, showing phase diagrams in semi-quantitative agreement;44 the relative binding free energy of a systematic series of pentapeptides to the water/lipid interface,45 consistent with the experimental hydrophobicity scale derived from these peptides; dimerization free energies of transmembrane (TM) helices within the range of experimental values from FRET data;462H-NMR quadrupolar splittings of WALP peptides,47 reproduced to a better extent than atomistic simulations; and the structure of the glycophorin A dimer,48 compared to NMR data.Many more critical tests have been performed in several of the papers listed below in the Application section. Sometimes it turns out that the set of standard Martini beads is not sufficient to reproduce the desired accuracy, and further optimizations are made. One of the advantages of Martini is its limited set of parameters, which makes it relatively easy to adjust or optimize the interactions. We quote the original characterization of the Martini model:13 “how a few simple ingredients (read: chemical building blocks) can be endlessly varied to create a complex palette of taste”. Cheers.
Martini based applications
The first applications of the Martini model, dating back 10 years, concerned the self-assembly49 and fusogenicity50 of small lipid vesicles. Since these first studies, the list of applications has grown dramatically, reflecting the flexibility and transferability underlying our coarse-graining protocol. Currently, applications can be grouped as: characterization of lipid membrane properties; lipid polymorphism; protein–lipid interplay; membrane protein oligomerization; self-assembly of soluble peptides and proteins; protein conformational changes; binding and pore-formation in membranes by membrane active peptides; design of drug and gene delivery systems; structure and dynamics of lipoprotein particles; membrane fusion; compression and expansion of monolayers; self-assembly of surfactants; characterization of carbohydrate based systems; structure and dynamics of polymers; and interaction of nanoparticles with membranes. From this overview it is apparent that lipid membranes are central in many applications. Given the origin of the Martini force field for simulations of lipid systems this is not surprising. Below we discuss the main application areas to date.Lipid membrane characterization
Although simple lipid membranes can be modelled at the fully atomistic level, Martini offers three significant advantages given the current state of atomistic simulations: (i) the use of a CG model allows many independent simulations in which state conditions are systematically varied, e.g. the in silico design of robust membranes,51 tethered or supported membranes,52–54 or correlating lipid type and membrane properties;55,56 (ii) direct, unbiased, sampling of rare processes becomes possible, such as lipid flip–flop and lipid desorption,57,58 deformation of asymmetric membranes,59–62 or the membrane binding and permeation of drugs and amphiphiles;63–67 and (iii) the Martini model allows simulations requiring large system sizes, e.g. modelling of lipid vesicles49,68–73 and calculation of membrane bending moduli74–77 and viscosity.78 As an example of the latter category, Baoukina et al.79 simulated the formation of membrane tethers (Fig. 2A), mimicking micropipette aspiration experiments. The largest systems studied contained close to 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 CG particles, making it one of the biggest Martini-based simulations so far.
000000 CG particles, making it one of the biggest Martini-based simulations so far. | ||
| Fig. 2 Large scale membrane remodelling. (A) Pulling of a membrane tether from a planar lipid bilayer.79 Lipid head groups are coloured grey, lipid tails green. The tether is partly cut to view the inside. (B) Stabilization of an inverted cubic phase by fusion peptides.91 Fusion peptides are depicted as red cylinders, the water/lipid interface is rendered as a green surface. The space occupied by the lipids is coloured black. | ||
Lipid polymorphism
Another important area of applications of the Martini model is lipid phase behaviour, requiring both large systems due to the collective nature of phase transitions and long simulation times to observe the critical nucleation events. Examples include the formation of gel phases80–86 and inverted hexagonal26,87–89 and cubic phases,90,91 or transitions between micelles, bicelles, and vesicles.28 A snapshot of an inverted cubic phase stabilized by fusion peptides, obtained by Fuhrmans and Marrink91 using a self-assembly approach, is shown in Fig. 2B. An important breakthrough in our ability to model realistic, heterogeneous, membranes was reported by Risselada and Marrink,44 who simulated the spontaneous formation of liquid-ordered (Lo) and liquid-disordered (Ld) domains in ternary mixtures of saturated and unsaturated lipids together with cholesterol. Probing the structural and dynamical properties of these fluid domains has received a lot of attention, as it is presumably linked to the formation of lipid nano-domains (“rafts”) in real cells. Follow up studies have further explored the properties of these domains in a number of ways, including: systematic evaluation of composition,29,92–96 studying partitioning of lipids and amphiphiles at Ld/Lo domain boundaries,97,98 probing conditions for domain registration,99 observing sorting anomalies of domains under stress,100 looking into the effect of immobilization on domain formation,101 and analysis of raft dynamics.102–105 A recent review by Bennett and Tieleman covers in more detail simulations of membrane domains and membrane asymmetry, including many Martini simulations.106Membrane protein–lipid interplay
Martini has proven to be very useful to probe protein–lipid interactions of membrane embedded proteins. Recent applications can be broadly classified into six categories: (i) predicting binding modes of proteins to membranes as well as membrane adaptation around proteins, as exemplified by many simulations of a variety of peptides46,107–112 and proteins.113–118 These kinds of simulations can be efficiently done in high-throughput mode using a self-assembly approach pioneered by Sansom and coworkers.119,120 3D stress profiles around membrane proteins121–123 have been resolved using Martini as well; (ii) simulating protein sorting, notably the propensity of TM peptides and membrane proteins to partition in either Ld or Lo domains.124–129 Domanski et al.,129 for instance, modelled the sorting of TM model peptides (the synthetic WALP peptides) under crowding conditions. They found that, at lipid/protein ratios characteristic of real membranes, the peptides induced Ld/Lo domain segregation (Fig. 3C). Unravelling the complicated dynamics of membrane proteins in homogeneous membranes is a related area of interest;129–134 (iii) identifying specific lipid binding sites on membrane proteins is a rapidly growing area. Current examples include the enrichment of short-tail lipids around OmpA,135 phosphatidylinositolphosphate (PIP) binding to the pleckstrin homology domain136 and to inwardly rectifying potassium (Kir) channels,137,138 cardiolipin binding to mitochondrial creatine kinase (MtCK),139 cytochrome c oxidase,140 and cytochrome bc1,141 cholesterol binding to the serotonin (1A) receptor,142 enrichment of anionic lipids at the gap junction hemichannel connexion-26,143 at potassium channels KcsA and chimeric KcsA-Kv1.3,144 and at TM and juxtamembrane domains of cytokines,145 specific interactions of phosphatidic acid (PA) and PIP2 with the actin capping protein (CP),146 and binding of cholesteryl esters to cholesteryl ester transfer protein;147 (iv) simulating protein mediated membrane remodelling, i.e. the deformation of membranes under the action of proteins, is also suitable for ‘Martinization’ as proven by an increasing number of applications. A fine example is the large-scale simulation of Davies et al.,148 probing the role of F1F0-ATP synthase dimers in shaping the mitochondrial cristae. Based on these simulations, the authors propose that the assembly of ATP synthase dimer rows is driven by the reduction in the membrane elastic energy, rather than by direct protein contacts, and that the dimer rows enable the formation of highly curved ridges in mitochondrial cristae (Fig. 3A). Other examples in the category of membrane remodelling are simulations of the curvature field induced by α-synuclein,149 and the shaping of membranes by BAR domains.150 Studies on (v) membrane-mediated protein clustering and (vi) gating are described in sections below. | ||
| Fig. 3 Membrane protein sorting and clustering. (A) Formation of membrane folds (cristae) by ATP synthase dimers148 (courtesy of W. Kühlbrandt). Membrane surface rendered blue, proteins in yellow. Adapted with permission from ref. 148. Copyright (2012) National Academy of Sciences. (B) Aggregation of membrane proteins due to hydrophobic mismatch152 (courtesy M. S. P. Sansom). Membrane viewed from top in blue, with barrel proteins depicted yellow. Adapted with permission from ref. 152. Copyright (2011) Elsevier. (C) Sorting of transmembrane helices into liquid-disordered domains.129 TM helices in yellow, unsaturated lipids red, saturated lipids blue, cholesterol grey with the white hydroxyl group. | ||
Membrane protein oligomerization
Many integral membrane proteins assemble in oligomeric structures in biological membranes. The Martini force field makes it possible to study these self-assembly processes at near-atomic detail over time scales of micro- to milliseconds. The first study on large scale protein assembly, by Periole et al.,151 considered rhodopsin, a prototypical G-protein coupled receptor (GPCR). Starting from random initial positions, the proteins formed linear aggregates due to competition of non-specific lipid mediated forces and specific sidechain–sidechain interactions at the protein surface. This explained in detail previous experimentally observed linear aggregates. An extensive systematic study on aggregation of model membrane proteins, modulated by hydrophobic mismatch, membrane curvature, and protein class, was performed by Parton et al.152 An example from this study is presented in Fig. 3B, showing clustering of generalized β-barrel proteins under mild mismatch conditions. Other examples of protein–protein self-assembly simulated with Martini include the dimerization studies of glycophorin A,48,153 the repressor of primer protein,23 the c0-subunit of the ATP synthase complex,154 TM WALP peptides,46,47,124,129 gramicidin channels,155 the delta opioid receptor,156 TM domains of ErbB2 receptors157 and Fukutin-I,158 integrin TM helix heterodimers,159,160 the integrin–talin complex,161,162 the cohesion-dockerin system,163 the TM domain of T cell receptor complex,164 the bacterial stress-response peptide TisB165 and the serine receptor Tsr,166 as well as tetramerization of the TM domain of the M2 channel167 and modelling of caseinolytic peptidase B (ClpB) hexameric ring structures.168 The fast sampling speed of Martini also allows computation of the dimerization free energy of membrane proteins; recent studies of GPCRs169,170 and OmpF171 reveal specific, favorable, association interfaces.Self-assembly of soluble peptides and proteins
In addition to membrane proteins, aggregation of soluble proteins is a potentially large field of Martini applications. So far, however, self-assembly simulations have been restricted mainly to small peptides. A number of these studies deal with dipeptides. In a very nice high-throughput study by Frederix et al.,172 the Martini force field was used to screen all 400 dipeptide combinations and predict their ability to aggregate as a potential precursor to their self-assembly. Systems that showed strong aggregation tendencies were selected for longer simulations in which supramolecular structures were formed consistent with known aggregation states of dipeptides reported in the literature (Fig. 4A). Related studies show self-assembly of diphenylalanine peptides into a range of morphologies,173 and of N-(fluorenyl-9-methoxycarbonyl)-dialanine peptides into hydrogels.174 Aggregation of larger peptides has also been the subject of a number of Martini studies. Lee et al.175 addressed the self-assembly of peptide amphiphiles, composed of a 13-residue hydrophilic peptide connected to a hydrophobic tail. In line with experimental data, they observed spontaneous formation of fibres (Fig. 4B). Likewise, self-assembly of amphiphilic peptides into peptide-vesicles (“peptosomes”)176 or micelles177 has been simulated. A particular challenging case for CG models such as Martini, which lack directional hydrogen bonds, is the formation of amyloid fibrils. For a realistic simulation of amyloid formation, a more detailed description of the peptide backbone is probably required;178 more qualitative studies, however, can be performed. For instance, Sorensen et al.179 modelled the assembly of amylin (20–29) peptides, pre-assembled into protofibril fragments. The protofibril fragments were kept together with an elastic network using the ElNeDyn23 approach. The simulations pointed to an elongation growth mechanism of the protofibrils into fibres (Fig. 4C). Another example is the study on the effect of lipid concentration on the aggregation propensity of the amyloidogenic peptide apolipoprotein C-II (60–70).180 Martini studies of interactions between soluble proteins are still limited, but do exist, for example on the stability of the spider silk's N-terminal protein domain181 and of keratin filaments,182 the stability of engineered nano-fibres,183 the mechanical properties of protein filaments,184,185 and dissociation of gas phase protein complexes.186,187 A related application of the Martini model is protein adsorption on solid supports188,189 or at fluid interfaces.190 Martini could also be used to screen for protein–ligand interactions in principle. Thus far, we are aware of one example, in which peptide binding to the OppA transporter was studied.191 | ||
| Fig. 4 Self-assembled peptide aggregates. (A) Nanotube formed by diphenylalanine peptides172 (courtesy of P. W. J. M. Frederix). Peptide backbone in red, side chains grey, and water inside the tube blue. (B) Fibre formed by a peptide amphiphile175 (courtesy of G. C. Schatz). IKVAV epitope, blue; rest of PA, grey. (C) Fibre composed of amylin (20–29) peptide fragments.179 Phenylalanine side chains, purple; other side chains, yellow; backbone, cyan. | ||
Conformational changes in proteins
Although secondary structure changes of proteins cannot be modelled in Martini yet, changes in tertiary structure are unrestricted and in principle realistic within the general approximations underlying our CG model. A fine example is the gating process of the mechano-sensitive channel of large conductance (MscL). When the membrane is put under tension, MscL channels undergo significant conformational changes in accordance with an iris-like expansion mechanism and reach a conducting state on a microsecond timescale.192,193 The gating of an MscL channel embedded in a liposome has been simulated in a large-scale study by Louhivuori et al.194 After putting the liposome under stress by adding excess water, mimicking hypoosmotic shock conditions, the MscL channel opened to release excess solvent (Fig. 5A). The energetics of the gating process have been analysed in more detail elsewhere.195 Full closure of the channel has also been achieved in simulations of the reversible gating of MscL by Ingolfsson et al.196 (Fig. 5B). Structural transitions of other channel proteins that have been simulated with Martini include voltage-gated potassium channels,197 the SecY channel,198 and Ca-ATPase.199 Furthermore, acyl carrier protein (ACP) substrate-shuttling within the fatty acid synthase reaction chamber has been modelled,200 providing valuable insight into the role of linkers and crowding in the shuttling process. The dynamics of membrane-bound and soluble cytochrome P450 has also been studied, revealing correlations between opening and closing of different tunnels from the enzyme's buried active site.201 Another study addresses collective motions of RNA polymerases that might contribute significantly to the conformational transition between the open and closed states.202 A final example is a Martini study on the dynamics of TM helices of bacterial chemoreceptors, supporting a piston model of signalling.203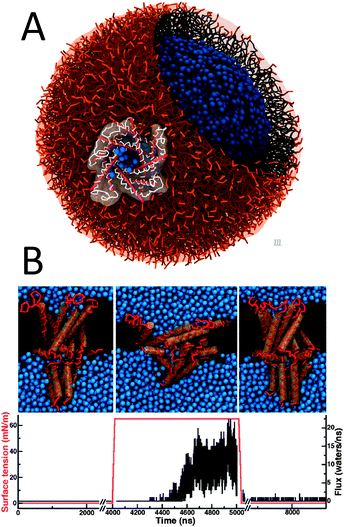 | ||
| Fig. 5 Proteins in action. (A) Gating of a mechanosensitive protein channel, MscL, in a liposome under hypoosmotic shock.194 (B) Reversible gating of MscL in a planar membrane196 (courtesy H. Ingolfsson). Upper panel shows snapshots of the closed starting structure, the open structure under tension, and the closed structure after tension has been released. Lower graphs shows the applied surface tension (red) and water flux (black) as a function of time. | ||
Peptide induced membrane permeabilization
Antimicrobial peptides (AMPs) are still in the spotlight as potential new antibiotics essential to overcome bacterial resistance to conventional antibiotics, while similar peptides are studied as anti-cancer and anti-fungal agents as well as vehicles for drug delivery. At a certain threshold concentration, AMPs permeabilize the membrane, either by forming a discrete pore or by disrupting the bilayer structure. Using the Martini model it is in principle possible to simulate AMP induced membrane poration. A first example was shown as validation example of the protein force field.14 Magainin peptides stabilized a toroidal pore with a structure similar to those seen with atomistic models. In Martini simulations, spontaneous pore formation typically has been observed only under rather favourable conditions, such as in very thin bilayers,204 at very high peptide concentrations,205,206 or with highly asymmetric bilayers.207 Other attempts to simulate AMP-induced membrane poration revealed potential problems with Martini, such as the formation of dehydrated pores208,209 or membrane buckling,210,211 see the section on ‘Limitations’ below. Stabilization of pores by human islet amyloid polypeptides has also been investigated,212 as well as pores stabilized by fusion peptides.90,91,213 Likewise, Martini allows for systematic calculations of the binding and translocation free energy of AMPs214,215 and other membrane active peptides such as caveolin,216 but also in these cases the shortcoming of Martini in modelling lipid-lined membrane pores has to be kept in mind. The ability of AMPs in a surface bound state to cluster charged lipids does not suffer from this problem, and is therefore expected to be a real phenomenon well suited for Martini.217,218Drug and gene delivery systems
Membranes pose a barrier for typical drug molecules and gene fragments and hinder their use in practice. Therefore, design of delivery vehicles that help these molecules to cross the cell membrane is an active area of research in which Martini has found a growing number of applications. A series of simulation studies of polyamidoamine (PAMAM) dendrimers show these nanoparticles are able to porate membranes.40,41 PAMAMs can be functionalized to optimize drug loading.219 Other polymer-based delivery systems that have been investigated using the Martini model are poly(gamma-glutamyl-glutamate) paclitaxel (PGG-PTX), a series of semiflexible polymer–drug constructs varying in PTX loading fraction220–222 and ABA type triblock copolymers.223 The design of self-assembling nanofibres composed of amphiphilic peptide derivatives for delivery of therapeutic peptides has also been addressed.224 Other examples of Martini-based studies of drug-delivery systems include simulations of encapsulation of propofol in quaternary ammonium palmitoyl glycol chitosan micelles,225 investigation of the permeability of lysolipid-incorporated liposomes for enhanced permeation of anticancer drugs,226 and fusion of perfluorocarbon-based nanoemulsions to target liposomes.227 Considering gene delivery, the structure of lipoplexes (DNA–lipid complexes) has been modelled.228 A recent large scale Martini application by Leung et al.229 involves modelling of lipid nanoparticles containing ionizable cationic lipids, enabling therapeutic applications of siRNA (Fig. 6A).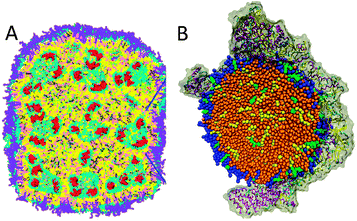 | ||
| Fig. 6 Molecular view on nanoparticles. (A) Lipid nanoparticle used for gene delivery,229 composed of ionisable lipids (yellow), cholesterol (pink), phospholipid (grey; lipid polar moiety in cyan), PEG–lipid (violet) and 12 bp DNA (red). (B) Low density lipoprotein particle234 (courtesy I. Vattulainen), consisting of a droplet of lipids (blue), cholesterol (green), triglycerides (yellow), and cholesterylester (brown), bound to lipoprotein ApoB-100. Adapted with permission from ref. 229. Copyright (2012–2013) American Chemical Society. | ||
Lipoprotein particles
Low-density and high-density lipoproteins (LDL/HDL) transport cholesterol in the bloodstream and play an important role in the development of atherosclerosis. They consist of a mixture of components including phospholipids, triglycerides, cholesterol, and associated proteins. The Martini model has proven useful to study the structure and dynamics of these particles, starting with the self-assembly of model HDL by Shih et al.230 using the Martini variant developed by the Schulten group. Subsequently, the structure and dynamics of HDL231,232 with realistic size and lipid composition have been modelled with standard Martini. The interfacial tension and surface pressure of various lipid droplets have also been investigated.233 A model for LDL was also developed, by Murtola et al.,234 and consists of an ApoB-100 protein wrapped around a 20 nm thick lipid droplet (Fig. 6B). The simulations reveal a complex distribution of the lipid constituents in the particle, and point to biologically relevant contacts between the surrounding protein and some of the core molecules. Another type of lipoprotein particle is the complex formed by Saposin A, a protein involved in lipid transfer. Simulation studies show that the protein can solubilize various phospholipids into small discs.235 Martini based simulations have been helpful to reveal the molecular packing of artificial lipoprotein particles, so-called nanodiscs, which are increasingly used by experimentalists to characterize membrane proteins. Examples are nanodiscs stabilized by amphipathic polymers (amphipols)236 or engineered proteins.237Membrane fusion
Membrane fusion was one of the very first applications of the Martini model50 and is still a hot topic where simulation studies can resolve details that are not easily probed experimentally. Possible fusion pathways between lamellar membranes and between vesicles are now quite well established,50,87,238,239 and current efforts are directed to calculate the energetics of the various intermediates240–244 and to investigate the role of fusion promoting molecules such as PEG.245 An important new direction is peptide and protein mediated fusion. Baoukina and Tieleman246 simulated the fusion of small unilamellar vesicles mediated by lung surfactant protein B (SP-B). They found SP-B monomers capable of triggering fusion events, consistent with the experimentally observed fusogenic effect of SP-B, by anchoring two vesicles and facilitating the formation of a lipid bridge between the proximal leaflets. Another recent example is the Martini simulation of neuronal SNARE-mediated membrane fusion by Risselada et al.247 (Fig. 7A). The simulations reveal that SNARE complexes mediate membrane fusion in a cooperative and synchronized way, requiring at least one single SNARE complex consistent with single-molecule fluorescence studies. After fusion, the zipping of the SNAREs extends into the membrane region, in agreement with the recently resolved X-ray structure of the fully assembled state. Force transduction of the SNARE protein synaptobrevin has also been looked into with Martini.248 Furthermore, wild-type hemagglutinin (HA) influenza fusion peptides were shown to stabilize a highly fusogenic pre-fusion structure, i.e. a peptide bundle formed by four or more TM fusion peptides.213 Studies on mutant peptides provided a rationale for experimentally observed incomplete fusion pathways. Related studies also point to the ability of HA fusion peptides to stabilize fusion intermediates.90,91 For a recent review of simulations on fusion, including many Martini-based works, see Markvoort and Marrink.249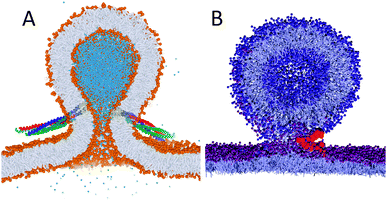 | ||
| Fig. 7 Examples of protein mediated fusion. (A) Fusion of vesicles induced by SNARE proteins247 (courtesy H. J. Risselada). Lipids are coloured with orange heads and grey tails, interior water in blue, and the SNARE complexes with yellow TM helices and red/green/blue soluble parts. (B) Fusion of vesicles and monolayers mediated by lung surfactant protein SP-B258 (courtesy S. Baoukina). Lipids are shown in grey with blue (vesicle) or purple (monolayer) heads, protein backbone in red. | ||
Lipid monolayers
At polar–apolar interfaces, lipids form monolayers that reduce the surface tension. Besides being of fundamental interest for surface science, lipid monolayer collapse is crucial for maintaining low surface tension at the gas-exchange interface in the lungs during breathing. The Martini force field can be used to study the behaviour of lipid monolayers in detail, both for pure lipid systems and in combination with peptides and proteins. Although quantitative reproduction of the pressure–area isotherms at low surface pressure remains challenging,250 the model is well suited to study properties of monolayers in the liquid-expanded and liquid-condensed phases,250,251 and to study for instance the collapse of lipid monolayers at high surface pressure.252 Mixed monolayers have also been studied, e.g. to model the tear fluid lipid layer,253–255 or to study phase transitions256 and phase coexistence,103,104 and the effect of the presence of lung surfactant proteins.257 In a large scale study Baoukina and Tieleman simulated fusion of lipid reservoirs to monolayers mediated by lung surfactant protein SP-B.258 They found SP-B induced establishment of a lipid-lined connection similar to the hemifusion stalk (Fig. 7B) allowing lipid flow in a surface-tension-dependent manner. These findings are in line with existing hypotheses on SP-B activity in lung surfactant and explain its molecular mechanism.Surfactant self-assembly
The Martini force field is ideally suited to study formation of micelles, as it enables simulations that are long enough to equilibrate many surfactant solutions. However, the ability to obtain an equilibrated micellar size distribution depends on the type of surfactant; exchange of surfactants between micelles, and fusion and fission of small micelles are only observed for surfactants with a relatively high CMC. For instance, in the first study on surfactants using the Martini model, DPC (dodecyl-phosphatidylcholine) could be observed to self-assemble into micelles, but convergence of the size distribution was very slow and not achieved on the sub-microsecond time scale.12 Recent Martini simulation studies, involving acyl-trimethylammonium chloride,259,260 dodecyltrimethylammonium bromide (DTAB),73 cetyltrimethylammonium bromide (CTAB),261 sodium dodecyl sulphate (SDS),262 dihexanoyl-PC (DHPC),263 and pentaethylene glycol monododecyl ether (C12E5),264 however, achieve longer (multi-microsecond) simulation times and approach equilibrium distributions. Sampling of interesting phenomena, such as the sphere-to-rod transition,259,261,264 becomes possible. In a study comparing a number of surfactants,265 the Martini model reproduced experimental CMCs and aggregation numbers for non-ionic surfactants reasonably well. The temperature dependence of these properties, however, is not correct. Reproducing the correct temperature dependence is a known challenge for CG models in general (see ‘Limitation’ section below). Martini simulations of micelles can also be used to provide starting structures for fine-grained models, as shown in a recent study on lyso-phospholipids,266 and to provide details on the absorption and desorption process of non-ionic surfactants.267 Bicelles, composed of double tail and single or short tail lipids, have also been the topic of a number of studies.28,268,269Carbohydrates
Carbohydrates (saccharides) constitute a fundamental class of biomolecules and are present in a variety of emerging classes of biomimetic materials. The large size of most polysaccharides warrants the use of a coarse-grained model, yet the complexity of carbohydrate physico-chemical properties makes this a very challenging undertaking. Recently, common mono- and disaccharides have been parameterized for Martini,31 providing a basis for further carbohydrate modelling. Simulations of glucose and trehalose interacting with a DPPC membrane show suppression of gel phase formation,31 in agreement with the cryoprotective properties of these sugars. Based on the Martini carbohydrate model, oligosaccharides such as amylose and curdlan,31 and cellulose32,270 have also been parameterized. Martini parameters for the important class of glyco-lipids were developed as well,27 including biologically relevant lipids for the thylakoid membrane, signalling lipids such as PI and its phosphorylated analogues PIPn, as well as gangliosides. A pioneering study on multi-component membranes containing the ganglioside monosialotetrahexosylganglioside (GM1) shows formation of GM1 enriched nano-domains that act as protein shuttles between membrane regions with different degrees of order.127Polymers
Coarse-graining has been of fundamental importance in the field of polymer modelling. In order to study the large size of polymer chains and the associated slow relaxation processes, a reduction of the degrees of freedom has proven absolutely necessary. Although the Martini model was designed primarily with biomolecular applications in mind, there is no reason why the same philosophy could not be extended to soft matter in general. Indeed, there is a growing number of basic polymer systems for which Martini parameters have been derived, currently including polyethyleneglycol (PEG),33,271 polystyrene,34 triblock copolymers polyethyleneoxide–polypropyleneoxide–polyethyleneoxide (PEO–PPO–PEO),223,272 polyurea,273 Nafion ionomers,274 polyester coatings composed of two dicarboxylic acids and a diol, esterified to neopentyl glycol monomers and crosslinked by hexa(methoxymethyl)melamine,275 polymer nanofibres composed of nylon-6 (polycaprolactam),276 PAMAM dendrimers40,41,219 and PEG-conjugated PAMAM dendrimers,277 and amphipols.236 Two recent examples are illustrative of the type of applications for which the Martini model can be used. Simulations of nanocoating of a polymer matrix composed of polymethylmethacrylate (PMMA), by Uttarwar et al.,278 show the effect of nanoparticle (NP) concentration, size, and polydispersity on the behaviour of the polymer matrix. For instance, the presence of NPs leads to elongation of the polymer chains as the radius of gyration is increased (Fig. 8A). Simulations of grafted polystyrene brushes in good solvent (benzene), by Rossi et al.,279 explore the structural and dynamical configuration of the brushes as a function of their grafting density in a high grafting density regime (0.1–0.3 chains nm−2) (Fig. 8B). The presence of metastable collapsed states, with free chain ends trapped close to the substrate, is revealed at all grafting densities. These collapsed states are shown to be long-lived, surviving over a time scale of several microseconds.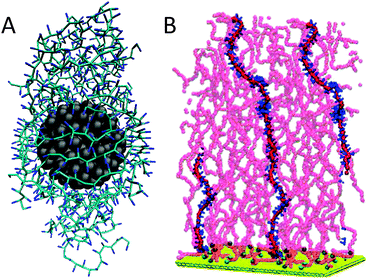 | ||
| Fig. 8 Polymeric systems. (A) Nanoparticle coated with PMMA resin278 (courtesy Y. Huang). Nanoparticle is shown in grey, the polymer in blue. (B) Grafted polystyrene chains in good solvent279 (courtesy G. Rossi). Support is shown in yellow, polymer backbone in red with one polymer highlighted with blue phenyl rings. Adapted with permission from ref. 278 and 279. Copyright (2012–2013) American Chemical Society. | ||
Nanoparticles
The two-dimensional carbon sheet graphene and its three dimensional cousins fullerene and carbon nanotubes (CNTs) enjoy a huge scientific interest due to their interesting mechanical and electrical properties, with many realised and potential applications. The interaction of these nanoparticles with biological material is of specific importance with respect to their toxicity. To address these concerns, a number of groups have started developing Martini models for fullerene,37,39,280 graphene,281,282 and CNTs,38,283–287 and explore how they interact with lipids and surfactants. For instance, in a breakthrough study by Wong-Ekkabut et al.,37 the lipid membrane was shown to be a very good solvent for fullerenes (Fig. 9A). Clustering in the aqueous phase, the fullerene aggregates spontaneously enter the membrane and disperse on a microsecond time scale. Likewise, as demonstrated in a pioneering study by Titov et al.,281 graphene nanoflakes dissolve into a lipid membrane and adopt a flat orientation in between the membrane leaflets (Fig. 9B). The affinity of graphene-based particles for lipids is also elegantly shown in a study by Wallace and Sansom38 on CNTs. Dispersing lipids or surfactants randomly in the aqueous solution, self-assembly of a CNT–lipid complex is observed (Fig. 9C). In addition, the interaction of other nanoparticles such as gold clusters,288–292 nanocrystals,293 and coated nanoparticles294,295 with lipid membranes has been studied using Martini. As an example, in the work of Lin et al.,288 the interaction of functionalized gold particles with DPPC/DPPG membranes was systematically investigated, showing penetration, disruption, pore formation or wrapping (Fig. 9D) depending on the surface charge of both the nanoparticle and the membrane. In a related study,296 the aggregation of monolayer-protected gold nanoparticles in various solvents was investigated. We expect many more of such studies in the near future, for instance along the lines of a very recent study of the effect of PEG functionalized phospholipids on nanotube bundling297 and on morphological characterization of polymer-functionalized gold nanoparticles.298 Finally, we mention three other non-biological applications, namely nanopore inhibition,299,300 surface wetting,301 and physisorption of organic molecules on graphite.302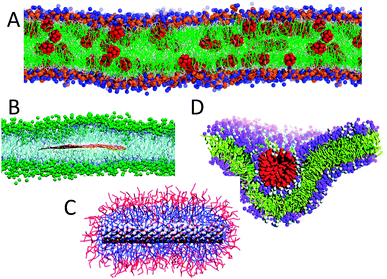 | ||
| Fig. 9 Nanoparticles interacting with lipids. (A) Fullerene dissolved in a lipid membrane37 (courtesy L. Monticelli), with fullerenes in red, lipid tails green, lipid heads blue/orange. (B) Graphene sandwiched in between the bilayer leaflets281 (courtesy P. Kral), with graphene flake in orange, lipid tails blue, lipid heads green. (C) Carbon nanotube wrapped by surfactants38 (courtesy M. S. P. Sansom), with CNT in grey, surfactant heads red and tails blue. (D) Wrapping of a cationic gold nanoparticle by a negatively charged lipid membrane288 (courtesy H. W. Zhang), showing the cationic coat in red, the lipid tails in yellow, and lipid heads in purple. Adapted with permission from ref. 38, 281 and 288. Copyright (2007–2011) American Chemical Society. | ||
Limitations of Martini
Martini, as any other model, has a number of limitations. It is obviously important to know these limitations, both to make sure the model is used appropriately and to further improve the model. Some of Martini's limitations are shared with coarse-grained models at a fundamental level, such as the chemical and spatial resolution, which are both limited compared to atomistic models; a shifted balance between entropy and enthalpy due to the reduced number of degrees of freedom; and kinetics that are modified in unpredictable ways. Other limitations are more tightly linked to Martini itself and could be improved in the future at the level of coarse-graining inherent in Martini, or by accepting more detailed descriptions in parts of the system of interest. This presents an interesting philosophical problem, as the essence of a coarse-grained model is that it contains less detail than a particular reference model (atomistic models, in Martini's case). While it is clear that adding back details in such a model can improve its accuracy, this also negates (some of) its computational advantages. Below we discuss a number of known limitations of Martini.Model resolution and accuracy
With its 4-1 mapping and its particular range of interactions, Martini can reproduce the thermodynamics of a large number of different organic compounds. This mapping clearly limits the chemical resolution. Phosphatidylcholine lipids can be used to illustrate this in a biological context. Key properties of such lipids, including melting temperature and bilayer stability and thickness depend strongly on the length of the acyl chains. The lipid dilauroylphosphatidylcholine (DLPC) has two acyl chains with 12 carbon atoms, represented in principle by 3 Martini beads, while the lipid dipalmitoylphosphatidylcholine (DPPC) has two acyl chains with 16 carbon atoms, represented by 4 Martini beads. The common lipid dimyristoylphosphatidylcholine (DMPC) with 14 carbon atoms falls somewhere in the middle. Thus the mapping to specific lipids is not unique. Martini reproduces properties such as lipid bilayer density profiles, but within the limitations of this mapping and the spatial resolution afforded by the size of Martini particles. Similar limitations occur in common biomolecules such as sterols, where small changes in ring structure or tail can have large effects on the thermodynamics of membranes that contain these sterols; carbohydrates, where stereochemistry and other details that are too subtle to easily represent in a coarse-grained model occur; and proteins, where all amino acids have a different representation but it is unlikely subtle differences between similar side chains are accurately represented.Martini in many cases is comparable in accuracy to atomistic models, particularly in thermodynamics, but it also amplifies a number of limitations in atomistic models, where lack of electronic polarizability in the standard force field is one of the main limitations. Important interactions such as cation–pi interactions in proteins or the strong electrostatically driven interactions between benzene molecules are difficult to include in a coarse-grained force field to reproduce both the strength of these interactions and, more challenging, the peculiar geometries resulting from these interactions. However, the wide use of Martini and extensive testing in the original papers clearly shows the degree of agreement with experiments and atomistic simulations and allows an assessment of whether Martini is accurate enough for a particular application. Recent progress in linking Martini more closely to atomistic simulations through backmapping and through hybrid simulations appears promising in terms of extending the use of Martini simulations to problems that may be outside the current resolution (see ‘Outlook’ section).
Effective time scale
In atomistic simulations the time scale is well-defined and properties such as water diffusion coefficients are used to judge the quality of the underlying model. In coarse-grained simulations this is not normally the case. Coarse-graining involves modifying the energy landscape to become smoother, which effectively results in more sampling of the energy landscape in a given time period, speeding up the kinetics of the system. This is one of the main advantages of coarse-grained models, but the speed-up is not easily predictable and is not likely to be the same for all degrees of freedom.303 In Martini the current best estimate of a semi-universal factor of speed-up compared to atomistic simulations is about 4, based on lateral diffusion coefficients of lipids13 and TM peptides130 in membranes. Some papers therefore report an effective simulation length, which is 4 times the formal simulation length. However, the use of a global speed-up factor has to be considered with care. In a systematic study by de Jong304 on a range of compounds, speed-up factors based on self-diffusion ranged between 1.2 and 22 comparing Martini to experiments and 0.79 and 17 comparing Martini to atomistic simulations. The speed-up is dependent on the type of molecule: Martini alkanes have a small speed-up compared to experiments or even a reduced speed compared to atomistic simulations, whereas alcohols show large speed-up factors. This can be rationalized by the lack of explicit hydrogen bonds in the CG model. Water, although highly polar, shows a relatively small speed-up (≈2.5), but this is due to the bundling of four molecules together. Lipids show a large speed-up, comparable to polar fluids suggesting that their diffusion is mainly controlled by the polar head groups.These estimates of speed-up factors are based on single-molecule diffusion, but the underlying mechanism has not been carefully investigated and may be different for tracer diffusion, viscosity, collective diffusion, etc. For instance, the viscosity of standard Martini water has been determined78,305 at η = 7 × 10−4 Pa s (at 323 K) in close agreement with the experimental value of 5.5 × 10−4 Pa s and actually closer to the experimental value than the widely used SPC water model. For lipid bilayers, apparent surface shear viscosity has been calculated by Baoukina et al.79 and is similar to experimental estimates; for lipid monolayers, the apparent surface shear viscosity was similar to previously reported atomistic simulations but much smaller than measured experimentally for monolayers.252 However, a direct comparison for surface viscosities is difficult because experimental shear rates are much lower and simulation results are expected to depend on the system size. Baoukina et al.103 also calculated the effect of the presence of domains on monolayer surface viscosity and found a clear increase in viscosity in the presence of domains or at higher compression levels, in qualitative agreement with experiment. Thus although there is some data on viscosities, more thorough investigations would be welcome. In addition, density fluctuations (collective diffusion) have not been carefully analyzed yet. Another example of comparing effective time scales is the recent study on the dynamics of the onset of phase separation in membranes composed of ternary lipid mixtures.105 Comparing atomistic and Martini simulations, the authors conclude that, while sharing the structural features of phase separation in the CG model, the onset of demixing for the atomistic model is 40 times slower.
In addition to the effect of a simplified energy landscape, there is also a much smaller source of uncertainty in kinetics due to the choice of masses of the Martini particles. The actual time step used in molecular dynamics depends on the masses of the particles involved, which correspond to real atomic masses in atomistic simulations but to effective masses in coarse-grained simulations. All Martini particles have a mass of 72 amu for full particles or 45 amu for scaled particles, regardless of the underlying atoms. Effectively this scales the simulation time differently for groups with different underlying masses. This could be corrected by using more accurate masses but since the masses do not affect the ensemble obtained from a molecular dynamics simulation this is not critical. As atomistic simulations increase in reach we expect more detailed comparisons of kinetics between atomistic and Martini simulations, which would help calibrate the effective timescale of Martini simulations. One useful application of Martini is to assess the amount of sampling required to study a particular phenomenon, such as lipid mixing, before attempting this with atomistic simulations.
Free energies, enthalpies, entropies
A third category of properties that are fundamentally affected in CG models are thermodynamic properties, particularly the balance between enthalpy and entropy. Martini is parameterized to reproduce accurate free energies, with a reduced number of degrees of freedom compared to atomistic simulations. Reducing the number of degrees of freedom affects the entropy of the simulation system, which is compensated for by reduced enthalpic terms in the model. However, this means that the temperature dependence of coarse-grained models is a priori not correct. It also means that although free energy differences are accurate, a break-down of free energies into enthalpies and entropies may not be accurate. The classical example of this is the potential of the mean force (PMF) between two hydrophobic molecules in solution. MacCallum et al. performed detailed all-atom calculations of the PMF between two hydrophobic helices, showing a deep well in the free energy, despite an enthalpic barrier at a slighter higher separation than the equilibrium conformation.306 Aggregation was driven by a favorable entropy, related to the structure of water around the hydrophobic helices and the release of water trapped between the two helices at near-contact. Corresponding Martini simulations with the standard water model showed the right free energy profile, but the basis for this profile is almost entirely enthalpic.307 The polarizable Martini water model shows a bit more water-ordering effects, but still did not reproduce the atomistic simulations. The BMW water model did at least reproduce the correct sign although the features of the entropy/enthalpy decomposition still differed from the atomistic simulations. This water model is not a universal solution due to its interactions with other parts of the Martini force field, but does point out the importance of water structure if one wants to reproduce more accurately the underlying thermodynamics. A recent study compared atomistic simulations with a different CG model in their ability to reproduce different components of the thermodynamic driving force for a ligand binding to a protein.308 The authors concluded that an anisotropic component to the water model interactions combined with translational entropy was sufficient to retain a reasonable enthalpy/entropy balance and temperature dependence of ligand binding.Thus in solution, PMF decompositions based on CG models should be used with great caution and in many cases results with both the standard and polarizable water models in Martini will be unrealistic. In hydrophobic solvents or membranes, the story may be different as water ordering is probably one of the least accurate components of Martini, while lipid ordering or hydrophobic solvents are likely to be represented much more accurately. PMFs for dimerization of two fullerene molecules in octane gave good agreement between atomistic and coarse-grained simulations, including decomposition in entropy and enthalpy profiles (Monticelli, personal communication). A study of the aggregation of two TM helices gave thermodynamic parameters, including enthalpy and entropy, in reasonable agreement with experiment,47 although obtaining these parameters from atomistic simulations is out of reach at the moment. Thus it seems entropy/enthalpy decompositions in apolar environments give at least qualitatively useful results and warrant further testing.
In a number of studies, Baron et al. compared thermodynamic properties of alkanes and lipids in atomistic and coarse-grained simulations, including configurational entropies of alkanes309 and lipids,310 as well as a larger range of thermodynamic properties in liquid alkanes.18 In general, both models showed similar behaviour, although the temperature dependence of alkanes and lipids modelled with Martini was notably weaker than with the atomistic force field or compared to experiment.
Other properties related to strong water interactions and water ordering are also challenging to reproduce in coarse-grained models like Martini. Free energies of hydration and transfer of groups with full charges are so large that they are difficult to fit in the range of interactions Martini is capable of reproducing based on the current LJ and limited Coulomb interactions. Charge–charge interactions in apolar solvents are too weak because the implicit screening in Martini and the size of the Martini particles reduce interactions too much. One way to improve this may be to place charges off-centre to allow a closer interaction, analogous to recent changes in the protein force field.15
The functional form of the non-bonded potential
The use of a LJ 12-6 potential to describe the non-bonded interactions in Martini is, in hindsight, not the best choice. The steep repulsion leads to overstructuring of fluids compared to atomistic models, as evidenced for instance in the radial distribution functions (RDFs) for simple alkanes.309 Furthermore, the LJ 12-6 potential has only a limited fluid range. Consequently, Martini water is prone to freezing even at room temperature, although the switch function reduces attraction and increases the melting temperature compared to a pure LJ function. The melting temperature of Martini water is 290 ± 5 K.13 In confined geometries freezing is more pronounced. A practical, if chemically difficult to justify, solution is the use of ‘anti-freeze’ particles, water particles with a slightly larger radius.13 Switching to a different non-bonded interaction potential could, in principle, improve the relative stability of the fluid phase (see ‘Outlook’ section). A related property that is not accurate in current versions of Martini is the surface tension of the air/water interface, which is much too low. This results in unrealistic behaviour in monolayers at the air/water interface, where lipids do not spread homogeneously at high surface tensions but rather form pores in tighter packed monolayers.250 It is also apparent in water/oil interfaces, which show properties of consisting of two interfaces (water/air and air/oil) close to each other, rather than a single interface.251 Using the polarizable water model,21 these properties improve but only to a limited amount.Secondary structure constraints
Realistic modelling of conformational changes in proteins and peptides in biomolecular force fields is essential for many problems, but is a major challenge in CG simulation. Martini is unusual among CG protein force fields in that it has rather sophisticated side chains but a very simple backbone, consistent with the overall design philosophy of Martini. However, that means that conformational changes with the simplified description of Martini cannot be accurately reproduced in general. In the current version of the protein force field, we apply secondary structure constraints to maintain extended and helical secondary structures by strengthening bonded interactions to maintain a preset secondary structure. The secondary structure elements are able to move relative to each other, which in general does not lead to accurate protein structures in solution but enables a number of applications of primarily TM proteins like MscL mentioned above. These proteins are constrained by the membrane environment and consist mainly of helices with short loops where details of the conformations are in some cases not important. Situations where proteins or peptides change structure considerably cannot be reliably handled. A few examples that are hindered by this but otherwise seem very suitable for Martini are membrane-binding peptides that may be unstructured in solution but adopt specific conformations when binding to the membrane, and may adopt yet different structures in aggregates or channels; membrane-binding proteins with loops that may fold into helices or unfold from a protected, perhaps multimeric, structure into a membrane-binding state, and other systems with similar types of conformational changes. These changes can be incorporated as assumptions in Martini simulations, of course, but this is typically less desirable. Perhaps a combination of backmapping and coarse-graining steps with atomistic and coarse-grained models may be useful in some of these cases. Further work in this direction would be interesting. In a more experimental approach, we tried to include backbone pseudo-dihedral potentials that reproduce backbone conformations of atomistic simulations.178 This method enables some new applications but requires atomistic simulations as input and is not generally transferable between sequences or between chemical environments.Testing the limits: simulations of pore formation
We discussed a range of limitations of Martini, most of which have their origins clearly in the choice of approximations, including the resolution of the model and the functional form of the potential. However, Martini can also break down in cases where there is no obvious reason. One example of this is pore formation in lipid bilayers. This is a particularly interesting example because lipid-lined pores occur in a wide range of problems for which Martini has been used or could be used. Pore formation, or more generally defects, in lipid bilayers occur in processes such as permeabilization of bilayers by antimicrobial peptides, transport of polar molecules across membranes, fusion intermediates, and lipid flip–flop from one membrane leaflet to another. These defects involve water penetrating into the membrane, followed by rearrangements of phospholipids. This is an interesting test case for Martini because of the relatively small characteristic size at the level of water molecules and the complex environment of water and polar head groups in a low-dielectric membrane interior. We have studied membrane defects in great detail by atomistic simulations and tried to reproduce this data with Martini.311Fig. 10A shows a snapshot of a DLPC bilayer in which we have restrained a DLPC headgroup in the centre of the bilayer. Atomistically this leads to pore formation and an extensive structural defect, but with Martini (Fig. 10C) the structure is very different although the free energy cost of placing the lipid at the centre is similar. To test whether this is due to the simple water model, we repeated the calculation with the polarizable Martini water, but with the same result (Fig. 10B). To test whether the difference is due to the large size of a Martini water particle compared to an atomistic water molecule, we tethered atomistic water molecules together in tightly-bound groups of four, but this gave nearly the same results as the regular atomistic simulations. Increasing the interaction between water and headgroups did give structures closer to the pore observed in atomistic simulations (Fig. 10D), but worsened other important bilayer properties such as the area/lipid. We will continue to use a system like this for tests of future refinements of Martini.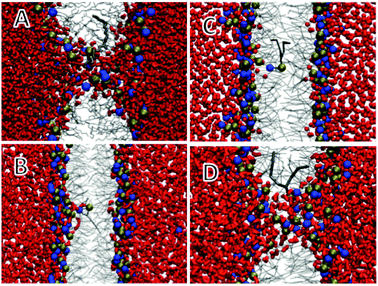 | ||
| Fig. 10 Snapshots of a DLPC phospholipid restrained at the centre of a DLPC bilayer.311 Water is shown as red licorice, lipid tails as grey lines, restrained lipid as thick grey lines, and headgroup phosphate (phosphorus) and choline (nitrogen) as balls. (A) Atomistic, (B) polarizable Martini water, (C) standard Martini water, (D) polarizable Martini water with increased attraction between water and lipid headgroups. Reprinted with permission from ref. 311. Copyright (2011) American Chemical Society. | ||
Outlook
Given the large interest of the modelling community to use Martini on the one hand, and the clear limitations of the model on the other hand, there is a lot of work to be done. Below we sketch the main developments we foresee in the near future.The next generation, Martini 3.x
The use of a LJ 12-6 potential to describe the non-bonded interactions in Martini leads to overstructuring as discussed above. To reproduce the ‘softness’ of real systems a smaller power for the repulsive term appears more appropriate, as in the recently reparameterized CG model of Klein and co-workers.312 The other drawback of the LJ 12-6 potential is the limited fluid range. Possible solutions are given in the literature,313,314 including switching to a power 4 attraction or to a different potential such as the Morse potential. For the next generation of the Martini force field, we aim to change the non-bonded form to a softer potential with longer-ranged attraction. We are currently exploring the use of the Mie potential,315 a generalized LJ potential with exponents n and m. Preliminary results304 indicate that key properties can be reproduced with Mie 5-4, 5-3, 4-3, and 4-2 potentials in their shifted form. Another major direction forward is the development of a foldable protein backbone. The lack of structural flexibility of the Martini proteins obviously limits the applicability range of the model. While aiming at a CG model capable of folding a protein from its sequence may be overly ambitious, describing the backbone in a more realistic manner would be an important improvement. One solution is the use of a specific set of bonded parameters optimized to reproduce an ensemble obtained from atomistic simulations or NMR experiments, an approach already used to model amyloids178 and fusion peptides.91 For a less biased approach, the directionality of H-bonds needs to be restored. Available coarse-grained models for protein folding represent the backbone H-bonding in quite different manners and generally use a specific potential.316–320 In contrast we want to keep a simple and generic approach. For instance, we have been exploring the possibility of using a fixed dipole to represent the backbone polarity, see Fig. 11. The dipoles interact with other dipoles and particles through regular Coulomb and LJ potentials. Importantly, this description restores the directionality of backbone interactions, and has a number of added benefits: the overall dipole along the axis of an α-helix is represented, and the charges are natural sensors of the dielectric environment. A similar approach was used to model polar side chains more realistically in Martini 2.2 and may prove to be an effective way to model polar chemical building blocks in general.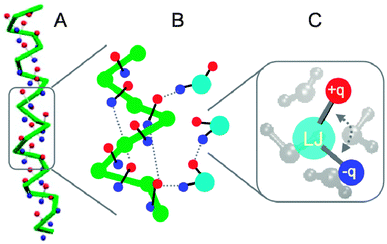 | ||
Fig. 11 Toward a foldable Martini protein backbone (courtesy X. Periole). (A) Stabilization of an α-helix due to added degrees of freedom in the form of charged particles (red +q, blue −q) attached to the backbone (green), mimicking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H hydrogen acceptor and donors and providing an overall dipole moment. (B) Zoomed view of the interaction of the peptide backbone with the polarizable water model. Effective H-bonds along the backbone and with the water particles are shown with dashed lines. (C) Close up of the polarizable water model, consisting of a central LJ bead with two charged particles attached, representing four real waters. O and N–H hydrogen acceptor and donors and providing an overall dipole moment. (B) Zoomed view of the interaction of the peptide backbone with the polarizable water model. Effective H-bonds along the backbone and with the water particles are shown with dashed lines. (C) Close up of the polarizable water model, consisting of a central LJ bead with two charged particles attached, representing four real waters. | ||
More Martini molecules
Nucleotides, RNA and DNA, constitute the major class of biomolecules that has not yet been thoroughly parameterized for Martini. Although there is a Martini model available for a small piece of DNA,321 a more generic approach is required to make the model applicable to all nucleic acids. We are currently developing parameters for single and double stranded DNA, using a systematic approach analogous to our work on carbohydrates.31 A preliminary model is shown in Fig. 1F. Another class of biomolecules that requires careful parameterization are glycans (oligosaccharides), e.g. peptidoglycans that are the building block of the bacterial cell wall, or the glycans attached to proteins in the process of glycosylation, or lipopolysaccharides that are the major component of the outer membrane of Gram-negative bacteria. The Martini carbohydrate31 and glycolipid27 force fields can serve as starting points. Apart from biomolecules, other molecular systems are in principle amenable to the Martini CG approach. We encourage parameter development for many more polymers and nanoparticles, and expect a growing number of applications involving hybrid molecules; combining bio- and synthetic molecules into hybrid molecules is a booming research field in the era of synthetic biology. And why not a Martini force field for ionic liquids? Using polarized or polarizable CG particles this might well turn out to be a fruitful approach. Attempts in this direction have recently been undertaken.322Algorithmic improvements affecting Martini
A number of algorithmic developments are worthwhile to mention as they are likely to improve the accuracy, speed, and stability of Martini simulations in the near future. A first issue is the choice of thermostat. Although Martini was developed with the widely-used Berendsen weak-coupling scheme for temperature and pressure control, recent developments promise efficient temperature control methods that conserve momentum,323 which may be important in future work on time and length scales where hydrodynamic effects play a role. It remains to be seen how accurately the Martini model can capture hydrodynamic effects; the fact that the solvent viscosity is actually reproduced quite well is promising in that respect (see the discussion on the effective time scale above). Second, to improve speed, the buffered Verlet pairlist scheme recently implemented in GROMACS shows promise. Instead of a switch function, the Verlet scheme uses a straight cut-off in combination with a shift of the potential to zero at the cut-off. Neighbour list searching is much faster with this method. However, as this involves a change in the overall shape of the non-bonded potential, it might affect system properties. Preliminary results (de Jong and Marrink, unpublished) show that key properties of the Martini model remain unaffected when using a slightly reduced cut-off (1.1 nm instead of the current 1.2 nm) in combination with an optimized neighbour list cut-off. These settings result in a speed-up of almost 100%. Third, improving numerical stability is also deemed important, especially in light of high-throughput applications (see below). One common reason for Martini simulations to crash is that dihedrals, formed by four particles, are not defined when three of these particles are co-linear. In atomistic models this does not occur but at the coarser level of Martini this is difficult to avoid. We are working on alternative definitions of local geometry to avoid this numerical problem, making use of combined bending-torsion potentials or dummy-assisted dihedrals.324Mixing Martinis
One of the current challenges in biomolecular simulation is to develop effective multi-scale methods,325–329 which combine the advantages of atomistic and coarse-grained models. Multi-resolution methods can either use a static division as in QM/MM (denoted “hybrid” models), or allow particles to change resolution on the fly (“adaptive” models). Recently we introduced a straightforward scheme to perform hybrid simulations, making use of virtual sites to couple the two levels of resolution.327 With the help of these virtual sites interactions between molecules at different levels of resolution, i.e. between CG and atomistic molecules, are treated in the same way as the pure CG–CG interactions. As a proof of principle, hybrid GROMOS/Martini simulations of simple liquids like butane and small peptides were shown to behave well. However, in an extension of this work330 challenges are pointed out in regard to the electrostatic coupling between the two levels of resolution. In particular, a proper description of the interaction between polar AA molecules suffers from the poor short range screening behaviour of the CG solvent. Applications of this multi-scale method dealing with less polar solvents, e.g. a lipid bilayer, are more promising. For instance, the gating of AA mechanosensitive channels in CG bilayers is currently being studied by our group (Fig. 12A). To achieve a quantitatively more accurate method, cross optimization of the interactions between the Martini and the atomistic force field is probably necessary, as has been recently attempted in the PACE force field.331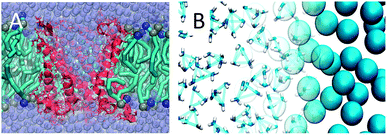 | ||
| Fig. 12 Mixing Martini and AA force fields. (A) Hybrid simulation of an atomistic membrane protein, MscL, in CG environment330 (courtesy H. Ingolfsson). Protein channel depicted in red, lipid bilayer with cyan tails and blue/ochre heads, and water beads purple. (B) Adaptive resolution simulation of CG Martini and atomistic water using the AdResS scheme (courtesy M. de Melo). The right hand side shows CG water beads, the left hand side shows bundled clusters of four atomistic SPC water molecules. | ||
A breakthrough implementation of an adaptive multi-scale method is the AdResS (Adaptive Resolution Simulation) approach, developed by Kremer and co-workers.332 In this method, a transition region allows molecules to pass from atomistic to CG resolution and vice versa as a function of the position of the molecule in the simulation box. The coupling of resolutions is achieved through the use of a thermodynamic force in the transition region that compensates for the chemical potential difference between the two resolutions. We are currently exploring the coupling of Martini to atomistic resolution using the AdResS scheme (Fig. 12B). Within the field of biomolecules, Martini based multi-scale methods appear ideally suited to study e.g. protein–ligand binding, where the active site and ligand are modelled in atomistic detail and the rest of the protein together with the solvent is coarse-grained, or aggregation of membrane proteins where the surface of the proteins is treated at a finer grained level compared to the interior and lipids may increase their resolution in close contact with proteins. Another potentially powerful approach to multi-scale simulations is based on Hamiltonian Replica Exchange MD (HREMD). In HREMD, multiple simulations are run at the same time with systematic differences in the Hamiltonian, in this case the atomistic and CG Hamiltonians. Coupling of CG models to AA models using REMD has been pioneered by several groups,333,334 but has not yet been used in combination with the Martini model. Not really multi-scale, but worth mentioning as an interesting method that opens a range of new problems that can be studied, is the use of constant pH algorithms with Martini.335
At the other end of the scale, there is promise in combining Martini with self-consistent field theory,336 to couple Martini to multi-domain elastic network models,337,338 and to combine Martini with protein–protein docking approaches. Another scale bridging opportunity is the ability of Martini to yield converged PMFs between proteins.169 These PMFs may subsequently be used to describe the effective interactions between simplified proteins in a solvent free approach, a method coined Atomistic Resolution Brownian Dynamics (ARBD).339 In a related line of research, we are currently parameterizing a variant of the Martini force field that is solvent free (“Martini dry”).
Automated high throughput
World-wide the trend remains to invest in massively parallel computing with petaflop clusters currently topping the list of the fastest computers (see http://www.top500.org). Within the foreseeable future, the exascale will be reached, with clusters composed of millions of cores. Even local facilities will approach the petascale soon. This opens the way to simulations of either very huge systems or massively high throughput. The largest systems simulated with Martini to date include systems of a few million particles, e.g. a huge membrane patch from which a tether was pulled (cf.Fig. 2A), totalling almost 4000000 particles in a box with dimensions 86 × 86 × 65 nm3 and simulated for 1 μs;79 a lipid nanoparticle for gene delivery (cf.Fig. 6A) with 1400000 particles in total, a box size of 54 × 54 × 54 nm3 and simulated for 10 μs;229 and a tetrameric row of F1F0 ATPase dimers (cf.Fig. 3A) composed of 4635000 particles in a box of 84 × 336 × 19 nm3 and simulated for 100 ns.148 Examples of high throughput studies are: scoring of the aggregation propensity of all 400 dipeptides based on 400 × 100 ns simulations of a box of 300 solvated dipeptides;172 construction of a CG database for the positioning of 91 membrane proteins in a lipid bilayer, based on hundreds of self-assembly simulations;119 and performing 10000 separate simulations by using the Folding@Home distributed computing project to model membrane fusion.239 A recent example of high-throughput simulations is the systematic sampling of protein–protein interfaces (Fig. 13). In the forthcoming exascale era, and with the aid of the continuous improvement of simulation software, we can expect system sizes to grow to 109 particles, timescale reaching seconds, and high throughput studies systematically exploring 1000 s of conditions. To harvest this enormous power, automated workflows should be designed and optimized. Efforts in that direction are already undertaken in a number of groups, for example in the grid-enabled web portal for MD simulations,340 automated topology builder341 and automatic embedding of membrane proteins.342 To facilitate the Martini workflow, we have developed the Martinize script15 which generates GROMACS topology files for proteins from coordinate input files. We are currently developing scripts to automatically perform and analyse simulations of heterogeneous membranes composed of arbitrary lipids and proteins.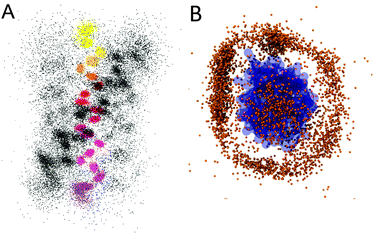 | ||
| Fig. 13 Examples of high-throughput Martini based screening assays. (A) Dimerization interfaces of TM glycophorin A helices (courtesy T. Wassenaar). Results of 1000 × 500 ns self-assembly dimerization events are shown, with the position of backbone of the reference helix as coloured dots, and the other helix in grey. Two distinct populations are clearly visible. (B) Prediction of protein–protein complexes (courtesy D. de Jong). Reference protein (GYF-domain of the CD2 receptor) in blue, centre-of-mass of the complexing protein (U5-52K) as brown dot. Based on 1000 × 1000 ns self-assembly simulations preferential contact interfaces are revealed. | ||
Conclusions
In the ten years since its initial publication, the Martini model has developed from a model for simulations of lipids and surfactants to the most widely-used coarse-grained force field for biomolecular simulation and increasingly in synthetic biology. It has been used in a broad range of applications and been subjected to many critical tests, resulting both in exciting scientific results and clear routes towards further development.Acknowledgements
We greatly appreciate the support and feedback from the global Martini user community. SJM acknowledges funding from the Netherlands Organisation for Scientific Research (NWO). Work in DPT's group is supported by the Canadian Institutes of Health Research (grant MOP-62690) and the Natural Sciences and Engineering Research Council. DPT is an Alberta Innovates Health Solutions Scientist and Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation.Notes and references
- A. P. Lyubartsev and A. Laaksonen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 52, 3730 CrossRef CAS.
- D. Reith, M. Putz and F. Muller-Plathe, J. Comput. Chem., 2003, 24, 1624 CrossRef CAS.
- S. Izvekov, A. Violi and G. A. Voth, J. Phys. Chem. B, 2005, 109, 17019 CrossRef CAS.
- W. G. Noid, J. W. Chu, G. S. Ayton, V. Krishna, S. Izvekov, G. A. Voth, A. Das and H. C. Andersen, J. Chem. Phys., 2008, 128, 244114 CrossRef CAS.
- A. Chaimovich and M. S. Shell, J. Chem. Phys., 2011, 134, 094112 CrossRef.
- E. Brini and N. F. A. van der Vegt, J. Chem. Phys., 2012, 137, 154113 CrossRef CAS.
- G. A. Voth, Coarse-Graining of Condensed Phase and Biomolecular Systems, CRC Press, Boca Raton, 2008 Search PubMed.
- J. T. Padding and W. J. Briels, J. Phys.: Condens. Matter, 2011, 23, 233101 CrossRef CAS.
- M. Deserno, Macromol. Rapid Commun., 2009, 30, 752 CrossRef CAS.
- C. Clementi, Curr. Opin. Struct. Biol., 2008, 18, 10 CrossRef CAS.
- M. L. Klein and W. Shinoda, Science, 2008, 321, 798 CrossRef CAS.
- S. J. Marrink, A. H. de Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812 CrossRef CAS.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S. J. Marrink, J. Chem. Theor. Comput., 2008, 4, 819 CrossRef CAS.
- D. H. de Jong, G. Singh, W. F. D. Bennett, C. Arnarez, T. A. Wassenaar, L. V. Schäfer, X. Periole, D. P. Tieleman and S. J. Marrink, J. Chem. Theor. Comput., 2013, 9, 687 CrossRef CAS.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701 CrossRef CAS.
- A. Y. Shih, A. Arkhipov, P. L. Freddolino and K. Schulten, J. Phys. Chem. B, 2006, 110, 3674 CrossRef CAS.
- R. Baron, D. Trzesniak, A. H. de Vries, A. Elsener, S. J. Marrink and W. F. van Gunsteren, ChemPhysChem, 2007, 8, 452 CrossRef CAS.
- K. J. Bowers, E. Chow, H. F. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06) Tampa, Florida, 2006.
- P. J. Bond, J. Holyoake, A. Ivetac, S. Khalid and M. S. P. Sansom, J. Struct. Biol., 2007, 157, 593 CrossRef CAS.
- S. O. Yesylevskyy, L. V. Schafer, D. Sengupta and S. J. Marrink, PLoS Comput. Biol., 2010, 6, e1000810 Search PubMed.
- Z. Wu, Q. Cui and A. Yethiraj, J. Phys. Chem. B, 2010, 114, 10524 CrossRef CAS.
- X. Periole, M. Cavalli, S. J. Marrink and M. Ceruso, J. Chem. Theor. Comput., 2009, 5, 2531 CrossRef CAS.
- S. J. Marrink, X. Periole, D. P. Tieleman and A. H. de Vries, Phys. Chem. Chem. Phys., 2010, 12, 2254 RSC.
- M. Winger, D. Trzesniak, R. Baron and W. F. van Gunsteren, Phys. Chem. Chem. Phys., 2009, 11, 1934 CAS.
- M. Dahlberg, J. Phys. Chem. B, 2007, 111, 7194 CrossRef CAS.
- C. A. Lopez, Z. Sovova, F. J. van Eerden, A. H. de Vries and S. J. Marrink, J. Chem. Theor. Comput., 2013, 9, 1694 CrossRef CAS.
- H. Lee and R. W. Pastor, J. Phys. Chem. B, 2011, 115, 7830 CrossRef CAS.
- J. D. Perlmutter and J. N. Sachs, J. Am. Chem. Soc., 2009, 131, 16362 CrossRef CAS.
- A. Gautieri, A. Russo, S. Vesentini, A. Redaelli and M. J. Buehler, J. Chem. Theor. Comput., 2010, 6, 1210 CrossRef CAS.
- C. A. Lopez, A. J. Rzepiela, A. H. de Vries, L. Dijkhuizen, P. H. Hunenberger and S. J. Marrink, J. Chem. Theor. Comput., 2009, 5, 3195 CrossRef CAS.
- J. Wohlert and L. A. Berglund, J. Chem. Theor. Comput., 2011, 7, 753 CrossRef CAS.
- H. Lee, A. H. de Vries, S. J. Marrink and R. W. Pastor, J. Phys. Chem. B, 2009, 113, 13186 CrossRef CAS.
- G. Rossi, L. Monticelli, S. R. Puisto, I. Vattulainen and T. Ala-Nissila, Soft Matter, 2011, 7, 698 RSC.
- M. Hatakeyama and R. Faller, Phys. Chem. Chem. Phys., 2007, 9, 4662 RSC.
- A. Milani, M. Casalegno, C. Castiglioni and G. Raos, Macromol. Theory Simul., 2011, 20, 305 CrossRef CAS.
- J. Wong-Ekkabut, S. Baoukina, W. Triampo, I. M. Tang, D. P. Tieleman and L. Monticelli, Nat. Nanotechnol., 2008, 3, 363 CrossRef CAS.
- E. J. Wallace and M. S. P. Sansom, Nano Lett., 2007, 7, 1923 CrossRef CAS.
- L. Monticelli, J. Chem. Theor. Comput., 2012, 8, 1370 CrossRef CAS.
- H. Lee and R. G. Larson, J. Phys. Chem. B, 2008, 112, 7778 CrossRef CAS.
- H. Lee and R. G. Larson, J. Phys. Chem. B, 2006, 110, 18204 CrossRef CAS.
- J. L. MacCallum, W. F. D. Bennett and D. P. Tieleman, Biophys. J., 2008, 94, 3393 CrossRef CAS.
- D. H. de Jong, X. Periole and S. J. Marrink, J. Chem. Theor. Comput., 2012, 8, 1003 CrossRef CAS.
- H. J. Risselada and S. J. Marrink, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17367 CrossRef CAS.
- G. Singh and D. P. Tieleman, J. Chem. Theor. Comput., 2011, 7, 2316 CrossRef CAS.
- L. Monticelli, D. P. Tieleman and P. F. J. Fuchs, Biophys. J., 2010, 99, 1455 CrossRef CAS.
- N. Castillo, L. Monticelli, J. Barnoud and D. P. Tieleman, Chem. Phys. Lipids, 2013, 69, 95 CrossRef.
- D. Sengupta and S. J. Marrink, Phys. Chem. Chem. Phys., 2010, 12, 12987 RSC.
- S. J. Marrink and A. E. Mark, J. Am. Chem. Soc., 2003, 125, 15233 CrossRef CAS.
- S. J. Marrink and A. E. Mark, J. Am. Chem. Soc., 2003, 125, 11144 CrossRef CAS.
- M. Bulacu, X. Periole and S. J. Marrink, Biomacromolecules, 2012, 13, 196 CrossRef CAS.
- S. Wang and R. G. Larson, Soft Matter, 2013, 9, 480 RSC.
- C. Liu and R. Faller, Langmuir, 2012, 28, 15907 CrossRef CAS.
- C. Xing, O. H. S. Ollila, I. Vattulainen and R. Faller, Soft Matter, 2009, 5, 3258 RSC.
- N. Kucerka, J. Gallova, D. Uhrikova, P. Balgavy, M. Bulacu, S. J. Marrink and J. Katsaras, Biophys. J., 2009, 97, 1926 CrossRef CAS.
- J. D. Perlmutter and J. N. Sachs, Biochim. Biophys. Acta Biomembr., 2009, 1788, 2284 CrossRef CAS.
- W. F. D. Bennett, J. L. MacCallum, M. J. Hinner, S. J. Marrink and D. P. Tieleman, J. Am. Chem. Soc., 2009, 131, 12714 CrossRef CAS.
- F. Ogushi, R. Ishitsuka, T. Kobayashi and Y. Sugita, Chem. Phys. Lett., 2012, 522, 96 CrossRef CAS.
- S. Esteban-Martin, H. J. Risselada, J. Salgado and S. J. Marrink, J. Am. Chem. Soc., 2009, 131, 15194 CrossRef CAS.
- J. Yoo and Q. Cui, Biophys. J., 2009, 97, 2267 CrossRef CAS.
- S. O. Yesylevskyy and A. P. Demchenko, Eur. Biophys. J. Biophys. Lett., 2012, 41, 1043 CrossRef CAS.
- N. Maftouni, M. Amininasab, M. Vali, M. Ejtehadi and F. Kowsari, J. Membr. Biol., 2013, 246, 67 CrossRef CAS.
- M. J. Hinner, S. J. Marrink and A. H. de Vries, J. Phys. Chem. B, 2009, 113, 15807 CrossRef CAS.
- M. Klacsová, M. Bulacu, N. Kučerka, D. Uhríková, J. Teixeirad, S. J. Marrink and P. Balgavý, Biochim. Biophys. Acta Biomembr., 2011, 808, 2136 CrossRef.
- H. Khandelia, L. Duelund, K. I. Pakkanen and J. H. Ipsen, PLoS One, 2010, 5, e12811 Search PubMed.
- M. Pickholz and G. Giupponi, J. Phys. Chem. B, 2010, 114, 7009 CrossRef CAS.
- R. Notman, M. G. Noro and J. Anwar, J. Phys. Chem. B, 2007, 111, 12748 CrossRef CAS.
- H. J. Risselada and S. J. Marrink, Soft Matter, 2009, 5, 4531 RSC.
- H. J. Risselada and S. J. Marrink, Phys. Chem. Chem. Phys., 2009, 11, 2056 RSC.
- H. J. Risselada, A. E. Mark and S. J. Marrink, J. Phys. Chem. B, 2008, 112, 7438 CrossRef.
- C. Y. Leung, L. C. Palmer, B. F. Qiao, S. Kewalramani, R. Sknepnek, C. J. Newcomb, M. A. Greenfield, G. Vernizzi, S. I. Stupp, M. J. Bedzyk and M. Olvera de la Cruz, ACS Nano, 2012, 6, 10901 CrossRef CAS.
- H. Lee, H. R. Kim, R. G. Larson and J. C. Park, Macromolecules, 2012, 45, 7304 CrossRef CAS.
- R. L. Wu, M. L. Deng, B. Kong and X. Z. Yang, J. Phys. Chem. B, 2009, 113, 15010 CrossRef CAS.
- M. Hu, D. H. de Jong, S. J. Marrink and M. Deserno, Faraday Discuss., 2013, 161, 365 RSC.
- M. C. Watson, E. S. Penev, P. M. Welch and F. L. H. Brown, J. Chem. Phys., 2011, 135, 244701 CrossRef.
- E. G. Brandt, A. R. Braun, J. N. Sachs, J. F. Nagle and O. Edholm, Biophys. J., 2011, 100, 2104 CrossRef CAS.
- E. R. May, A. Narang and D. I. Kopelevich, Mol. Simul., 2007, 33, 787 CrossRef CAS.
- W. K. den Otter and S. A. Shkulipa, Biophys. J., 2007, 93, 423 CrossRef CAS.
- S. Baoukina, S. J. Marrink and D. P. Tieleman, Biophys. J., 2012, 102, 1866 CrossRef CAS.
- S. J. Marrink, J. Risselada and A. E. Mark, Chem. Phys. Lipids, 2005, 135, 223 CrossRef CAS.
- R. Faller and S. J. Marrink, Langmuir, 2004, 20, 7686 CrossRef CAS.
- Q. Waheed, R. Tjornhammar and O. Edholm, Biophys. J., 2012, 103, 2125 CrossRef CAS.
- J. M. Rodgers, J. Sørensen, F. J. M. de Meyer, B. Schiøtt and B. Smit, J. Phys. Chem. B, 2012, 116, 1551 CrossRef CAS.
- T. Nagai, R. Ueoka and Y. Okamoto, J. Phys. Soc. Jpn., 2012, 81, 024002 CrossRef.
- K. Lai, B. Wang, Y. Zhang and Y. W. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 5744 RSC.
- N. D. Winter and G. C. Schatz, J. Phys. Chem. B, 2010, 114, 5053 CrossRef CAS.
- S. J. Marrink and A. E. Mark, Biophys. J., 2004, 87, 3894 CrossRef CAS.
- S. Sun, J. T. Y. Wong and T. Y. Zhang, Soft Matter, 2011, 7, 9307 RSC.
- E. R. May, D. I. Kopelevich and A. Narang, Biophys. J., 2008, 94, 878 CrossRef CAS.
- M. Fuhrmans, V. Knecht and S. J. Marrink, J. Am. Chem. Soc., 2009, 131, 9166 CrossRef CAS.
- M. Fuhrmans and S. J. Marrink, J. Am. Chem. Soc., 2012, 134, 1543 CrossRef CAS.
- S. J. Marrink, A. H. de Vries, T. A. Harroun, J. Katsaras and S. R. Wassall, J. Am. Chem. Soc., 2008, 130, 10 CrossRef CAS.
- C. Rosetti and C. Pastorino, J. Phys. Chem. B, 2012, 116, 3525 CrossRef CAS.
- N. Kucerka, D. Marquardt, T. A. Harroun, M. P. Nieh, S. R. Wassall, D. H. de Jong, L. V. Schäfer, S. J. Marrink and J. Katsaras, Biochemistry, 2010, 49, 7485 CrossRef CAS.
- C. Rosetti and C. Pastorino, J. Phys. Chem. B, 2011, 115, 1002 CrossRef CAS.
- R. S. Davis, P. B. Sunil Kumar, M. M. Sperotto and M. Laradji, J. Phys. Chem. B, 2013, 117, 4072 CrossRef CAS.
- L. V. Schäfer and S. J. Marrink, Biophys. J., 2010, 99, L91 CrossRef.
- H. S. Muddana, H. H. Chiang and P. J. Butler, Biophys. J., 2012, 102, 489 CrossRef CAS.
- J. D. Perlmutter and J. N. Sachs, J. Am. Chem. Soc., 2011, 133, 6563 CrossRef CAS.
- H. J. Risselada, S. J. Marrink and M. Muller, Phys. Rev. Lett., 2011, 106, 148102 CrossRef.
- T. Fischer, H. J. Risselada and R. L. C. Vink, Phys. Chem. Chem. Phys., 2012, 14, 14500 RSC.
- T. Apajalahti, P. Niemela, P. N. Govindan, M. Miettinen, E. Salonen, S. J. Marrink and I. Vattulainen, Faraday Discuss., 2010, 144, 411 RSC.
- S. Baoukina, E. Mendez-Villuendas and D. P. Tieleman, J. Am. Chem. Soc., 2012, 134, 17543 CrossRef CAS.
- S. Baoukina, E. Mendez-Villuendas, W. F. D. Bennett and D. P. Tieleman, Faraday Discuss., 2013, 161, 63 RSC.
- D. Hakobyan and A. Heuer, J. Phys. Chem. B, 2013, 117, 3841 CrossRef CAS.
- W. F. D. Bennett and D. P. Tieleman, Biochim. Biophys. Acta Biomembr., 2013, 1828, 1765 CrossRef.
- B. A. Hall, A. P. Chetwynd and M. S. P. Sansom, Biophys. J., 2011, 100, 1940 CrossRef CAS.
- J. M. Crowet, D. L. Parton, B. A. Hall, S. Steinhauer, R. Brasseur, L. Lins and M. S. P. Sansom, J. Phys. Chem. B, 2012, 116, 13713 CrossRef CAS.
- D. A. Holdbrook, Y. M. Leung, T. J. Piggot, P. Marius, P. T. F. Williamson and S. Khalid, Biochemistry, 2010, 49, 10796 CrossRef CAS.
- S. Sobhanifar, B. Schneider, F. Lohr, D. Gottstein, T. Ikeya, K. Mlynarczyk, W. Pulawski, U. Ghoshdastider, M. Kolinski, S. Filipek, P. Guntert, F. Bernhard and V. Dotsch, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9644 CrossRef CAS.
- V. V. Vostrikov, B. A. Hall, D. V. Greathouse, R. E. Koeppe and M. S. P. Sansom, J. Am. Chem. Soc., 2010, 132, 5803 CrossRef CAS.
- H. M. Nguyen, C. A. Galea, G. Schmunk, B. J. Smith, R. A. Edwards, R. S. Norton and K. G. Chandy, J. Biol. Chem., 2013, 288, 6451 CrossRef CAS.
- J. W. Klingelhoefer, T. Carpenter and M. S. P. Sansom, Biophys. J., 2009, 96, 3519 CrossRef CAS.
- K. Balali-Mood, P. J. Bond and M. S. P. Sansom, Biochemistry, 2009, 48, 2135 CrossRef CAS.
- G. Khelashvili, P. B. C. Albornoz, N. Johner, S. Mondal, M. Caffrey and H. Weinstein, J. Am. Chem. Soc., 2012, 134, 15858 CrossRef CAS.
- C. N. Lumb and M. S. P. Sansom, Biophys. J., 2013, 104, 613 CrossRef CAS.
- S. S. Qin, Y. X. Yu, Q. K. Li and Z. W. Yu, Biochemistry, 2013, 52, 1477 CrossRef CAS.
- R. Tjörnhammar and O. Edholm, Chem. Phys. Lipids, 2013, 169, 2 CrossRef.
- M. S. P. Sansom, K. A. Scott and P. J. Bond, Biochem. Soc. Trans., 2008, 36, 27 CrossRef CAS.
- P. J. Bond and M. S. P. Sansom, J. Am. Chem. Soc., 2006, 128, 2697 CrossRef CAS.
- O. H. S. Ollila, H. J. Risselada, M. Louhivuori, E. Lindahl, I. Vattulainen and S. J. Marrink, Phys. Rev. Lett., 2009, 102, 078101 CrossRef.
- J. Yoo and Q. Cui, Biophys. J., 2013, 104, 117 CrossRef CAS.
- N. Maftouni, M. Amininasab, M. R. Ejtehadi, F. Kowsari and R. Dastvan, J. Chem. Phys., 2013, 138, 065101 CrossRef.
- L. V. Schäfer, D. H. de Jong, A. Holt, A. J. Rzepiela, A. H. de Vries, B. Poolman, J. A. Killian and S. J. Marrink, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1343 CrossRef.
- G. van den Bogaart, K. Meyenberg, H. J. Risselada, H. Amin, K. I. Willig, B. E. Hubrich, M. Dier, S. W. Hell, H. Grubmuller, U. Diederichsen and R. Jahn, Nature, 2011, 479, 552 CrossRef CAS.
- L. Janosi, Z. Li, J. F. Hancock and A. A. Gorfe, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8097 CrossRef CAS.
- D. H. de Jong, C. A. Lopez and S. J. Marrink, Faraday Discuss., 2013, 161, 347 RSC.
- D. L. Parton, A. Tek, M. Baaden and M. S. P. Sansom, PLoS Comput. Biol., 2013, 9, e1003034 CAS.
- J. Domanski, S. J. Marrink and L. V. Schäfer, Biochim. Biophys. Acta Biomembr., 2012, 1818, 984 CrossRef CAS.
- S. Ramadurai, A. Holt, L. V. Schäfer, V. V. Krasnikov, D. T. S. Rijkers, S. J. Marrink, J. A. Killian and B. Poolman, Biophys. J., 2010, 99, 1447 CrossRef CAS.
- P. S. Niemela, M. S. Miettinen, L. Monticelli, H. Hammaren, P. Bjelkmar, T. Murtola, E. Lindahl and I. Vattulainen, J. Am. Chem. Soc., 2010, 132, 7574 CrossRef CAS.
- M. Javanainen, H. Hammaren, L. Monticelli, J. H. Jeon, M. S. Miettinen, H. Martinez-Seara, R. Metzler and I. Vattulainen, Faraday Discuss., 2013, 161, 397 RSC.
- J. E. Goose and M. S. P. Sansom, PLoS Comput. Biol., 2013, 9, e1003033 CAS.
- B. A. Camley and F. L. H. Brown, Soft Matter, 2013, 9, 4767 RSC.
- F. Yin and J. T. Kindt, Biophys. J., 2012, 102, 2279 CrossRef CAS.
- C. N. Lumb and M. S. P. Sansom, PLoS Comput. Biol., 2012, 8, e1002617 CAS.
- P. J. Stansfeld, R. Hopkinson, F. M. Ashcroft and M. S. P. Sansom, Biochemistry, 2009, 48, 10926 CrossRef CAS.
- M. R. Schmidt, P. J. Stansfeld, S. J. Tucker and M. S. P. Sansom, Biochemistry, 2013, 52, 279 CrossRef CAS.
- J. Karo, P. Peterson and M. Vendelin, J. Biol. Chem., 2012, 287, 7467 CrossRef CAS.
- C. Arnarez, S. J. Marrink and X. Periole, Sci. Rep., 2013, 3, 1263 CAS.
- C. Arnarez, J. P. Mazat, J. Elezgaray, S. J. Marrink and X. Periole, J. Am. Chem. Soc., 2013, 135, 3112 CrossRef CAS.
- D. Sengupta and A. Chattopadhyay, J. Phys. Chem. B, 2012, 116, 12991 CrossRef CAS.
- A. Hung and I. Yarovsky, Biochemistry, 2011, 50, 1492 CrossRef CAS.
- M. Weingarth, A. Prokofyev, E. van der Cruijsen, D. Nand, A. M. J. J. Bonvin, O. Pongs and M. Baldus, J. Am. Chem. Soc., 2013, 135, 3983 CrossRef CAS.
- H. Koldsø and M. S. P. Sansom, J. Phys. Chem. Lett., 2012, 3, 3498 CrossRef.
- R. Pleskot, P. Pejchar, V. Žárský, C. J. Staiger and M. Potocký, PLoS Comput. Biol., 2012, 8, e1002765 CAS.
- A. Koivuniemi, T. Vuorela, P. T. Kovanen, I. Vattulainen and M. T. Hyvönen, PLoS Comput. Biol., 2012, 8, e1002299 CAS.
- K. M. Davies, C. Anselmi, I. Wittig, J. D. Faraldo-Gomez and W. Kuhlbrandt, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13602 CrossRef CAS.
- A. R. Braun, E. Sevcsik, P. Chin, E. Rhoades, S. Tristram-Nagle and J. N. Sachs, J. Am. Chem. Soc., 2012, 134, 2613 CrossRef CAS.
- A. Arkhipov, Y. Yin and K. Schulten, Biophys. J., 2008, 95, 2806 CrossRef CAS.
- X. Periole, T. Huber, S. J. Marrink and T. P. Sakmar, J. Am. Chem. Soc., 2007, 129, 10126 CrossRef CAS.
- D. L. Parton, J. W. Klingelhoefer and M. S. P. Sansom, Biophys. J., 2011, 101, 691 CrossRef CAS.
- E. Psachoulia, D. P. Marshall and M. S. P. Sansom, Acc. Chem. Res., 2010, 43, 388 CrossRef CAS.
- D. Sengupta, A. Rampioni and S. J. Marrink, Mol. Membr. Biol., 2009, 26, 422 CrossRef.
- J. Yoo and Q. Cui, Biophys. J., 2013, 104, 117 CrossRef CAS.
- J. M. Johnston, M. Aburi, D. Provasi, A. Bortolato, E. Urizar, N. A. Lambert, J. A. Javitch and M. Filizola, Biochemistry, 2011, 50, 1682 CrossRef CAS.
- A. Prakash, L. Janosi and M. Doxastakis, Biophys. J., 2011, 101, 1949 CrossRef CAS.
- P. Marius, Y. M. Leung, T. J. Piggot, S. Khalid and P. T. F. Williamson, Eur. Biophys. J., 2012, 41, 199 CrossRef CAS.
- C. P. Chng and S. M. Tan, Proteins, 2011, 79, 2203 CrossRef CAS.
- A. C. Kalli, B. A. Hall, I. D. Campbell and M. S. P. Sansom, Structure, 2011, 19, 1477 CrossRef CAS.
- A. C. Kalli, I. D. Campbell and M. S. P. Sansom, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11890 CrossRef CAS.
- A. C. Kalli, K. L. Wegener, B. T. Goult, N. J. Anthis, I. D. Campbell and M. S. P. Sansom, Structure, 2010, 18, 1280 CrossRef CAS.
- B. A. Hall and M. S. P. Sansom, J. Chem. Theor. Comput., 2009, 5, 2465 CrossRef CAS.
- S. Sharma and A. H. Juffer, J. Am. Chem. Soc., 2013, 135, 2188 CrossRef CAS.
- T. Steinbrecher, S. Prock, J. Reichert, P. Wadhwani, B. Zimpfer, J. Burck, M. Berditsch, M. Elstner and A. S. Ulrich, Biophys. J., 2012, 103, 1460 CrossRef CAS.
- B. A. Hall, J. P. Armitage and M. S. P. Sansom, PLoS Comput. Biol., 2012, 8, e1002685 CAS.
- T. Carpenter, P. J. Bond, S. Khalid and M. S. P. Sansom, Biophys. J., 2008, 95, 3790 CrossRef CAS.
- Y. Oguchi, E. Kummer, F. Seyffer, M. Berynskyy, B. Anstett, R. Zahn, R. C. Wade, A. Mogk and B. Bukau, Nat. Struct. Mol. Biol., 2012, 19, 1338 CAS.
- X. Periole, A. M. Knepp, T. P. Sakmar, S. J. Marrink and T. Huber, J. Am. Chem. Soc., 2012, 134, 10959 CrossRef CAS.
- J. M. Johnston, H. Wang, D. Provasi and M. Filizola, PLoS Comput. Biol., 2012, 8, e1002649 CAS.
- I. Casuso, J. Khao, M. Chami, P. Paul-Gilloteaux, M. Husain, J. P. Duneau, H. Stahlberg, J. N. Sturgis and S. Scheuring, Nat. Nanotechnol., 2012, 7, 525 CrossRef CAS.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380 CrossRef CAS.
- C. Guo, Y. Luo, R. Zhou and G. Wei, ACS Nano, 2012, 6, 3907 CrossRef CAS.
- H. Zhang, H. Wang, G. Xu and S. Yuan, Colloids Surf., A, 2013, 417, 217 CrossRef CAS.
- O. S. Lee, V. Cho and G. C. Schatz, Nano Lett., 2012, 12, 4907 CrossRef CAS.
- S. Gudlur, P. Sukthankar, J. Gao, L. A. Avila, Y. Hiromasa, J. Chen, T. Iwamoto and J. M. Tomich, PLoS One, 2012, 7, e45374 CAS.
- J. Mondal and A. Yethiraj, J. Chem. Phys., 2012, 136, 084902 CrossRef.
- M. Seo, S. Rauscher, R. Pomès and D. P. Tieleman, J. Chem. Theor. Comput., 2012, 8, 1774 CrossRef CAS.
- J. Sorensen, X. Periole, K. K. Skeby, S. J. Marrink and B. Schiott, J. Phys. Chem. Lett., 2011, 2, 2385 CrossRef CAS.
- A. Hung and I. P. P. Yarovsky, J. Mol. Graphics Modell., 2011, 29, 597 CrossRef CAS.
- G. Gronau, Z. Qin and M. J. Buehler, Biomater. Sci., 2013, 1, 276 RSC.
- C. F. Cruz, M. M. Fernandes, A. C. Gomes, L. Coderch, M. Martí, S. Méndez, L. Gales, N. G. Azoia, U. Shimanovich and A. Cavaco-Paulo, Int. J. Cosmet. Sci., 2013, 34, 244 CrossRef.
- Y. Komatsu, M. Fukuda, H. Yamada, S. Kawamoto, T. Miyakawa, R. Morikawa, M. Takasu, S. Yokojima, S. Akanuma and A. Yamagishi, Int. J. Quantum Chem., 2012, 112, 3750 CrossRef CAS.
- A. Gautieri, A. Russo, S. Vesentini, A. Redaelli and M. J. Buehler, J. Chem. Theor. Comput., 2010, 6, 1210 CrossRef CAS.
- T. Li, Y. T. Gu, A. Oloyede and P. K. D. V. Yarlagadda, Comput. Methods Biomech. Biomed. Eng., 2012 DOI:10.1080/10255842.2012.706279.
- S. K. Fegan and M. Thachuk, J. Chem. Theor. Comput., 2012, 8, 1304 CrossRef CAS.
- S. K. Fegan and M. Thachuk, J. Chem. Theor. Comput., 2013 DOI:10.1021/ct300906a.
- L. Zhang, S. Bai and Y. Sun, J. Mol. Graphics Modell., 2011, 29, 906 CrossRef CAS.
- A. Hung, S. Mwenifumbo, M. Mager, J. J. Kuna, F. Stellacci, I. Yarovsky and M. M. Stevens, J. Am. Chem. Soc., 2011, 133, 1438 CrossRef CAS.
- S. R. Euston, Biomacromolecules, 2010, 11, 2781 CrossRef CAS.
- R. P. A. Berntsson, M. K. Doeven, F. Fusetti, R. H. Duurkens, D. Sengupta, S. J. Marrink, A. M. W. H. Thunnissen, B. Poolman and D. J. Slotboom, EMBO J., 2009, 28, 1332 CrossRef CAS.
- S. Yefimov, P. R. Onck, E. van der Giessen and S. J. Marrink, Biophys. J., 2008, 94, 2994 CrossRef CAS.
- E. Deplazes, M. Louhivuori, D. Jayatilaka, S. J. Marrink and B. Corry, PLoS Comput. Biol., 2012, 8, e1002683 CAS.
- M. Louhivuori, H. J. Risselada, E. van der Giessen and S. J. Marrink, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19856 CrossRef CAS.
- O. H. S. Ollila, M. Louhivuori, S. J. Marrink and I. Vattulainen, Biophys. J., 2011, 100, 1651 CrossRef.
- H. Ingolfsson, M. de Melo and S. J. Marrink, 2013, in preparation.
- W. Treptow, S. J. Marrink and M. Tarek, J. Phys. Chem. B, 2008, 112, 3277 CrossRef CAS.
- J. A. Lycklama, M. Bulacu, S. J. Marrink and A. J. M. Driessen, J. Biol. Chem., 2010, 285, 23747 CrossRef.
- A. Nagarajan, J. P. Andersen and T. B. Woolf, Proteins, 2012, 80, 1929 CAS.
- C. Anselmi, M. Grininger, P. Gipson and J. D. Faraldo-Gomez, J. Am. Chem. Soc., 2010, 132, 12357 CrossRef CAS.
- V. Cojocaru, K. Balali-Mood, M. S. P. Sansom and R. C. Wade, PLoS Comput. Biol., 2011, 7, e1002152 CAS.
- H. Shen, I. M. Moustafa, C. E. Cameron and C. M. Colina, J. Phys. Chem. B, 2012, 116, 14515 CrossRef CAS.
- B. A. Hall, J. P. Armitage and M. S. P. Sansom, PLoS Comput. Biol., 2011, 7, e1002204 CAS.
- A. Rzepiela, D. Sengupta, N. Goga and S. J. Marrink, Faraday Discuss., 2010, 144, 431 RSC.
- K. P. Santo and M. L. Berkowitz, J. Phys. Chem. B, 2012, 116, 3021 CrossRef CAS.
- K. P. Santo, S. J. Irudayam and M. L. Berkowitz, J. Phys. Chem. B, 2013, 117, 5031 CrossRef CAS.
- Z. Li, H. Ding and Y. Ma, Soft Matter, 2013, 9, 1281 RSC.
- L. Thogersen, B. Schiott, T. Vosegaard, N. C. Nielsen and E. Tajkhorshid, Biophys. J., 2008, 95, 4337 CrossRef CAS.
- P. J. Bond, D. L. Parton, J. F. Clark and M. S. P. Sansom, Biophys. J., 2008, 95, 3802 CrossRef CAS.
- A. Khalfa and M. Tarek, J. Phys. Chem. B, 2010, 114, 2676 CrossRef CAS.
- H. J. Woo and A. Wallqvist, J. Phys. Chem. B, 2011, 115, 8122 CrossRef CAS.
- W. X. Xu, G. H. Wei, H. B. Su, L. Nordenskiold and Y. G. Mu, Phys. Rev. E, 2011, 84, 051922 CrossRef.
- H. J. Risselada, G. Marelli, M. Fuhrmans, Y. G. Smirnova, H. Grubmüller, S. J. Marrink and M. Muller, PLoS One, 2012, 7, e38302 CAS.
- C. I. E. von Deuster and V. Knecht, Biochim. Biophys. Acta Biomembr., 2011, 1808, 2867 CrossRef CAS.
- P. Gkeka and L. Sarkisov, J. Phys. Chem. B, 2010, 114, 826 CrossRef CAS.
- D. Sengupta, J. Phys. Chem. B, 2012, 116, 14556 CrossRef CAS.
- A. A. Polyansky, P. E. Ramaswarny, P. E. Volynsky, I. F. Sbalzarini, S. J. Marrink and R. G. Efremov, J. Phys. Chem. Lett., 2010, 1, 3108 CrossRef CAS.
- J. N. Horn, J. D. Sengillo, D. Lin, T. D. Romo and A. Grossfield, Biochim. Biophys. Acta Biomembr., 2012, 1818, 212 CrossRef CAS.
- H. Lee, J. S. Choi and R. G. Larson, Macromolecules, 2011, 44, 8681 CrossRef CAS.
- L. X. Peng, L. Yu, S. B. Howell and D. A. Gough, J. Chem. Inf. Model., 2011, 51, 3030 CrossRef CAS.
- L. L. X. Peng, A. Ivetac, A. S. Chaudhari, S. Van, G. Zhao, L. Yu, S. B. Howell, J. A. McCammon and D. A. Gough, Biopolymers, 2010, 93, 936 CrossRef CAS.
- L. L. X. Peng, L. Yu, S. B. Howell and D. A. Gough, Mol. Pharmacol., 2012, 9, 37 CrossRef CAS.
- S. Hezaveh, S. Samanta, A. De Nicola, G. Milano and D. Roccatano, J. Phys. Chem. B, 2012, 116, 14333 CrossRef CAS.
- M. Mazza, R. Notman, J. Anwar, A. Rodger, M. Hicks, G. Parkinson, D. McCarthy, T. Daviter, J. Moger, N. Garrett, T. Mead, M. Briggs, A. G. Schatzlein and I. F. Uchegbu, ACS Nano, 2013, 7, 1016 CrossRef CAS.
- S. Ahmad, B. F. Johnston, S. P. Mackay, A. G. Schatzlein, P. Gellert, D. Sengupta and I. F. Uchegbu, J. R. Soc. Interface, 2010, 7, S423 CrossRef CAS.
- N. D. Winter, R. K. J. Murphy, T. V. O'Halloran and G. C. Schatz, J. Liposome Res., 2011, 21, 106 CrossRef CAS.
- S. J. Lee, P. H. Schlesinger, S. A. Wickline, G. M. Lanza and N. A. Baker, Soft Matter, 2012, 8, 3024 Search PubMed.
- J. Corsi, R. W. Hawtin, O. Ces, G. S. Attard and S. Khalid, Langmuir, 2010, 26, 12119 CrossRef CAS.
- A. K. K. Leung, I. M. Hafez, S. Baoukina, N. M. Bellinveau, I. Zhigaltsev, E. Afshinmanesh, D. P. Tieleman, C. L. Hansen, M. J. Hope and P. R. Cullis, J. Phys. Chem. C, 2012, 116, 18440 CAS.
- A. Y. Shih, P. L. Freddolino, A. Arkhipov and K. Schulten, J. Struct. Biol., 2007, 157, 579 CrossRef CAS.
- A. Catte, J. C. Patterson, D. Bashtovyy, M. K. Jones, F. Gu, L. Li, A. Rampioni, D. Sengupta, T. Vuorela, P. Niemela, M. Karttunen, S. J. Marrink, I. Vattulainen and J. P. Segrest, Biophys. J., 2008, 94, 2306 CrossRef CAS.
- T. A. Vuorela, A. Catte, P. S. Niemela, A. Hall, M. T. Hyvonen, S. J. Marrink, M. Karttunen and I. Vattulainen, PLoS Comput. Biol., 2010, 6, e1000964 Search PubMed.
- O. H. S. Ollila, A. Lamberg, M. Lehtivaara, A. Koivuniemi and I. Vattulainen, Biophys. J., 2012, 103, 1236 CrossRef CAS.
- T. Murtola, T. A. Vuorela, M. T. Hyvonen, S. J. Marrink, M. Karttunen and I. Vattulainen, Soft Matter, 2011, 7, 8135 RSC.
- K. Popovic, J. Holyoake, R. Pomès and G. G. Privé, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2908 CrossRef.
- J. D. Perlmutter, W. J. Drasler, W. Xie, J. L. Gao, J. L. Popot and J. N. Sachs, Langmuir, 2011, 27, 10523 CrossRef CAS.
- A. Y. Shih, P. L. Freddolino, S. G. Sligar and K. Schulten, Nano Lett., 2007, 7, 1692 CrossRef CAS.
- P. M. Kasson and V. S. Pande, PLoS Comput. Biol., 2007, 3, 2228 CAS.
- P. M. Kasson, N. W. Kelley, N. Singhal, M. Vrljic, A. T. Brunger and V. S. Pande, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11916 CrossRef CAS.
- Y. G. Smirnova, S. J. Marrink, R. Lipowsky and V. Knecht, J. Am. Chem. Soc., 2010, 132, 6710 CrossRef CAS.
- D. Mirjanian, A. N. Dickey, J. H. Hoh, T. B. Woolf and M. J. Stevens, J. Phys. Chem. B, 2010, 114, 11061 CrossRef CAS.
- J. Yoo, M. B. Jackson and Q. Cui, Biophys. J., 2013, 104, 841 CrossRef CAS.
- M. Nishizawa and K. Nishizawa, Biophys. J., 2013, 104, 1038 CrossRef CAS.
- J. Yoo, M. B. Jackson and Q. Cui, Biophys. J., 2013, 104, 841 CrossRef CAS.
- A. Raudino, M. Pannuzzo and M. Karttunen, J. Chem. Phys., 2012, 136, 055101 CrossRef.
- S. Baoukina and D. P. Tieleman, Biophys. J., 2010, 99, 2134 CrossRef CAS.
- H. J. Risselada, C. Kutzner and H. Grubmüller, ChemBioChem, 2011, 12, 1049 CrossRef CAS.
- M. Lindau, B. A. Hall, A. Chetwynd, O. Beckstein and M. S. P. Sansom, Biophys. J., 2012, 103, 959 CrossRef CAS.
- A. J. Markvoort and S. J. Marrink, Curr. Top. Membr., 2011, 68, 259 CrossRef CAS.
- S. Baoukina, L. Monticelli, S. J. Marrink and D. P. Tieleman, Langmuir, 2007, 23, 12617 CrossRef CAS.
- S. Baoukina, S. J. Marrink and D. P. Tieleman, Faraday Discuss., 2010, 144, 393 RSC.
- S. Baoukina, L. Monticelli, H. J. Risselada, S. J. Marrink and D. P. Tieleman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10803 CrossRef CAS.
- P. Kulovesi, J. Telenius, A. Koivuniemi, G. Brezesinski, I. Vattulainen and J. M. Holopainen, Biophys. J., 2010, 99, 2559 CrossRef CAS.
- J. Telenius, A. Koivuniemi, P. Kulovesi, J. M. Holopainen and I. Vattulainen, Langmuir, 2012, 28, 17092 CrossRef CAS.
- P. Kulovesi, J. Telenius, A. Koivuniemi, G. Brezesinski, A. Rantamaki, T. Viitala, E. Puukilainen, M. Ritala, S. K. Wiedmer, I. Vattulainen and J. M. Holopainen, Biophys. J., 2010, 99, 2559 CrossRef CAS.
- S. Duncan, I. S. Dalal and R. G. Larson, Biochim. Biophys. Acta Biomembr., 2011, 1808, 2450 CrossRef CAS.
- S. L. Duncan and R. G. Larson, Biochim. Biophys. Acta Biomembr., 2010, 1798, 1632 CrossRef CAS.
- S. Baoukina and D. P. Tieleman, Biophys. J., 2011, 100, 1678 CrossRef CAS.
- A. V. Sangwai and R. Sureshkumar, Langmuir, 2011, 27, 6628 CrossRef CAS.
- S. V. Burov, N. P. Obrezkov, A. A. Vanin and E. M. Piotrovskaya, Colloid J., 2008, 70, 1 CrossRef CAS.
- G. Perez-Sanchez, J. R. B. Gomes and M. Jorge, Langmuir, 2013, 29, 2387 CrossRef CAS.
- S. Jalili and M. Akhavan, Colloids Surf., A, 2009, 352, 99 CrossRef CAS.
- J. F. Kraft, M. Vestergaard, B. Schiøtt and L. Thøgersen, J. Chem. Theor. Comput., 2012, 8, 1556 CrossRef CAS.
- M. Velinova, D. Sengupta, A. V. Tadjer and S. J. Marrink, Langmuir, 2011, 27, 14071 CrossRef CAS.
- S. A. Sanders and A. Z. Panagiotopoulos, J. Chem. Phys., 2010, 132, 114902 CrossRef.
- P. Brocos, P. Mendoza-Espinosa, R. Castillo, J. Mas-Oliva and A. Pineiro, Soft Matter, 2012, 8, 9005 RSC.
- Y. N. Ahn, G. Mohan and D. I. Kopelevich, J. Chem. Phys., 2012, 137, 164902 CrossRef.
- Y. Jiang and J. T. Kindt, J. Chem. Phys., 2007, 126, 045105 CrossRef.
- J. de Joannis, F. Y. Jiang and J. T. Kindt, Langmuir, 2006, 22, 998 CrossRef CAS.
- S. Wu, H. Zhan, H. Wang and Y. Ju, Chin. J. Chem. Phys., 2012, 25, 191 CrossRef CAS.
- G. Rossi, P. François, J. Fuchs, J. Barnoud and L. Monticelli, J. Phys. Chem. B, 2012, 116, 14353 CrossRef CAS.
- M. Hatakeyama and R. Faller, Phys. Chem. Chem. Phys., 2007, 9, 4662 RSC.
- M. Grujicic and B. Pandurangan, J. Mater. Sci., 2012, 47, 3876 CrossRef CAS.
- K. Malek, M. Eikerling, Q. P. Wang, Z. S. Liu, S. Otsuka, K. Akizuki and M. Abe, J. Chem. Phys., 2008, 129, 204702 CrossRef.
- G. Rossi, I. Giannakopoulos, L. Monticelli, N. K. J. Rostedt, S. R. Puisto, C. Lowe, A. C. Taylor, I. Vattulainen and T. Ala-Nissila, Macromolecules, 2011, 44, 6198 CrossRef CAS.
- A. Milani, M. Casalegno, C. Castiglioni and G. Raos, Macromol. Theory Simul., 2011, 20, 305 CrossRef CAS.
- H. Lee and R. G. Larson, J. Phys. Chem. B, 2009, 40, 13202 CrossRef.
- R. G. Uttarwar, J. Potoff and Y. Huang, Ind. Eng. Chem. Res., 2013, 52, 73 CAS.
- G. Rossi, I. G. Elliott, T. Ala-Nissila and R. Faller, Macromolecules, 2012, 45, 563 CrossRef CAS.
- R. S. G. D'Rozario, C. L. Wee, E. J. Wallace and M. S. P. Sansom, Nanotechnology, 2009, 20, 115102 CrossRef.
- A. V. Titov, P. Kral and R. Pearson, ACS Nano, 2010, 4, 229 CrossRef CAS.
- D. Wu and X. Yang, J. Phys. Chem. B, 2012, 116, 12048 CrossRef CAS.
- E. J. Wallace and M. S. P. Sansom, Nano Lett., 2008, 8, 2751 CrossRef CAS.
- N. Patra and P. Kral, J. Am. Chem. Soc., 2011, 133, 6146 CrossRef CAS.
- H. Lee and H. Kim, J. Phys. Chem. C, 2012, 116, 9327 CAS.
- H. Lee, J. Phys. Chem. B, 2013, 117, 1337 CrossRef CAS.
- M. Lelimousin and M. S. P. Sansom, Small, 2013 DOI:10.1002/smll.201202640.
- L. Q. Lin, Y. G. Zheng, H. W. Zhang and Z. Chen, Langmuir, 2011, 27, 8323 CrossRef.
- S. Nangia and R. Sureshkumar, Langmuir, 2012, 28, 17666 CrossRef CAS.
- A. Kyrychenko, G. V. Karpushina, S. I. Bogatyrenko, A. P. Kryshtal and A. O. Doroshenko, Comput. Theor. Chem., 2011, 977, 34 CrossRef CAS.
- B. Song, H. Yuan, S. V. Pham, C. J. Jameson and S. Murad, Langmuir, 2012, 28, 16989 CrossRef CAS.
- E. L. da Rocha, G. F. Caramori and C. R. Rambo, Phys. Chem. Chem. Phys., 2013, 15, 2282 RSC.
- B. Song, H. J. Yuan, C. J. Jameson and S. Murad, Mol. Phys., 2011, 109, 1511 CrossRef CAS.
- J. P. P. Ramalho, P. Gkeka and L. Sarkisov, Langmuir, 2011, 27, 3723 CrossRef.
- P. Gkeka and P. Angelikopoulos, Curr. Nanosci., 2011, 7, 690 CrossRef CAS.
- J. Q. Lin, H. W. Zhang, Z. Chen, Y. G. Zheng, Z. Q. Zhang and H. F. Ye, J. Phys. Chem. C, 2011, 115, 18991 CAS.
- S. Aslan, J. Määttä, B. Z. Haznedaroglu, J. P. M. Goodman, L. D. Pfefferle, M. Elimelech, E. Pauthe, M. Sammalkorpi and P. R. van Tassel, Soft Matter, 2013, 9, 2136 RSC.
- J. Dong and J. Zhou, Macromol. Theory Simul., 2013, 22, 174 CrossRef CAS.
- M. R. Stukan, P. Ligneul, J. P. Crawshaw and E. S. Boek, Langmuir, 2010, 26, 13342 CrossRef CAS.
- M. R. Stukan, P. Ligneul and E. S. Boek, Oil Gas Sci. Technol., 2012, 67, 737 CrossRef CAS.
- D. Sergi, G. Scocchi and A. Ortona, J. Chem. Phys., 2012, 137, 094904 CrossRef.
- C. Gobbo, I. Beurroies, D. de Ridder, R. Eelkema, S. J. Marrink, S. de Feyter, J. van Esch and A. H. de Vries, J. Phys. Chem. C, 2013 Search PubMed , in press.
- D. Fritz, K. Koschke, V. A. Harmandaris, N. van der Vegt and K. Kremer, Phys. Chem. Chem. Phys., 2011, 22, 10412 RSC.
- D. H. de Jong, PhD thesis, Univ. of Groningen, 2013.
- M. Fuhrmans, B. P. Sanders, S. J. Marrink and A. H. de Vries, Theor. Chem. Acc., 2010, 125, 335 CrossRef CAS.
- J. L. MacCallum, M. S. Moghaddam, H. S. Chan and D. P. Tieleman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6206 CrossRef CAS.
- Z. Wu, Q. Cui and A. Yethiraj, J. Phys. Chem. Lett., 2011, 2, 1794 CrossRef CAS.
- R. Baron and V. Molinero, J. Chem. Theory Comput., 2012, 8, 3696 CrossRef CAS.
- R. Baron, A. H. de Vries, P. H. Huenenberger and W. F. van Gunsteren, J. Phys. Chem. B, 2006, 110, 8464 CrossRef CAS.
- R. Baron, A. H. de Vries, P. H. Huenenberger and W. F. van Gunsteren, J. Phys. Chem. B, 2006, 110, 15602 CrossRef CAS.
- W. F. D. Bennett and D. P. Tieleman, J. Chem. Theor. Comput., 2011, 7, 2981 CrossRef CAS.
- W. Shinoda, R. Devane and M. L. Klein, Mol. Simul., 2007, 33, 27 CrossRef CAS.
- C. C. Chiu, R. DeVane, M. L. Klein, W. Shinoda, P. B. Moore and S. O. Nielsen, J. Phys. Chem. B, 2010, 114, 6394 CrossRef CAS.
- B. van Hoof, A. J. Markvoort, R. A. van Santen and P. A. J. Hilbers, J. Phys. Chem. B, 2011, 115, 10001 CrossRef CAS.
- G. Mie, Ann. Phys., 1903, 8, 657 CrossRef.
- G. H. Wei, N. Mousseau and P. Derreumaux, Proteins, 2004, 56, 464 CrossRef CAS.
- T. Bereau and M. Deserno, J. Chem. Phys., 2009, 130, 235106 CrossRef.
- G. G. Maisuradze, P. Senet, C. Czaplewski, A. Liwo and H. A. Scheraga, J. Phys. Chem. A, 2010, 114, 4471 CrossRef CAS.
- T. Ha-Duong, J. Chem. Theor. Comput., 2010, 6, 761 CrossRef.
- D. Alemani, F. Collu, M. Cascella and M. Dal Peraro, J. Chem. Theor. Comput., 2010, 6, 315 CrossRef CAS.
- S. Khalid, P. J. Bond, J. Holyoake, R. W. Hawtin and M. S. P. Sansom, J. R. Soc. Interface, 2008, 5, S241 CrossRef CAS.
- D. Sun and J. Zhou, AIChE J., 2013 DOI:10.1002/aic.14009.
- N. Goga, A. J. Rzepiela, A. H. de Vries, S. J. Marrink and H. J. C. Berendsen, J. Chem. Theor. Comput., 2012, 8, 3637 CrossRef CAS.
- M. Bulacu, N. Goga, X. Periole, G. Rossi, W. Zhao, L. Monticelli, D. P. Tieleman and S. J. Marrink, 2013, in preparation.
- J. W. Chu, G. S. Ayton, S. Izvekov and G. A. Voth, Mol. Phys., 2007, 105, 167 CrossRef CAS.
- S. Nielsen, R. Bulo, P. Moore and B. Ensing, Phys. Chem. Chem. Phys., 2010, 12, 12401 RSC.
- A. J. Rzepiela, M. Louhivuori, C. Peter and S. J. Marrink, Phys. Chem. Chem. Phys., 2011, 13, 10437 RSC.
- C. Peter and K. Kremer, Soft Matter, 2009, 5, 4357 RSC.
- S. C. L. Kamerlin and A. Warshel, Phys. Chem. Chem. Phys., 2011, 13, 10401 RSC.
- T. A. Wassenaar, H. I. Ingólfsson, M. Prieß, S. J. Marrink and L. V. Schäfer, J. Phys. Chem. B, 2013, 117, 3516 CrossRef CAS.
- W. Han and K. Schulten, J. Chem. Theor. Comput., 2012, 8, 4413 CrossRef CAS.
- M. Praprotnik, L. Delle Site and K. Kremer, J. Chem. Phys., 2005, 123, 224106 CrossRef.
- E. Lyman, F. M. Ytreberg and D. M. Zuckerman, Phys. Rev. Lett., 2006, 96, 028105 CrossRef.
- M. Christen and W. F. van Gunsteren, J. Chem. Phys., 2006, 124, 154106 CrossRef.
- W. F. D. Bennett, A. Chen, S. Donnini, G. Groenhof and D. P. Tieleman, Can. J. Chem., 2013 DOI:10.1139/cjc-2013-0010.
- A. De Nicola, Y. Zhao, T. Kawakatsu, D. Roccatano and G. Milano, Theor. Chem. Acc., 2012, 131, 1167 CrossRef.
- I. A. Siuda and L. Thøgersen, 2013, in preparation.
- C. Globisch, V. Krishnamani, M. Deserno and C. Peter, PLoS One, 2013, 8, e60582 CAS.
- J. Corner and A. Aksimentiev, J. Phys. Chem. C, 2012, 116, 3376 Search PubMed.
- M. van Dijk, T. A. Wassenaar and A. M. J. J. Bonvin, J. Chem. Theor. Comput., 2012, 8, 3463 CrossRef CAS.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theor. Comput., 2011, 7, 4026 CrossRef CAS.
- T. H. Schmidt and C. Kandt, J. Chem. Inf. Model., 2012, 52, 2657 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |