 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of solid/vapor interfaces using ambient pressure X-ray photoelectron spectroscopy
D. E.
Starr
a,
Z.
Liu
b,
M.
Hävecker
cd,
A.
Knop-Gericke
c and
H.
Bluhm
*e
aSolar Energy Research, Energy Materials In-situ Laboratory (EMIL), Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, D-12489 Berlin, Germany
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cFritz Haber Institute of the Max Planck Society, Department of Inorganic Chemistry, 14195 Berlin, Germany
dCatalysis for Energy, Group E-GKAT, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, D-12489 Berlin, Germany
eChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: hbluhm@lbl.gov
First published on 19th April 2013
Abstract
Heterogeneous chemical reactions at vapor/solid interfaces play an important role in many processes in the environment and technology. Ambient pressure X-ray photoelectron spectroscopy (APXPS) is a valuable tool to investigate the elemental composition and chemical specificity of surfaces and adsorbates on the molecular scale at pressures of up to 130 mbar. In this review we summarize the historical development of APXPS since its introduction over forty years ago, discuss different approaches to minimize scattering of electrons by gas molecules, and give a comprehensive overview about the experimental systems (vapor/solid interfaces) that have been studied so far. We also present several examples for the application of APXPS to environmental science, heterogeneous catalysis, and electrochemistry.
 D. E. Starr | David Starr obtained his PhD in physical chemistry in 2001 from the University of Washington under the direction of Prof. Charlie Campbell. After post-doctoral stays at the Free University Berlin with Prof. Martin Wolf as an Alexander von Humboldt fellow, Prof. Hajo Freund at the Fritz-Haber-Institute and Dr Hendrik Bluhm at Lawrence Berkeley National Laboratory he joined the Center for Functional Nanomaterials at Brookhaven National Laboratory in 2007. In 2012 he returned to Berlin after accepting a position in the Division of Solar Energy Research in the Helmholtz Center Berlin. |
 Z. Liu | Zhi Liu is a Staff Scientist at the Advanced Light Source, Lawrence Berkeley National Laboratory. He received a BSc from Beijing University, and MSc in Electrical Engineering as well as a PhD in Physics from Stanford University. Before joining the ALS he was a research associate at Stanford University and Stanford Synchrotron Radiation Laboratory. His research interests include in situ characterization of chemical reactions at the gas–solid and liquid–solid interfaces, particularly at the interfaces of electrochemical devices, using ambient pressure photoelectron spectroscopy and other synchrotron based techniques. |
 M. Hävecker | Michael Hävecker is a researcher in the group “Catalysis for Energy” at the synchrotron source BESSY II/Helmholtz-Zentrum Berlin (HZB), Germany. Before joining the HZB he was a postdoctoral researcher in the Department of Inorganic Chemistry/Fritz-Haber-Institute of the Max-Planck-Society, where he still holds a guest appointment. He received his MSc degree in Physics from the University of Hamburg and his PhD degree in Physics from the TU Berlin. His work is centred on the development of novel in situ electron spectroscopy instruments, in particular synchrotron based NAP-XPS and -XAS set-ups and their application in characterising functional materials, in particular catalysts. |
 A. Knop-Gericke | Axel Knop-Gericke is a physicist and leads the group of electronic structure in the Department of Inorganic Chemistry of the Fritz-Haber-Institut der Max-Planck-Gesellschaft in Berlin. His main scientific interests are in heterogeneous catalysis, electrochemistry and the application of synchrotron radiation to study the electronic structure of catalyst surfaces under reaction conditions. He has published about 150 papers and book contributions. He is a member of the editorial board of Catal. Sci. Technol. and a member of the beam time allocation panel of the European Synchrotron Radiation Facility (ESRF) and of the synchrotron radiation facility BESSY. |
 H. Bluhm | Hendrik Bluhm is a Senior Scientist in the Chemical Sciences Division at Lawrence Berkeley National Laboratory. He obtained his MSc in Crystallography from the University of Leipzig and his PhD in Physics from the University of Hamburg. During his Postdoc in the group of Miquel Salmeron at LBNL he worked on the investigation of ice surfaces using scanning probe microscopy as well as the development of synchrotron-based ambient-pressure X-ray photoelectron spectroscopy, a work that he continued during his subsequent stay at the Fritz Haber Institute of the Max Planck Society in Berlin. His current research focuses on the interfacial chemistry of liquid and solid surfaces (e.g., metal oxides, ice) under environmentally relevant conditions of water vapor pressure und trace gas concentration. |
Introduction
The interfaces between gases and solids govern many processes in the environment, energy generation, and heterogeneous catalysis. Examples include the removal of harmful components from automotive exhaust streams,1 the reaction of fuels and oxidizers at the electrodes of solid oxide fuel cells,2 cloud droplet nucleation on atmospheric aerosols particles,3 as well as the uptake and release of trace gases by polar snow packs.4 There are a number of surface sensitive spectroscopies and microscopies that can be used to study vapor/solid interfaces, such as infrared spectroscopy (IR);5,6 vibrational sum-frequency generation (VSFG);7,8 X-ray emission spectroscopy (XES);9 surface X-ray diffraction (SXRD);10 scanning force microscopy (SFM) in both contact11 and non-contact12 modes; scanning tunneling microscopy (STM);13 as well as transmission electron microscopy14 and scanning electron microscopy.15X-ray photoelectron spectroscopy (XPS) is one of the most versatile methods for the investigation of surfaces on the atomic scale.16 It provides quantitative information about the elemental composition and chemical specificity (e.g., oxidation state) of the surface. Due to the strong interaction of electrons with atoms at typical electron energies used in XPS (100 eV–1000 eV), the mean free path of the electrons is only on the order of several monolayers, giving XPS exquisite surface sensitivity.17 However, photoelectrons are also strongly scattered by gas molecules, which complicates the application of XPS at elevated pressures. For instance, the inelastic mean free path of electrons with 100 eV kinetic energy in 1 mbar water vapor is about 1 mm, much shorter than the typical working distance between the sample and the entrance to the electrostatic lens system of an electron analyzer, which is a few centimeters. The attenuation of electrons by gas molecules can be overcome by differential pumping schemes; the most commonly used approaches are discussed in the next section. The use of differential pumping has led to the development of a variety of photoelectron spectrometers that can now operate at up to 130 mbar. This technique is known as ambient pressure or high pressure XPS to distinguish it from vacuum-based X-ray photoelectron spectroscopy.
A timeline for the development of ambient pressure X-ray photoelectron spectroscopy (APXPS) is shown in Fig. 1. Shortly after introducing vacuum-based XPS, Kai Siegbahn's group at Uppsala University built the first APXPS instruments in the early 1970s.18,19 These instruments featured several differential pumping stages between the sample compartment and the electrostatic lens system of a hemispherical analyzer and were mainly used for pioneering investigations of vapor/liquid interfaces. At the end of the 1970s, Joyner and Roberts developed an instrument with a similar differential pumping scheme for measurements of vapor/solid interfaces.20 Two of these systems were built, one located at Cardiff and the other in Novosibirsk in the mid-1980s. Shortly thereafter Grunze and collaborators developed an APXPS instrument which was installed at the University of Maine.21 All of the aforementioned systems used laboratory X-ray sources (anodes). The first synchrotron-based APXPS instrument was developed at Lawrence Berkeley National Laboratory (LBNL) at the end of the 1990s. This instrument featured a differentially-pumped electrostatic lens system, which increases the collection efficiency for electrons without sacrificing differential pumping performance (see next section). With few exceptions most of the instruments that were introduced over the last decade utilize some version of a differentially-pumped electrostatic lens system. The first instrument of this kind was installed at the Advanced Light Source (ALS), beamline 9.3.2,22 and was followed by a second generation of instruments, developed jointly by the Fritz Haber Institute, Berlin, LBNL and Specs GmbH, Berlin, with one instrument installed at Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung m.b.H. (BESSY II, Berlin)23 and a second at the ALS beamline 11.0.2.24 The use of synchrotron-based X-rays has many advantages (increased photon flux, smaller spot size, tunable photon energy) and thus there are now a number of APXPS instruments already operational (ALS,22,24–26 Bessy,23 SSRL,27 MAX-lab,28 NSLS, SLS, Photon Factory29), in commissioning (SOLEIL, ALBA) or under development (Shanghai, Diamond, SPring-8) at synchrotrons around the world. The proliferation of APXPS systems was greatly helped by the recent availability of commercial versions.30–32 With the improvement of small-spot, high flux, monochromatized laboratory X-rays sources a renaissance of laboratory-based APXPS instruments has begun several years ago33–36 (see Fig. 1); this is likely where the strongest growth in this field will be in the future.
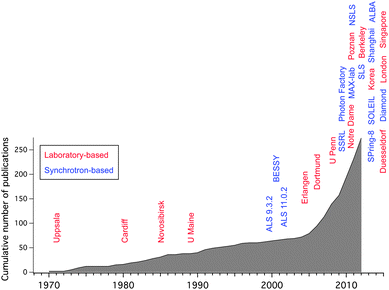 | ||
| Fig. 1 Ambient pressure XPS timeline, showing both the cumulative number of publications and the installation of new instruments. Red labels denote laboratory-based, blue labels synchrotron-based instruments. The dates for the installation of the instruments are approximate and to the best of our knowledge. | ||
Fig. 1 also shows the cumulative number of APXPS publications over time. The increase in the publication rate after the installation of the first synchrotron-based instruments reflects the wider user base that these instruments in general provide (as opposed to a lab-based instrument which is usually used by a single or just a few groups), but it also suggests that APXPS measurements are part of a larger trend in surface science, namely the increasing importance that investigations of surfaces under operating conditions have gained over the last decade. In this review we will give examples for APXPS investigations of vapor/solid interfaces in fields as diverse as environmental science, electrochemistry, and heterogeneous catalysis. We will start with a review of the basic design principles of APXPS instruments.
Technical aspects
In this section we will review the design principles for APXPS instruments on a general level. For a more detailed discussion the reader is pointed to recent review papers on the subject.37–42(A) Differential pumping
The principle obstacle to performing XPS experiments under elevated pressure conditions is scattering of electrons by gas molecules. Elastic scattering dominates at electron kinetic energies below ∼100 eV, while inelastic scattering is the main contribution to signal attenuation above ∼100 eV. The attenuation of the signal I at pressure p compared to the signal I0 at pressure p0 under vacuum conditions is proportional to exp-(σdp), with d the distance that the electrons travel in a gas at pressure p, and σ the scattering cross section, which depends on the chemical composition of the gas phase. Since the gas phase composition and thus the electron scattering cross section is a characteristic of a certain experiment, and the pressure in most cases is sought to be as high as possible, it follows that the distance, d, that the electrons travel through the gas phase needs to be limited to reduce loss of signal. Another requirement is to keep the electron detector and hemispherical analyzer under high vacuum (<10−7 mbar). Since each differential pumping stage provides pressure differentials of about 10−2 to 10−5 (depending on aperture size, pumping speed, and type of gas), it follows that several differential pumping stages are needed if the sample is to be measured at pressures in the mbar range. In addition the X-ray source, be it an X-ray anode or a synchrotron, also needs to remain under high vacuum; therefore the X-rays are admitted to the in situ cell through an X-ray transparent window, most commonly a silicon nitride or aluminum membrane (thickness ∼100 nm), but differential pumping stages between the X-ray source and in situ cell have also been used.The basic approach to all APXPS experiments, pioneered by Siegbahn et al. in their early designs, is the use of a differential pumping scheme, where the sample is located in an in situ measurement cell and is placed close to a differentially-pumped aperture. Since the pressure distribution in front of the aperture is not homogeneous and lower than the background pressure inside the in situ cell, the sample has to be placed at a distance of about two aperture diameters to ensure that the pressure drop across the aperture does not influence the heterogeneous reactions at the sample surface. From this consideration it follows that the size of the incident photon beam is the most important parameter for the determination of the pressure limit and signal strengths in APXPS experiments: a small incident photon spot allows a reduction of the aperture size (ideally matching the size of the photon spot on the sample), which in turn permits a smaller sample-aperture distance, thus reducing the path length of the electrons through the gas environment. A small entrance aperture to the differential pumping system also reduces the gas flow into the subsequent pumping stages and allows for larger secondary apertures with less detrimental effects on the electron collection efficiency.
Before we proceed to discuss various approaches to differential pumping in APXPS, a word is in order on the relative comparison of pressure limits in APXPS, of which there are two: (1) the threshold in situ chamber pressure for the pumping speed of the differential pumping system to cope with the gas flow, and (2) the threshold pressure for obtaining spectra with an acceptable signal-to-noise ratio at reasonable acquisition times. The answer to the second question is obviously the most important for APXPS measurements, and this pressure limit depends on a number of experimental parameters, in particular on the kinetic energy of the electrons (higher KE electrons are less scattered by the gas phase, but also provide less surface sensitivity), the type of gas or gas mixture in the experiment (e.g., the scattering cross sections for some selected gases increases in the order of H2 < He < O2 < CH3OH), the photoelectron emission cross section of the core level under investigation at the given photon energy, and the total flux as well as the beam size of the incident photon beam (the latter one determining the minimum distance between sample and aperture). All these parameters have a bearing on the detected signal, with each one of them easily changing the signal-to-noise levels by a factor of 10 or more. The question “What is the pressure limit in an APXPS experiment?” therefore requires a qualified answer which takes all of the above factors into account.
Fig. 2 shows differential pumping schemes that have been developed for APXPS (please note that these are schematic representations, and may not resemble the real electron trajectories or relative dimensions). The most basic differential pumping system inserts two or more differential pumping stages between the sample location and the entrance to a standard electron energy analyzer input lens (see Fig. 2a). The appeal of this scheme is its simplicity since it does not require any modifications to the electron optical components of a standard electron analyzer. On the other hand it requires a compromise between the differential pumping rates and the detection efficiency: smaller apertures lead to larger pressure differentials but also reduce the acceptance angle of the electrons. Therefore, aperture shapes and sizes are often adjusted to fit the electron trajectories. This approach was used in the APXPS systems developed by Siegbahn et al.,18,19 Joyner & Roberts,20 Grunze et al.,21 and Steinrück et al.34 All of these instruments use laboratory X-ray sources with spot sizes in the millimeter range, resulting in practical operating pressures of up to 1.3 mbar, which is a large step (indeed more than six orders of magnitude) in pressure towards more realistic operating conditions in XPS. A recently developed instrument by Nilsson et al. (SSRL) uses the same approach to differential pumping.27 In this case, however, the incident photon source is an undulator beamline, which provides a tightly focused, high flux photon source (50 μm × 10 μm) and the use of a matching front aperture size of 50 μm diameter. A reduction in the aperture diameter from 1 mm to 50 μm reduces the gas flow into the electrostatic lens system by a factor of 400 (from a purely geometrical point of view) and therefore increases the pressure limit in terms of differential pumping by the same amount (all other parameters, such as pumping speed and conductance, being equal). It also allows the sample to be brought closer to the front aperture and thus reduces the attenuation of the signal by electron scattering with gas molecules. Using this instrument, Pt 4f spectra were obtained at pressures of up to 130 mbar of O2 using photoelectron kinetic energies of ∼930 eV and an acquisition time of 1.5 hours.
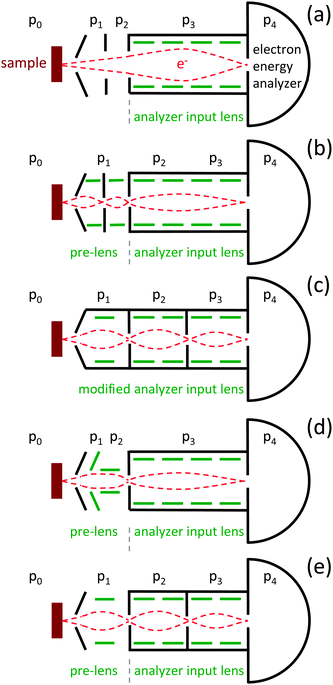 | ||
| Fig. 2 Principle layouts of differential pumping schemes for ambient pressure XPS. Schemes (b–e) use a variation of a differentially-pumped lens system, while scheme (a) uses a set of differentially-pumped apertures in front of a standard analyzer lens. For details see text. | ||
To overcome the trade-off between differential pumping and efficiency of electron detection, a differential pumping system with integrated electrostatic lenses was introduced by Ogletree et al. in 2000 (see Fig. 2b).22 A two stage differentially-pumped electrostatic lens transfers electrons from the sample plane onto the focal plane of a conventional hemispherical electron energy analyzer (Physical Electronics, Inc.). Since the exit aperture of the pre-lens is grounded, the effect of the pre-lens is to move the image plane farther away from the electron analyzer (in this case by ∼18 cm) without changing the electron kinetic energies. Due to the large separation between the differentially-pumped apertures, the pumping speed in the differential stages is sufficient to provide a pressure differential of 10−8 between the in situ cell and the hemisphere, using apertures with diameters of 0.9 mm, 1.5 mm and 3.0 mm (1st, 2nd, and 3rd, respectively). Using electrostatic lenses in between the apertures, electrons are focused onto the aperture planes, thus mostly preserving the acceptance angle of the standard electrostatic lens while at the same time keeping the aperture sizes small and thus increasing differential pumping. This instrument allowed to record spectra at up to 7 mbar of water vapor using 200 eV KE electrons. During the same period of time, Kelly et al. developed a two-stage differentially pumped system using electrostatic grid lenses, based on a laboratory X-ray source, with an upper pressure limit of ∼0.3 mbar.33 Most subsequently designed instruments have also employed differentially-pumped electrostatic lens stages.
The next generation of instruments, jointly developed by the Fritz Haber Institute in Berlin, LBNL, and Specs Surface Nano Analysis GmbH, Berlin, featured a modified electrostatic input lens (as opposed to a pre-lens; see Fig. 2c).23,39 The front lens elements (upstream of the intermediate image plane of the standard lens) of a Phoibos 150 hemispherical analyzer were replaced by two differential pumping/electrostatic lens stages, and the iris aperture (intermediate plane) replaced with a stationary aperture. Aperture sizes in this differentially pumped lens system are 0.9 mm, 2 mm, and 2 mm (1st, 2nd, and 3rd, respectively), each separated by about 25 cm, providing a pumping differential of 10−8 between the in situ cell and the hemisphere. These instruments are operating at BESSY II (ISISS beamline) and the ALS (beamline 11.0.2) and are also able to operate at water vapor pressures above 7 mbar for 200 eV kinetic energy electrons. Fully commercial systems became available in about 2005. While the Omicron analyzer uses scheme (a)34 in Fig. 2, the APXPS spectrometers by Scienta and Specs use differentially pumped electrostatic pre-lenses in front of a standard input lens, following schemes (d)25 and (e),36 respectively.
(B) In situ chambers
While the emphasis in the development of APXPS systems has until recently been mostly on improving differential pumping schemes, the design of precise sample environmental control (e.g., temperature, pressure, gas composition, irradiation with UV) has recently gained in importance. This is partly due to the ready availability of commercial spectrometers, but also due to the expansion of the user base for APXPS instruments to fields outside of traditional surface science, where non-standard UHV environments are required. Most APXPS experiments can be classified by the type of sample preparation into one of the following three categories: (1) in situ sample preparation, i.e., sputtering, annealing, and thin film growth. These are mostly single or polycrystalline samples that can be regenerated in an attached preparation chamber or through heating in certain gases inside the in situ cell. (2) Ex situ sample preparation, e.g., nanoparticles deposited onto a substrate, as well as powder catalysts. (3) Non-traditional samples, such as liquids, but also complex multicomponent devices (e.g., batteries, fuel cells).The basic layout of in situ cells currently in use at APXPS instruments is shown in Fig. 3. The most straightforward design is one where the in situ cell is also the vacuum chamber that separates the sample from the laboratory environment (Fig. 3a). During an experiment the whole chamber is exposed to the gas atmosphere. In this layout the in situ cell is usually connected to a load lock and/or preparation chamber. The advantage of this design is its simplicity; possible disadvantages are cross-contamination between different experiments and the relatively large volume and internal wall areas. It is also difficult to quickly switch between UHV experiments and measurements at elevated pressures, since, once the chamber is exposed to mbar pressures of a gas, without a bake-out it usually takes several hours or days to return to UHV conditions (depending on the type of gas).
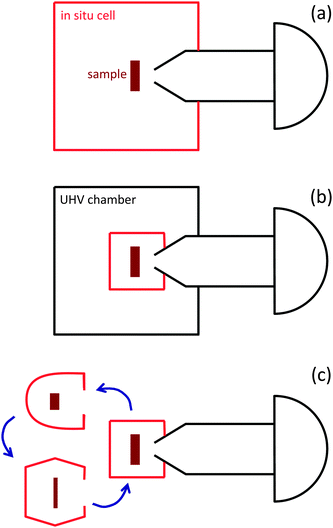 | ||
| Fig. 3 Principle layout of in situ measurement cells currently used in APXPS systems. (a) The analysis chamber/in situ cell is part of a vacuum system (often with load lock and preparation chamber). The whole chamber is exposed to gases during APXPS experiments. (b) The in situ cell is located inside a larger vacuum chamber. Only the in situ cell is exposed to gases during APXPS measurements. This enables to quickly switch between UHV-type and in situ experiments. (c) Exchangeable in situ cells, tailored to a particular experiment, are attached to the analyzer. This approach is best suited for complex or particularly “dirty” sample environments, such as in electrochemistry and investigations of liquid/vapor interfaces. | ||
To overcome this obstacle, a different design uses in situ cells that are placed inside a vacuum chamber and sealed against it during exposure of the sample to gases (Fig. 3b). The sample is transferred into the in situ cell using in-vacuum manipulators.28 This design enables to quickly switch between measurements at elevated pressures and UHV type experiments, and it also reduces the exposed chamber volume and wall area. It should be noted, however, that this is a more complex setup that requires precise manipulation of the sample for the safe transfer into and out of the in situ cell.
Another concept is the use of custom designed, tailor-made sample cells for specific applications (see Fig. 3c). This is particularly advantageous for liquid samples, where cross-contamination and easy cleanup after an experiment are important considerations, as well as for complex sample environments, such as electrochemical cells, where often numerous electrical contacts (in addition to heater and thermocouple) have to be made in a secure way on a small scale for the simultaneous measurement of electrical properties during the APXPS investigations.43 This concept also allows the design of cells with minimal volume and wall area.
APXPS experiments will increasingly be coupled with other characterization methods, which provide simultaneous information about the sample and gas phase. Already now many investigations in heterogeneous catalysis combine APXPS with gas phase analysis using, e.g., mass spectrometers. In those experiments it is important to reduce the rate of dark conversion reactions through the choice of the correct chamber and sample stage materials, as well as infrared heating which selectively only raises the temperature of the sample. Similar considerations will be becoming increasingly more important in other fields of APXPS research, raising the demands for the correct design of the in situ cells and sample environments.
APXPS has been applied to investigate a wide range of samples, including metals, metal oxides, alkali halides, and liquids (not discussed in this review). As in vacuum-based XPS, sample charging of insulating samples poses a significant challenge, in particular since the use of flood guns is not possible under elevated pressure conditions. This problem is partially mitigated by the generation of electrons in the gas phase in the volume that is illuminated by the incident photon beam, which can reduce charging under favorable conditions.40 Homogeneous charging of the surface can be corrected by using the BE of a well-known core level as an intrinsic BE reference. The situation is more complicated for powder samples, where the heterogeneous nature of the sample leads to inhomogeneous charging, which broadens photoelectron peaks and makes the analysis of core-level shifts virtually impossible. Due to the importance of powder samples in heterogeneous catalysis this is a pressing issue for APXPS. The design of differentially-pumped flood guns may be one strategy to overcome this problem.
Examples for the application of APXPS to solid/vapor interfaces
APXPS has been used to study a great variety of samples under a wide range of environmental conditions (e.g., temperature, pressure, gas composition, UV irradiation, electrical bias). In this review we will limit ourselves to the discussion of solid/vapor interfaces. Table 1 provides a list of peer-reviewed reports of APXPS investigations of solids in the presence of gases at pressures above 0.001 mbar.| Sample | Gases | Max. press. [mbar] | Temp. [K] | Year | Ref. |
|---|---|---|---|---|---|
| Pd | |||||
| - (100) | CO, O2 | 0.5 | 295–550 | 2011 | 44 |
| - (100) | CO, O2 | 0.1 | 300–640 | 2012 | 45 |
| - (100) | CO, O2 | 0.7 | 295–680 | 2013 | 46 |
| - (100) | C2H4, CO | 1.3 | 298 | 2013 | 47 |
| - Nanoparticles/SiOx | CO, O2 | 0.5 | 295–550 | 2011 | 44 |
| - Nanoparticles/SiOx | CO, O2 | 0.7 | 295–550 | 2012 | 48 |
| - (111) | O2 | 1.0 | 295–900 | 2005 | 49 |
| - (111) | H2 | 3.0 | 295–623 | 2005 | 50 |
| - (111) | Trans-2-pentene, H2 | 0.7 | 295–523 | 2005 | 51 |
| - (111) | CO | 0.7 | 300 | 2005 | 52 |
| - (111) | CO, CH3OH | 0.7 | 200–400 | 2003 | 53 |
| - (111) | O2, CH3OH | 0.07 | 300–600 | 2012 | 54 |
| - (111) | O2, C2H4 | 0.002 | 330–923 | 2006 | 55 |
| - (111) | O2 | 0.002 | 423–923 | 2006 | 56 |
| - (111) | O2 | 0.4 | 430–872 | 2006 | 57 |
| - (111) | H2, pentyne, pentene | 0.9 | 358–523 | 2006 | 58 |
| - (111) | CH4, O2 | 0.33 | 420–875 | 2007 | 59 |
| - (111) | CO, O2 | 0.7 | 470–770 | 2012 | 60 |
| - Particles/Ga2O3 | H2, O2 | 0.25 | 448–723 | 2012 | 61 |
| - PdZn near surface alloy | CH3OH, H2O | 0.36 | 300–623 | 2010 | 62 |
| - Polycrystalline foil | H2, C3H4 | 1.1 | 353–393 | 2010 | 63 |
| - Polycrystalline foil | H2, alkynes, alkenes | 1.0 | 343–353 | 2008 | 64 and 65 |
| - Polycrystalline foil | H2, alkynes | 7.5 | 343–353 | 2008 | 66 |
| - Polycrystalline foil | H2, pentyne, pentene | 0.9 | 358–523 | 2006 | 58 |
| - Particles on C nanotubes | H2, C3H4 | 1.1 | 353–393 | 2010 | 63 |
| - 5% Pd on C nanotubes | H2, pentyne, pentene | 0.9 | 358–523 | 2006 | 58 |
| - 5% Pd/CeO2 | CO, H2, O2 | 0.5 | 293–523 | 2006 | 67 |
| Pt | |||||
| - (557), (332) | CO, O2 | 0.5 | 295 | 2010 | 68 and 69 |
| - (533) | NH3, NO, O2 | 0.7 | 295–770 | 2008 | 70 |
| - (110) | CO, O2 | 0.5 | 295–473 | 2011 | 71 and 72 |
| - (111) | O2 | 0.5 | 295–620 | 2011 | 73 |
| - (111) | O2, NO, NO2 | 0.25 | 295–520 | 2009 | 74 |
| - (111) | NO | 1.3 | 295 | 2010 | 75 |
| - (111) | C2H4 | 1.3 | 295 | 2013 | 76 |
| - Pt precipitated on Mg(Al)O | H2O, H2, C2H6, O2 | 0.7 | 295–725 | 2007 | 77 |
| - Pt adatoms in FeOx/Pt(111) | CO | 1.0 | 295–510 | 2011 | 78 |
| - on CeO2 | O2, CO | 1.0 | 393–573 | 2007 | 79 |
| - 5% Pt/CeO2 | CO, H2, O2 | 0.5 | 293–523 | 2006 | 80 |
| - Nanoparticles/SiO2/Si(111) | H2, O2 | 0.5 | 373–873 | 2012 | 81 |
| - Nanoparticles on GaN | H2, O2 | 0.5 | 293–800 | 2012 | 82 |
| Au | |||||
| - Nanoparticles/TiO2(110) | O2, CO | 1.0 | 295 | 2010 | 83 |
| - Nanoparticles/TiO2 powder | O2, CO | 0.07 | 295–350 | 2006 | 84 |
| - Nanoparticles on SiO2 and TiO2 | O2, CO | 0.2 | 300–423 | 2009 | 85 |
| - Nanoparticles on SiO2 and TiO2- | O2, NO | 0.5 | 300–473 | 2011 | 86 |
| - Polycrystalline foil | O2, CO | 1.0 | 295 | 2010 | 83 |
| - Evaporated on TiO2(110) | O2, CO | 1.0 | 295 | 2011 | 87 |
| - (111), (310), (533) | NO | 0.005 | 300–500 | 2012 | 88 |
| Ag | |||||
| - Polycrystalline foil | C2H4, O2 | 0.7 | 295–520 | 2006 | 89 |
| - Polycrystalline foil | O2 | 0.4 | 473 | 1979 | 90 |
| - Polycrystalline foil | O2 | 0.01 | 300–700 | 1988 | 91 |
| - Foil, powder, (110), (111) | O2 | 0.2 | 300–773 | 2012 | 92 |
| - Nanoparticles on HOPG | C2H4, O2 | 0.4 | 423–483 | 2011 | 93 |
| - Nanoparticles on Si | C3H6, O2 | 0.5 | 293–493 | 2010 | 94 |
| Rh | |||||
| - Nanoparticles | CO, O2 | 0.5 | 295–550 | 2008 | 95 |
| - Nanoparticles/TiO2 | H2, O2 | 0.2 | 573 | 2011 | 96 |
| - (111) | CO, NO | 0.7 | 300–620 | 2004 | 97 |
| Bi | |||||
| - (0001) | O2 | 0.1 | 145–290 | 1981 | 98 |
| Ru | |||||
| - (0001) | CH3OH, O2 | 0.1 | 350–720 | 2007 | 99 |
| - (0001) | CO, O2 | 0.1 | 350–600 | 2006 | 100 |
| - (0001) | CO | 0.5 | 295–600 | 2013 | 101 |
| - (0001) | CH3OH, O2 | 0.3 | 320–620 | 2007 | 102 |
| - (10-10) | CH3OH, O2 | 0.3 | 320–620 | 2007 | 102 |
| - Polycrystalline foil | CH3OH, O2 | 0.3 | 320–620 | 2007 | 102 |
| - Nanoparticles | CO, O2 | 0.3 | 293–473 | 2012 | 103 |
| V | |||||
| - 8% V/alumina | n-Butane, O2 | 0.9 | 723 | 2008 | 65 |
| Mo | |||||
| - Polycrystalline foil | O2 | 0.2 | 700–900 | 1990 | 104 |
| Ta | |||||
| - Ta/SiO2 | C2H2, O2 | 0.04 | 293–920 | 2012 | 105 |
| - (100) and polycrystalline | O2 | 0.2 | 293–500 | 2010 | 106 |
| Fe | |||||
| - Fe/SiO2 | C2H2, O2 | 0.04 | 293–920 | 2012 | 105 |
| Ni | |||||
| - Nanoparticles on CeO2 | H2 | 1.3 | 293–773 | 2010 | 107 |
| - Polycrystalline foil | C3H8, O2 | 1.0 | 293–1000 | 2013 | 108 |
| Co | |||||
| - (0001) | H2, O2 | 0.2 | 295–650 | 2011 | 109 |
| - (0001) | CH3OH, O2 | 0.3 | 520 | 2010 | 110 |
| - Nanoparticles on carbon support | H2, O2 | 0.2 | 295–650 | 2011 | 109 |
| - Nanoparticles | H2 | 0.1 | 295 | 2011 | 111 |
| Cu | |||||
| - Polycrystalline foil; Zn/Cu | CO2, H2O | 0.2 | 295 | 2008 | 112 |
| - Polycrystalline foil | N2H4 (hydrazine) | 0.01 | 295–380 | 1986 | 113 |
| - Polycrystalline foil | CH3OH, O2 | 0.45 | 295–725 | 2004 | 23 |
| - Polycrystalline foil | CH3OH, O2 | 0.06 | 420–670 | 2003 | 114 |
| - (110) | H2O | 1.0 | 275–520 | 2008 | 115 |
| - (110) | H2O | 1.0 | 295 | 2007 | 116 |
| - (110) | CH3OH, O2 | 1.0 | 320–770 | 2006 | 117 |
| - (111) | H2O | 1.0 | 295 | 2007 | 118 |
| - (111) | SO2 | 0.1 | 173–473 | 1988 | 119 |
| - Ce/Cu(111) | CO2, H2O, CO, H2 | 0.4 | 300–573 | 2013 | 120 |
| - on ZnO/Al2O3 | H2 | 0.25 | 523 | 2012 | 121 |
| - on ZnO | H2 | 0.25 | 298–523 | 2008 | 38 |
| CuGaSe2 | |||||
| - Polycrystalline films | O2, H2O | 0.5 | 295–573 | 2005 | 122 |
| Si | |||||
| - Wafer | O2 | 2 × 10−4 | 295–775 | 2001 | 123 |
| - (100) | O2, H2O | 0.01–1 | 573–803 | 2008 | 124 and 125 |
| C | |||||
| - Nanotubes | C2H2, NH3 | 0.1 | 293–925 | 2011 | 126 and 127 |
| - Nanotubes on Ni | Ar, C2H2, H2 | 0.5 | 573–973 | 2011 | 128 |
| - Nanotubes | O2 | 0.5 | 293–723 | 2010 | 129 |
| - Nanotubes | Butane, O2 | 0.25 | 623–648 | 2008 | 130 |
| - Nanodiamonds | C6H5CH2CH3 | 0.25 | 293–723 | 2010 | 131 |
| - Nanotubes/(Au, Pd, Fe, Ni)/SiO2 | C2H2, H2, O2 | 0.005 | 773 | 2009 | 132 |
| - Nanotubes on CoSi2 | C2H2 | 0.2 | 873 | 2012 | 133 |
| - Nanotubes on Ta | C2H2 | 0.02 | 923 | 2011 | 134 |
| Alkane thiols | |||||
| - Self-assembled monolayer/Au | H2O | 1.0 | 295 | 2008 | 135 |
| POPC lipids | |||||
| - Self-assembled monolayer/SiO2 | H2O | 1.0 | 295 | 2008 | 135 |
| Glycine | |||||
| - on Cu(110) | H2O | 0.5 | 295–500 | 2011 | 136 |
| - on Pt(111) | H2O | 0.3 | 300–550 | 2012 | 137 |
| Alanine | |||||
| - on Cu(110) | H2O | 0.5 | 295–500 | 2011 | 136 |
| TiO2 | |||||
| - (110) (Rutile) | H2O | 1.5 | 265–800 | 2007 | 138 |
| - (110) (Rutile) | NO2, H2O | 0.2 | 300 | 2010 | 139 |
| - Polycrystalline (anatase) | H2O | 0.8 | 295 | 2009 | 140 |
| - (101) Anatase | NO2, H2O | 0.1 | 298 | 2013 | 141 |
| CuO2 | |||||
| - on polycrystalline Cu foil | H2O | 1.5 | 270–295 | 2008 | 142 |
| - on Cu(111) | SO2 | 0.1 | 173–673 | 1988 | 119 |
| CuO | |||||
| - on Cu(111) | SO2 | 0.1 | 173–473 | 1988 | 119 |
| - on Cu(110) | O2 | 1.3 | 700 | 2013 | 143 |
| - on polycrystalline foil | N2H4 (hydrazine) | 0.01 | 295–380 | 1986 | 113 |
| CoOx | |||||
| - CoO on Co(0001) | CH3OH, O2 | 0.3 | 520 | 2010 | 110 |
| - Co3O4 on Co(0001) | CH3OH, O2 | 0.3 | 520 | 2010 | 110 |
| WO3 | |||||
| - In situ grown on Si wafer | O2 | 0.001 | 295 | 2001 | 123 |
| MgO | |||||
| - (100) Thin film on Ag(100) | H2O | 0.5 | 263–573 | 2011 | 144 and 145 |
| In2O3 | |||||
| - Deposited on glass | H2, O2 | 5 × 10−4 | 373–773 | 2006 | 146 |
- ITO (In2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SnO2 = 90:10) :SnO2 = 90:10) |
H2, O2 | 5 × 10−4 | 373–773 | 2006 | 146 |
| (VO)2P2O7 | |||||
| - Pressed pellets | n-Butane, He, O2 | 1.5 | 423–673 | 2005 | 147 and 148 |
| - Pressed pellets | n-Butane, O2 | 0.5 | 593–673 | 2012 | 149 |
| LaxSr1−xCoO3−δ | |||||
| - La0.8Sr0.2CoO3−δ (100) film | O2 | 0.7 | 295–790 | 2012 | 150 |
| - La0.8Sr0.2CoO3−δ pressed pellet | O2 | 0.7 | 295–790 | 2012 | 150 |
| - La0.5Sr0.5CoO3−δ | O2 | 0.2 | 295–673 | 2009 | 151 |
| MoVTeNbO | |||||
| - M1 phase | C3H8, O2, H2O | 0.02 | 323–693 | 2012 | 152 |
| - M1 phase | C3H8, O2, H2O | 0.3 | 293–623 | 2010 | 153 |
| LixV2O5 | |||||
| - Li4+xTi5O12/LiPON/LixV2O5 cell | O2 | 0.66 | 298 | 2012 | 154 |
| CeO2 | |||||
| - (100), Sm doped | H2, H2O, O2 | 0.4 | 690–1000 | 2012 | 155 |
| - 5% Pd/CeO2 | CO, H2, O2 | 0.5 | 293–523 | 2006 | 67 |
| - 5% Pt/CeO2 | CO, H2, O2 | 0.5 | 293–523 | 2006 | 80 |
| - With Au, Pt, Pd Cu nanoparticles | CO/H2O/H2 | 3.0 | 540 | 2012 | 156 |
| FeOx | |||||
| - α-Fe2O3(0001) | H2O | 2.0 | 277–647 | 2010 | 157 |
| - α-Fe2O3 nanoparticles/SiO2 | O2, H2, CO | 0.3 | 295–673 | 2011 | 158 |
| - FeOx film on Pt(111) | CO | 1.0 | 295–510 | 2011 | 78 |
| - FeO nanoparticles on Au(111) | H2O | 0.1 | 295 | 2011 | 159 |
| - α-Fe2O3 particle/film on Au(111) | CO, H2O | 0.2 | 295–473 | 2010 | 160 |
| - Fe2O3; Fe2O3/Cu; Fe2O3/Cu/K/Si | CO, H2 | 0.4 | 293–623 | 2010 | 161 |
| - Fe3O4(001) | H2O | 1.0 | 264–533 | 2013 | 162 |
| SiO2 | |||||
| - Nanoparticles (unsupported) | H2O, N2 | 0.001 | 295 | 2010 | 163 |
| - Native oxide layer on Si(111) | H2O | 4.0 | 263–294 | 2007 | 164 |
| GeO2 | |||||
| - Grown on Ge(100) | H2O | 1.0 | 263–294 | 2012 | 165 |
| Al2O3 | |||||
| - Native oxide layer on Al foil | H2O | 1.5 | 260–295 | 2008 | 142 |
| KI | |||||
| - (100) | Butanol, H2O | 1.6 | 261–295 | 2007 | 166 |
| - (100) | Ozone, H2O, Ar | 0.4 | 295 | 2010 | 167 and 168 |
| KBr | |||||
| - (100) | H2O | 1.5 | 263–295 | 2005 | 169 |
| - Thin film on SiO2 | H2O | 1.5 | 258–295 | 2010 | 170 |
| NaCl | |||||
| - Frozen solutions | H2O | 2.3 | 242–286 | 2010 | 171 |
| - (100) | H2O | 2.0 | 261–308 | 2008 | 172 |
| - (100) | H2O | 1.6 | 263–295 | 2009 | 173 |
| - (100), with Br/Cl = 0.001–0.1 | H2O | 1.6 | 261–295 | 2008 | 174 |
| NaClO4 | |||||
| - Pressed pellets | H2O | 1.7 | 264–295 | 2009 | 173 |
| BaF2 | |||||
| - (111) | H2O | 1.5 | 259–300 | 2012 | 175 |
| RbCl | |||||
| - (100) | H2O | 1.6 | 261–295 | 2012 | 176 |
| RbBr | |||||
| - (100) | H2O | 1.6 | 261–295 | 2012 | 176 |
| Ice (H2O) | |||||
| - Polycrystalline | H2O | 3.9 | 234–271 | 2002 | 177 |
| - Polycrystalline | NO2, H2O | 0.2 | 230 | 2010 | 178 |
| - Polycrystalline | acetone, H2O | 0.3 | 218–243 | 2011 | 179 |
| - Polycrystalline | acetic acid, H2O | 1.0 | 230–240 | 2013 | 180 |
| Electrochemical cells | |||||
| - CeO2−x/Au/YSZ/Pt | H2, H2O | 1.0 | 875–1025 | 2010 | 181 and 182 |
| - CeO2−x/Au/YSZ/Pt | H2, H2O | 1.0 | 875–1025 | 2009 | 183 |
| - CeO2−x/Au/YSZ/Pt | H2, H2O | 0.6 | 973–1023 | 2012 | 184 |
| - Ni/Pt/YSZ | H2, H2O | 0.25 | 975 | 2010 | 43 and 185 |
| - Pt/YSZ | H2, H2O | 0.25 | 825–1025 | 2012 | 186 |
| - Ni/GDC/YSZ/Pt | CH4, H2, O2 | 0.2 | 973 | 2013 | 187 |
| Phtalocyanines | |||||
| - CoPc, FePc | H2, O2 | 0.4 | 293 | 2011 | 188 |
| Alloys | |||||
| - RhPd, RhPt, PdPt | O2, NO, CO, H2 | 0.2 | 573 | 2010 | 189 and 190 |
| - Rh1−xPdx nanoparticles | CO, O2 | 0.2 | 298–518 | 2011 | 191 |
| - RhPd crystal and nanoparticle | O2, NO, CO | 0.2 | 295–623 | 2010 | 192 |
| - AuPd nanoparticles on silicon | O2, CO | 0.3 | 298–473 | 2011 | 193 |
| - CoPt nanoparticles | O2, CO, H2 | 1.0 | 300–400 | 2012 | 194 |
| - Nanoporous RhPd powder | H2, O2 | 0.3 | 423 | 2012 | 195 |
| - PtSn, unsupported | H2, O2 | 0.6 | 393–573 | 2012 | 196 |
| - Pt3Sn(111) | CO, O2 | 0.7 | 300–573 | 2012 | 197 |
| - PtSn, unsupported | O2, H2, C6H12 | 0.5 | 573 | 2011 | 198 |
| - FeTa/SiO2 | C2H2, O2 | 0.04 | 293–910 | 2012 | 105 |
| - Zn/Cu near surface alloy | CH3OH, H2O | 0.35 | 300–693 | 2012 | 199 |
| - ZnPd, unsupported | CH3OH, H2O | 0.15 | 293–773 | 2012 | 200 |
| - PtCo nanoparticles | H2, O2 | 0.2 | 450–520 | 2011 | 201 |
| - PtCo nanoparticles on TiO2 | CH3OH, H2O, H2, O2 | 0.3 | 520–620 | 2012 | 202 |
| - Pd2Ga on carbon nanotubes | H2, C2H2 | 1.1 | 393 | 2011 | 203 |
| - PdGa powder pellets | H2, C2H2 | 1.1 | 400 | 2009 | 204 |
| - PdGa powder pellets | H2, C2H2 | 1.1 | 400 | 2007 | 205 |
| - PdGa near surface alloy | CH3OH, H2O, O2 | 0.3 | 298–573 | 2012 | 206 |
| - Cu2.75Ni0.25Fe | O2, C3H4, H2 | 1.0 | 523–783 | 2011 | 207 |
| - Cu3Fe | O2, C3H4, H2 | 1.0 | 523–783 | 2011 | 207 |
| - CuCo | H2, O2, CO | 6.5 | 523–623 | 2013 | 208 |
| - PtRuCo | CH3OH,H2O,CO,O2, | 0.5 | 570 | 2010 | 209 |
| - PdZn | H2 | 0.3 | 293–543 | 2010 | 210 |
| - PdZn | CH3OH, H2O, CO, O2 | 0.5 | 293–523 | 2012 | 211 |
| - PdZn near surface alloy | CH3OH, H2O | 0.4 | 293–673 | 2010 | 212 |
| - PdIn near surface alloy | CH3OH, H2O | 0.2 | 298–673 | 2012 | 213 |
| - AgCu | O2, C2H4 | 0.5 | 520 | 2010 | 214 |
| - Co0.5Pt0.5 nanoparticles | H2 | 0.1 | 293 | 2011 | 111 |
| - Co0.5Pt0.5 nanoparticles | CO, O2 | 1.0 | 293–418 | 2012 | 215 |
| - RhPd bilayers on SiO2 | CO, NO | 0.03 | 293–670 | 2012 | 216 |
| - PtAg on YSZ | O2, C2H4 | 0.25 | 650 | 2012 | 217 |
| - ZnNi, unsupported powder | CH3OH, H2O | 0.2 | 298–693 | 2012 | 218 |
| - RuCoOx | CO2, H2 | 0.7 | 373–773 | 2012 | 219 |
In the following we present examples of APXPS experiments on solid/vapor interfaces, from highly-ordered single crystals to supported nanoparticles and multicomponent model solid oxide fuel cell devices.
(A) AP-XPS experiments of adsorbates on single crystal surfaces
Surface science experiments on single crystal surfaces under ultra-high vacuum conditions have a long history of providing detailed molecular and atomic level information about adsorbate–surface interactions. For APXPS experiments on single crystal surfaces this level of detail can still be achieved, but in addition the elevated pressure conditions extend the thermodynamic phase-space that can be explored with photoelectron spectroscopy. The advantage of using single crystal substrates stems directly from their well-defined, periodic surface structures. Adsorption studies on single crystal surfaces provide information on site specific (e.g., a-top, bridge, or hollow) adsorption and occupation, as well as the formation of new chemical phases on surfaces upon exposure to gases. In addition, by changing surface orientation and/or miss-cut angle to form vicinal surfaces with varying step densities, factors such as face specificity (e.g. (100) versus (111)) of adsorption and reactions or the role of defects can be systematically addressed. APXPS experiments on single crystal surfaces allow investigation of these molecular-level properties as pressure is increased above UHV and therefore provide a direct connection between the vast knowledge gained from decades of research under UHV conditions to how these systems evolve at elevated pressures. Such detailed information may be more difficult to obtain on structurally more complex systems. Here we provide a few examples of recent APXPS results on well-defined single crystal surfaces.Recent APXPS experiments on the H2O–Cu(110) system have explored the adsorption of water at close to ambient relative humidities.115,116 O 1s spectra collected at 1.3 mbar water vapor pressure and temperatures ranging from 275 K to 520 K (corresponding to relative humidity, RH, from 19% to 0.003%) indicated the presence of pure OH (RH < ∼0.01%) and mixed H2O–OH phases (RH > ∼0.01%). The presence of water molecules at such low RH was attributed to H-bonding between the OH groups and water molecules. The results of both UHV studies and AP-XPS studies for H2O adsorption on Cu(110) have emphasized the important role that OH plays in stabilizing molecular water on the Cu(110) surface through hydrogen bond formation.
The importance of OH groups in stabilizing molecular water adsorption on Cu surfaces is directly illustrated by comparing APXPS results for the adsorption of water on the Cu(110) surface to those on Cu(111) (see Fig. 4).118 For a relative humidity up to 32% (1.3 mbar, 268 K) the Cu(111) surface remains free of both molecular H2O and OH. This is a direct consequence of the higher H2O dissociation barrier of ∼0.3 eV on the (111) surface compared to the (110) surface. The kinetically hindered dissociation of H2O on Cu(111) does not allow the formation of adsorbed OH which act as anchoring sites for molecular H2O adsorption. By pre-adsorbing atomic oxygen on Cu(111), OH groups can be formed on the surface upon exposure to water, which leads to the observation of both OH and molecular H2O at 1.3 mbar and 295 K (see Fig. 4). The difference in water adsorption properties on the Cu(111) and Cu(110) surfaces is a direct consequence of the different activation energies for water dissociation on these surfaces.
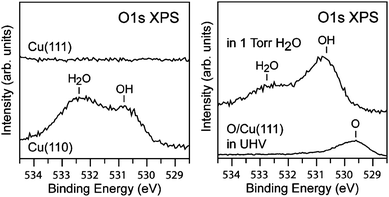 | ||
| Fig. 4 (left) O 1s spectra of Cu(110) and Cu(111) in 1.3 mbar of water at 298 K. The spectra for the Cu(110) surface shows the presence of both OH and H2O while the Cu(111) surface shows the presence of neither OH or H2O. (right) The adsorption of small amounts of O (0.12 ML) on Cu(111) causes the formation of OH groups and therefore H2O adsorption via hydrogen bonding at 1.3 mbar water and 298 K. Reproduced with permission from ref. 118. | ||
These studies highlight the influence of the surface crystallography on the surface chemistry. Depending on the specific catalytic reaction mechanism, these results may have profound implications for heterogeneous catalysis on Cu and other metal surfaces. For reactions that require the formation of OH groups, the reaction may be effectively poisoned if water molecules bind to OH and block access for other molecules to the adsorbed OH. Similarly, the lack of ability for Cu(111) to dissociate H2O to form OH without the presence of adsorbed oxygen may lead to decreased reactivity for those catalysts containing predominantly (111) facets.
Tao et al. recently investigated the restructuring of stepped Pt surfaces, (specifically (557) and (332)) at CO pressures up to 0.7 mbar.68 Using AP-XPS they determined that at 0.7 mbar the CO coverage is approximately one monolayer. This is nearly twice the amount of CO adsorbed on the Pt(557) surface at 7 × 10−9 mbar. Along with the increase in CO coverage, O 1s and Pt 4f spectra showed a substantial increase in intensity at 533.1 eV (O 1s) and 72.15 eV (Pt 4f) binding energies (see Fig. 5). These binding energies are higher than those observed for CO adsorbed in Pt a-top sites.241 In general, O 1s and C 1s binding energies shift to higher values as the coordination of CO to the surface decreases. For example, on Pt(111) the O 1s binding energy of CO bound to Pt bridge sites is 531.0 eV as compared to 532.7 eV for a-top bound CO.241 Similarly, a lower coordinated Pt atom should lead to higher Pt 4f binding energies. Thus, the higher binding energies observed by Tao et al. are consistent with CO bound to low-coordinated Pt sites. The additional features observed in the Pt 4f and O 1s spectra were reversible as evidenced by the consistent changes in peak intensity as the pressure was cycled between 7 × 10−9 mbar and 0.7 mbar. The observed CO coverage changed from ∼0.5 to 1.0, respectively, at those pressures. Complimentary STM experiments indicated a dramatic surface restructuring at pressures above 0.1 mbar and the formation of triangular nanoclusters of approximately 2.2 nm by 2.1 nm in size (see Fig. 5).68 The formation of these nanoclusters leads to an increase in the number of under-coordinated Pt atoms on the surfaces, which act as new adsorption sites for CO at elevated pressures. Such dramatic restructuring of the Pt(557) surface was proposed to be driven by a relaxation of repulsive CO–CO interactions as the CO coverage increased to nearly 1.0 and confirmed by DFT calculations. These results highlight the dynamic nature of Pt at elevated pressure conditions. Further, the level of detail in these studies provides a molecular-level understanding of the mechanism responsible (i.e., CO–CO repulsion) for dynamic changes at the surface under reaction conditions. Extending these kind of investigations to structurally more complex model and technical catalysts may provide new insights into the dynamic nature of the Pt surface and its role in the reactivity of supported Pt catalysts.
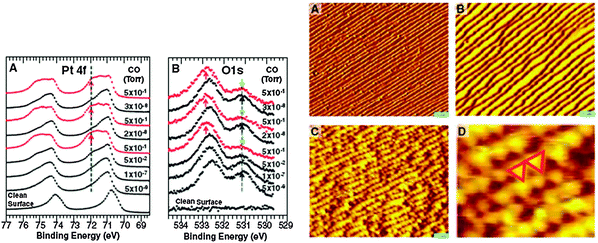 | ||
| Fig. 5 (left) Pt 4f and O 1s spectra for CO adsorption on Pt(557) at CO pressures indicated in the figure. The presence of high binding energy peaks at 72.15 eV and 533.1 eV in the Pt 4f and O 1s spectra respectively are likely due to the adsorption of CO at under-coordinated Pt atoms on the stepped surface. (right) STM images at UHV conditions (A), 7 × 1018 mbar CO (B) and 1.3 mbar CO pressure (C and D) showing the formation of Pt nanoclusters at elevated pressure conditions. Reproduced with permission from ref. 68. | ||
Another APXPS study has addressed CO oxidation over Pt(110) at elevated pressures, motivated by earlier high-pressure STM and gas analysis investigations by Hendriksen et al. who observed a roughening of the Pt(110) surface during CO oxidation, which was correlated to an enhanced rate of CO2 production.242 This roughening occurred at high O2/CO ratios of >45, pressures of ∼0.5 bar, and temperatures of 425 K and was therefore assumed to be associated with the formation of Pt-oxide. Hendriksen et al. concluded that, at high pressures and O2/CO ratios, CO oxidation may follow the Mars–Van Krevelen mechanism as opposed to the Langmuir–Hinshelwood mechanism. Recent APXPS experiments on this system, however, indicate that the actual mechanism responsible for CO-oxidation over Pt(110) may be sensitive to the precise conditions.71 Chung et al. used APXPS to study CO oxidation over Pt(110) at a variety of CO and O2 pressures and temperatures.71 When 0.26 mbar of CO is introduced into the chamber at room temperature both C 1s and O 1s spectra show the presence of CO adsorbed in both a-top and bridge sites in agreement with previous UHV studies. Upon addition of 0.26 mbar of O2 at room temperature the population of bridge sites decreased, and continued to decrease even further when the Pt(110) crystal was heated to 100 °C, when nearly all bridge-bound CO was removed. CO2 production began at about 120 °C. At 150 °C CO2 is still produced and the surface remained CO covered and there was no observation of either chemisorbed oxygen or Pt-oxide.
Chung et al. addressed the possibility of the presence of chemisorbed oxygen or Pt-oxide by exploring the effects of different O2/CO ratios at 150 °C on the CO coverage. Introduction of 0.23 mbar of O2 at 150 °C in the absence of CO created a Pt(110) surface covered with chemisorbed oxygen. Upon introduction of CO to 0.30 mbar, CO2 production instantly increased but then decreased with time. Once CO2 production stabilized, O 1s spectra revealed that the chemisorbed oxygen was completely removed. This demonstrates that an oxygen covered Pt(110) surface is not stable under CO rich conditions, but that the oxygen-covered surface is more reactive than a CO covered surface due to the higher rate of CO2 production at short time intervals following the introduction of CO. Upon reduction of the CO pressure to 0.18 mbar (i.e., in a more O2 rich environment), the rate of CO2 production increased but the Pt surface remained covered with CO, although at a slightly lower coverage. At these conditions no oxygen or Pt-oxide was observed in the O 1s spectra. These results demonstrate that even under O2 rich conditions at these pressures CO oxidation may still occur via a Langmuir–Hinshelwood mechanism. However, these results should not be taken as definitive evidence that CO-oxidation occurs via a Langmuir–Hinshelwood mechanism, but instead that the precise reaction mechanism is sensitive to the O2/CO ratio as well as total pressure.
Recent APXPS measurements have addressed these issues. The O 1s core level spectra of both a Ag(110) single crystal surface and of a Ag powder sample (nominal particle size 45 μm) are shown in Fig. 6.92 The oxygen species at the surface of both catalysts change with the sample temperature and, in the case of the powder sample, also with the exposure time to 0.25 mbar O2 at 180 °C. At the lowest temperature (150 °C) the most abundant species at both surfaces is Oα1, which is associated with the formation of a p(4 × 4) oxygen overlayer. At higher temperature three other oxygen species become more prominent.245 Oα2 (nucleophilic oxygen) is an oxide-like species located at steps on the surface.246 It is important for the activation of C–H bonds in hydrocarbons and is therefore involved in the total oxidation reaction. The peaks at the higher binding energy, Oα3 (electrophilic oxygen) and Oβ, are assigned to atomically adsorbed oxygen at the Ag surface89 and oxygen located in the subsurface region in the Ag catalysts,247 respectively. Electrophilic oxygen activated the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in olefins and is thus involved in selective oxidation reactions.
C bond in olefins and is thus involved in selective oxidation reactions.
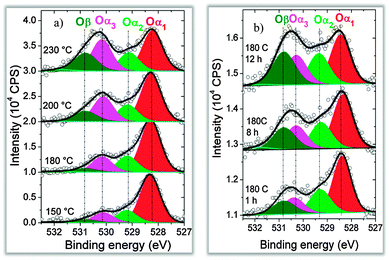 | ||
| Fig. 6 O 1s spectra following the formation kinetics of low temperature oxygen species on silver. Left: Ag(110) under 0.25 mbar O2 at different temperatures. Right: silver powder with 45 μm particle size at 180 °C after different reaction times with O2. Reproduced with permission from ref. 89. | ||
To characterize the nature of the different oxygen species, Ag 3d core level spectra were measured as well (Fig. 7). The preparation of the surfaces was done under conditions favoring mainly the formation of a single oxygen species in the O 1s spectrum. The amount of ionic silver (Ag+) as a function of the different oxygen species is shown in the bottom graph in Fig. 7. Different oxygen species clearly lead to different levels of charge transfer from the Ag to the oxygen. The degree of Ag+ formation in the presence of Oα1 and Oα2 is much higher than that for electrophilic oxygen, indicating different roles for the different oxygen species in the ethylene epoxidation reaction. The strongly charged oxygen species Oα1 and Oα2 activate the C–H bonds, leading to CO2 formation, while the less charged electrophilic oxygen activates the CC bond. The different electronic structure of nucleophilic oxygen (Oα2) and electrophilic oxygen (Oα3) is strongly influenced by the subsurface oxygen species Oβ. In the presence of Oβ there are fewer electrons available that can be transferred to an adsorbed oxygen species: thus, the adsorbed oxygen is less charged and Oα2 is formed. In the absence of subsurface oxygen, more electrons can be transferred from Ag to adsorbed oxygen atoms, leading to the formation of highly charged nucleophilic oxygen. Fig. 7 also shows the amount of Ag+ as a function of another oxygen species, Oγ, which is formed at about 500 °C and is assigned to oxygen atoms replacing Ag atoms in the surface. Due to the high formation temperature, Oγ is only relevant for methanol oxidation. The data in Fig. 6 and 7 demonstrate that under constant oxygen partial pressure and catalytically relevant temperatures the silver–oxygen system shows a dynamic behavior, with the formation of different oxygen species as a result of oxygen incorporation in the subsurface region.
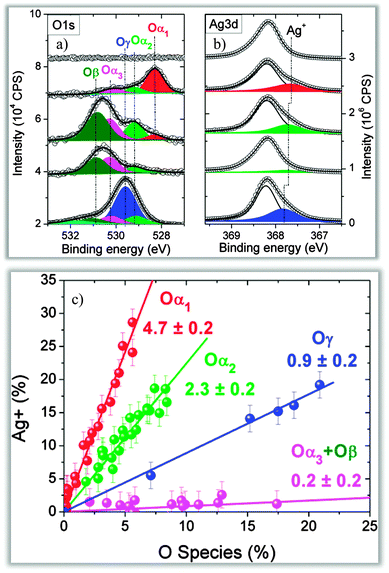 | ||
| Fig. 7 (a, b) O 1s and Ag 3d spectra showing the changes in the abundance of Ag+ depending on the oxygen species present on the silver surface. (c) Quantitative correlation of the amount of Ag+ related to different oxygen species. Reproduced with permission from ref. 89. | ||
To summarize the examples of APXPS measurements on single crystal samples, these studies highlight the utility of APXPS to provide detailed information on adsorbate-induced restructuring of surfaces, orientation-dependent adsorption properties of surfaces, and the pressure-dependence of surface reaction mechanisms. Studies on single crystal surfaces have the advantage of retaining the molecular or atomic level information available in traditional UHV surface science experiments. The level of detail provided by these studies may have been difficult to achieve on more structurally complex surfaces such as model or technical catalysts. Studies on single crystal surfaces at elevated pressures provide a direct means to bridge the pressure gap between UHV surface science experiments and more realistic catalytic operating conditions. We now proceed to discuss measurements on more complex systems.
(B) Investigation of nanoparticles used in CVD processes for carbon nanotube CNT growth
Carbon nanotubes (CNTs) have many potential applications, including supercapacitors,248 field-emission devices249 and vertical interconnects (vias) for integrated circuits,250 which require growth of vertically aligned CNTs on electrically conductive substrates. CNTs can be grown using chemical vapor deposition (CVD).251 CVD is an established technique to synthesize CNT “forests” (i.e., a dense layer of vertically aligned CNTs) on insulation oxide supports such as silica and alumina.252 The growth on conductive substrates such as metals, metal-nitrides, and metal-silicides is more difficult and less studied, mainly because of the much higher surface energy of metals compared to insulating oxides, which inhibits the catalyst film from transforming itself into nanoparticles during temperature treatment.253 In addition, the metallic support has to retain its conductivity and functionality during the CVD process at elevated temperatures in the presence of reactive gases. However, metals are often reactive under such conditions.127 Therefore the support has to fulfill the requirements of favorable surface energetics for high density nanoparticle formation and chemical stability against carbide-formation (from the gas that serves as the carbon source) or oxidation (from residual oxygen or water).Cobalt-silicides are promising catalysts for the synthesis of CNT forests and were recently investigated using APXPS and in situ X-ray diffraction (XRD) to study the evolution of the silicide-catalyst–gas system during CNT forest growth. The Co silicide was prepared as follows (Fig. 8): a 200 nm thick polycrystalline Si (“poly-Si”) film was deposited by CVD onto a crystalline Si(100) wafer (not shown). Then a 15 nm thin layer of Co was sputtered on top of the poly-Si. This structure was capped by a TiN layer and annealed at temperatures below 500 °C for less than three minutes, followed by the removal of the TiN capping layer. The annealing induces an inter-diffusion of Co and Si, resulting in the formation of CoSi. The TiN layer permits the development of a rather smooth silicide surface. Usually a second annealing step is required to transform CoSi into the highly conductive CoSi2.254 The second annealing step can be avoided here, since the transformation of CoSi to CoSi2 can be done simultaneously with the CNT growth over a pressure range from 0.1 mbar to 1 bar as described below.126
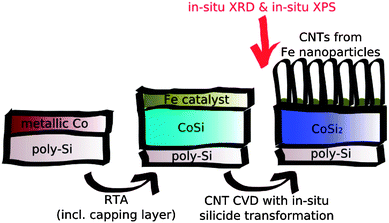 | ||
| Fig. 8 Schematic representation of the in situ preparation of CNT forests and the silicidation of high resistivity CoSi to low resistivity CoSi2 during CNT growth. Reproduced with permission from ref. 126. | ||
After the deposition of a 1 nm thick Fe layer the evolution of the sample was studied by APXPS during the CVD process in the mbar range. Fig. 9 shows XPS spectra (Si 2p, Co 2p and Fe 2p) during the CVD process. The as-loaded surface consists of Co oxide and Si oxide, since the samples were exposed to air during the transport between process steps. The Fe film is completely oxidized as well. The Si 2p spectra show an intensity increase when the sample is heated to 650 °C in NH3. This treatment is required in order to reduce the Fe nanoparticles, since only Fe metal catalyzes the growth of CNTs. The intensity increase is due to the reduction of the Co oxide and the removal of C contamination on the surface. A new peak at a BE of 99.7 eV indicates the formation of CoSi2. However, most of the Si remains oxidized. The Co is nearly completely reduced. A shoulder at the high binding energy side of the metallic Co peak at 778.9 eV indicates the formation of CoSi2 which forms nano-crystalline domains.132
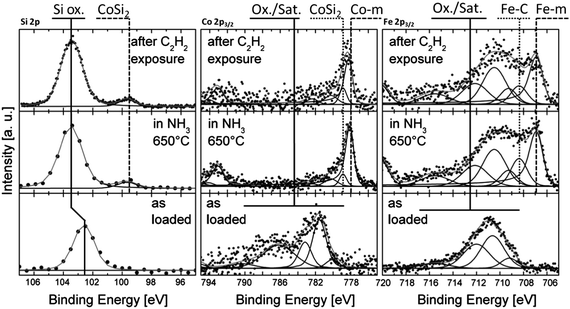 | ||
| Fig. 9 (From left to right) APXP spectra of the Si 2p, Co 2p3/2 and Fe 2p3/2 regions showing the evolution of the surface (Fe, Co; Ekin(e−) = 150 eV) and near surface (Si; Ekin(e−) = 1000 eV) chemistry under low pressure CVD. Reproduced with permission from ref. 126. | ||
During annealing the Fe film decomposes into nanoparticles. In contrast to the case where Fe is supported on and Al2O3 substrate, here only part of the Fe particles are reduced to metal by the treatment in NH3 (note the peak at 706.9 eV in the Fe 2p spectrum in Fig. 9). The Co silicide support keeps a portion of the Fe nanoparticles in their oxidized state. The combination of Co silicide substrate and Fe catalysts is one of the most promising methods for high density CNT growth on insulators to date. The interfacial oxide layer prevents the catalyst nanoparticles to diffuse onto the surface and to agglomerate to bigger clusters. An atomic force microscopy study has shown that CoSi2 inhibits sintering of the Fe nanoparticles, which facilitates efficient growth of CNT forests.255 The APXPS results shown in Fig. 9 indicate that the Fe nanoparticles are bound to the CoSi2 substrate through a similar interfacial interaction as observed for Fe nanoparticles supported on Al2O3.
The addition of 10% C2H2 to the NH3 (top spectra in Fig. 9) results in the fast evolution of sp2 and sp3 type bonds in the C 1s spectra, indicating CNT growth. The growth rate under the experimental conditions was too fast to measure high resolution APXP spectra during C2H2 exposure. Constant exposure of the sample to C2H2 leads to CNT forests with thicknesses of up to 40 μm, much thicker than the information depth in APXPS experiments. To monitor the chemical state of the interface during growth, short pulses of C2H2 (10−2 mbar for 10 s) were admitted to the chamber, which allows only sparse growth of CNTs, but enables probing the silicide surface. After a C2H2 pulse this surface is comprised of metallic Co, CoSi2, and some SiO2, with the state of Fe not affected by C2H2 exposure. XRD measurements (not shown) of the same sample reveal the exclusive presence of Si and CoSi2, thus proving that the oxides observed in the APXP spectra are located only at the surface and do not extend into the bulk of the film.
(C) Application of APXPS to electrochemistry
The need for clean, secure, and sustainable energy sources has created a surge in research and development of electrochemical devices, such as batteries, fuel cells, and super capacitors. Many roadblocks to higher performing electrochemical devices are not just due to engineering challenges, but also due to limited information on the fundamental processes in electrochemical devices at the molecular level, which requires experimental tools for observing electro-chemical processes directly at the interfaces where they occur in situ. Fuel cells, which were invented more than 100 years ago, are a case in point. Solid oxide fuel cells (SOFC) in particular offer several key advantages, including high efficiency, high tolerance to poisoning of the catalysts, reformation of hydrocarbon fuels, and the possibility of burning hydrocarbon fuels directly; however, despite these attractive features SOFCs have not yet found wide-spread use in everyday applications and devices.Traditional electrochemical evaluation of electrode overpotentials employs, e.g., voltammetry and electrochemical impedance spectroscopy. These techniques provide valuable information on the global electrode overpotentials and resistances in SOFCs. Despite these advances in electrochemical measurement and modeling, our understandings of the rate limiting steps in SOFCs, in particular the cathode oxygen reduction mechanism, the physics governing electrode overpotential losses, and dimensions of the electrochemically active regions of mixed ionic electronic conducting electrodes remain largely circumstantial to date. Many of these challenges are due to the inherently convoluted nature of electrochemical and chemical processes and the lack of suitable in situ techniques to probe these issues at relevant temperatures and pressures. As pointed out by Adler in 2004: “new in situ analytical techniques are needed, particularly which can be applied at ambient pressures, that can probe what is happening in an electrode as a function of temperature, PO2, polarization, local position, and time”.256
To address these challenges using photoelectron spectroscopy, scientists from the ALS, University of Maryland, and Sandia National Laboratory began using APXPS as an operando tool to study solid oxide electrochemical cells (SOCs) in 2008. APXPS allows the study of the surfaces in situ with elemental and chemical specificity. By scanning a focused X-ray spot across the surface or by using an imaging mode of the photoelectron spectrometer, local elemental and chemical information across the sample surface can be obtained. In addition, local electrical potential changes at the surface can be determined from the changes of the kinetic energy of core level photoelectron peaks. The correlation of local chemical processes with local electrical potentials under operating conditions is crucial for an understanding of fundamental processes in SOC devices.
The first experiments were performed on a SOC cell in which a Au–ceria working electrode (WE) and a Pt counter electrode (CE) were deposited on a single crystal YSZ electrolyte disk. In such a planar cell design, all components are exposed to the surrounding gas atmosphere and located on the same side of the electrolyte disk to enable APXPS access the electrode–electrolyte interfaces. Since the oxidizer and fuel are in the same volume, a bias is applied between the Pt CE and ceria WE to drive the electrochemical reactions. This cell was mounted inside the APXPS endstation at beamline 11.0.2 at the ALS24 and heated up to 750 °C in a 1:1 gas mixture of H2 and H2O at a total pressure of ∼1.3 mbar. The results of these measurements are shown in Fig. 10 and prove the validity of the experimental concept: a clear correlation between gradients in the electrical potential and changes in the surface chemistry (namely the Ce oxidation state) is observed.183
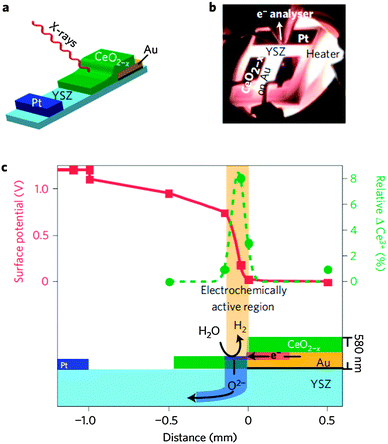 | ||
| Fig. 10 (a) Schematic layout of a solid oxide cells with a 200 nm thick Pt counter electrode, a 300 nm thick Au current collector on top of a 30 nm thick insulating alumina film (black), and a 50, 100 or 250 nm thick ceria working electrode patterned onto a polycrystalline YSZ substrate. This geometry exposes all cell components to the X-ray beam. The drawing is not to scale. (b) During operation, the cell is heated to ∼700 °C in the APXPS measurement position, close to the first aperture of the electrostatic lens system in a 1:1 gas mixture of H2 and H2O at a total pressure of about 1.3 mbar. (c) A 250 nm-thick ceria anode converts H2O to H2 and O2− in a 150 μm region at a cell potential of +1.2 V. APXPS reveals local surface potentials (red squares) and the relative change of the Ce oxidation state from equilibrium (green circles) in this region. Reproduced with permission from ref. 181. | ||
These techniques were subsequently applied to an new version of a model Ceria-YSZ-Pt SOC181 and Ni-YSZ-Pt SOC.185 A new endstation at ALS Beamline 9.3.225 and a special sample holder were utilized as well.43 In all of these experiments, the WE (ceria/Au or Ni) was grounded and the bias voltage was applied to the Pt CE using a potentiostat. Two-probe linear sweep voltammetry and electrochemical impedance spectroscopy experiments were conducted simultaneously to the APXPS measurements.
Fig. 10a shows the schematic layout of a planar ceria/YSZ/Pt cell geometry and simplified experimental setup.181 Ceria working electrodes of different thicknesses (50, 100, and 250 nm) are sputtered onto a gold current collector and only extend onto the YSZ electrolyte towards the Pt counter electrode. Such a cell design mandates that oxygen ions move in the vertical direction through ceria and electrons (polarons) move in the lateral direction across the ceria. Therefore, the ionic and electronic potential changes can be separated and measured individually. Using this specially fabricated single chamber SOC, the authors of ref. 181 have demonstrated that the active electrochemical region on ceria extends 150 μm away from the current collector and that significant shifts from the equilibrium surface Ce3+/Ce4+ concentrations are needed to drive the electro-oxidation of H2 and the electrolysis of H2O (see Fig. 10c). The correlation between local potential losses and local chemical state changes were obtained directly from working SOC devices.
Fig. 11 is taken from a study of a Ni-YSZ-Pt SOC,185 where the new endstation at ALS Beamline 9.3.2 was used.25 The spectrometer was optimized in this project to perform 1D spatially-resolved APXPS. These measurements probed the individual overpotentials (such as between Ni and YSZ, YSZ and Pt) in SOC devices, allowing a direct correlation of changes in the individual overpotentials with the applied bias in terms of the different electro-catalytic activities of Ni and Pt for the H2O splitting and H2 oxidation reactions. It was found that H2O splitting is faster than H2 oxidation on Ni, while on Pt the H2 oxidation reaction proceeds more rapidly than H2O splitting.
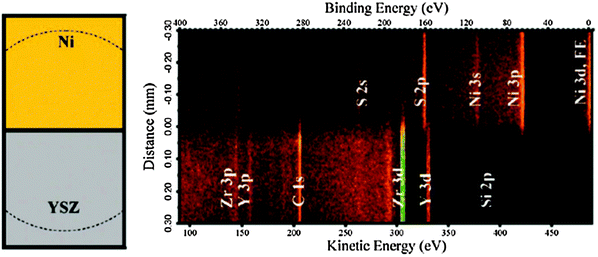 | ||
| Fig. 11 Left panel: top-view schematic of the SOEC Ni/YSZ interface measured in the APXPS image (right). The dashed lines shows the field-of-view (diameter ∼0.6 mm). Right panel: the photoelectron binding energy versus real-space distance around the cell's three-phase boundary during operation at zero bias. Core level XPS peaks of Ni, YSZ, their impurities and the Ni Fermi edge (FE) are labeled. Intensities (counts) are displayed using a false-color scale. The binding energy scale is referenced to the Fermi edge of the grounded Ni electrode. Reproduced with permission from ref. 185. | ||
APXPS is a unique non-contact tool to probe electrode/gas and electrode/electrolyte interfaces as a function of temperature, pressure, polarization, local position, and time, which makes it also an excellent method to study fundamental processes in model battery devices, such as Li–O2 battery cells.154 The combination of local measurements of the surface chemistry and electrical potentials using APXPS with bulk measurements of the device performance using voltammetry and electrochemical impedance spectroscopy is a promising strategy for gathering fundamental mechanistic information on electrochemical devices which may facilitate advances in the design of electrochemical devices.
Conclusions and outlook
Ambient pressure X-ray photoelectron spectroscopy provides a wealth of information on vapor/solid interfaces under reaction conditions, from the elemental composition and chemical specificity (oxidation state, functionalization), to the local electrical potentials and work functions. As the preceding examples and Table 1 demonstrate, this allows molecular scale investigations of interfacial phenomena in a wide range of scientific areas, including fundamental surface science, environmental science, electrochemistry, and industrial catalysis. As the technique has matured and broadened its user base over the last decade (both in total numbers of investigators as well as in the breadth of applications), mainly through the commissioning of new endstations at synchrotrons and now increasingly through the installation of new laboratory-based instruments, the task at hand is the further development of in situ cells that allow to measure samples under more realistic and complex experimental conditions, and combining APXPS with simultaneous measurements using other techniques to, e.g., monitor surface as well as bulk properties, and correlate the surface chemistry with the gas phase composition.Several new developments promise to expand APXPS to study phenomena that have hitherto been difficult or impossible to investigate:
(1) High kinetic energy APXPS (with photoelectron kinetic energies exceeding 5 keV) utilizes the increased mean free path of electrons with increasing KE. This will allow to study the chemistry of the subsurface region under reaction conditions, which may differ from that of the surface and plays an important role in heterogeneous catalysis and liquid/vapor reactions. Vacuum-based high kinetic energy XPS has already been used to investigate buried interfaces at depths larger than 10 nm.257 One of the most important buried interfaces is that between a solid and a liquid, which drives many processes in electrochemistry, corrosion, and environmental science.258 The inelastic mean free path of electrons in liquid water is about 20 nm at 10 keV kinetic energy, making it feasible to penetrate about 70 monolayers of water, thus approaching conditions at bulk water/solid interfaces.259 The preparation of thin water films with thicknesses of 10 to 20 nm is an experimental challenge, though. Another advantage of high kinetic energy APXPS is the reduced scattering of photoelectrons in the gas phase, which will indeed make it possible to obtain XPS spectra at atmospheric pressure.
(2) Increased spatial resolution in APXPS is crucial for the understanding of the complex chemistry at the surface of multi-component samples and devices, such as supported catalysts, electrochemical devices, as well as natural mineral and aerosols. The spatial resolution in APXPS experiments is in general determined by the dimension of the incident X-ray beam, which usually is on the order of several 10 to several 100 μm, or by the spatial resolution of an area detector (where spatial resolution is only available in one dimension). In general APXPS spectra average over the entire area that is illuminated by the incident X-rays or that is within the field of view of the electron spectrometer, thus convoluting contributions from different components of the heterogeneous surface, which complicates the determination of the roles of the various parts of the sample surface to the overall reactivity. A straightforward method for the improvement of the spatial resolution is to tightly focus the X-ray beam using either refocusing mirrors (such as Kirkpatrick–Baez type mirror pairs that have demonstrated a spatial resolution below 50 nm for hard X-rays260) or Fresnel zone plates with a spatial resolution of currently less than 10 nm.261 More tightly focused incident X-ray beams will increase the flux density at the sample surface, with a concomitant chance of beam-induced damage to the sample surface, which needs to be mitigated in those experiments.
(3) In addition to increased spatial resolution, the investigation of heterogeneous chemical processes at surfaces over a wide range of time scales will be of increasing importance in many fields of research. Areas of interest include the kinetics of low-temperature oxidation of metals and oxides (minutes to hours) on the slow side to the observation of intermediate species in heterogeneous catalytic reactions, which requires a temporal resolution on the nanosecond scale or better. The latter is particularly challenging since the time-averaged concentration of reaction intermediates is low under catalytically-relevant conditions, where it is difficult to observe these states using XPS, which has a sensitivity of usually not better than a few percent of a monolayer. Pump–probe experiments using, e.g., THz excitation, combined with fast probes (i.e., delay-line detectors) may provide a path to study these phenomena on the relevant time scales of catalytic reactions, opening up the possibility to detect the fundamental steps in a heterogeneous chemical reactions at relevant pressures and temperatures.
Acknowledgements
The Advanced Light Source and beamline 11.0.2 are supported by the Director, Office of Energy Research, Office of Basic Energy Sciences, and Chemical Sciences Division of the U.S. Department of Energy under contracts No. DE-AC02-05CH11231. This work was also supported by the Office of Science, Biological and Environmental Research, Environmental Remediation Sciences Division (ERSD), U.S. Department of Energy under Contract No. DE-AC02-05CH11231. DES acknowledges support through the Silicon In situ Spectroscopy@the Sychrotron (SISSY) project within the Bundesministerium für Bildung und Forschung (BMBF project number 03SF0403). The Helmholtz-Zentrum Berlin (Electron Storage Ring BESSY II) is acknowledged for provision of synchrotron radiation at beamline ISISS. Part of the work was supported by the European Union (Technotubes Project).References
- G. Ertl, Nobel Lecture, http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2007/ertl-lecture.html.
- High Temperature and Solid Oxide Fuel Cells, ed. C. S. Subhash and K. Keven, Elsevier, Oxford, U.K., 2003 Search PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts, Jr., Chemistry of the Upper and Lower Atmosphere, Academic Press, London, 2000 Search PubMed.
- F. Dominé and P. B. Shepson, Science, 2002, 297, 1506 CrossRef.
- S. J. Peters and G. E. Ewing, J. Phys. Chem. B, 1997, 101, 10880 CrossRef CAS.
- V. Sadtchenko, G. E. Ewing, D. R. Nutt and A. J. Stone, Langmuir, 2002, 18, 4632 CrossRef.
- Y. R. Shen, Nature, 1989, 337, 519 CrossRef CAS.
- G. Rupprechter, MRS Bull., 2007, 34, 1031 CrossRef.
- J. Forsberg, L. C. Duda, A. Olsson, T. Schmitt, J. Andersson, J. Nordgren, J. Hedberg, C. Leygraf, C. Asstrup, D. Wallinder and J.-H. Guo, Rev. Sci. Instrum., 2007, 78, 083110 CrossRef CAS.
- S. Ferrer, M. D. Ackermann and E. Lundgren, MRS Bull., 2007, 32, 1010 CrossRef CAS.
- G. Binnig, C. F. Quate and Ch. Gerber, Phys. Rev. Lett., 1986, 56, 930 CrossRef.
- J. Hu, X.-d. Xiao, D. F. Ogletree and M. Salmeron, Science, 1995, 268, 267 CAS.
- B. L. M. Hendriksen and J. W. M. Frenken, Phys. Rev. Lett., 2002, 89, 046101 CrossRef CAS.
- T. W. Hansen, J. B. Wagner, P. L. Hansen, S. Dahl, H. Topsøe and C. J. H. Jacobsen, Science, 2001, 294, 1508 CrossRef CAS.
- A. M. Donald, Nat. Mater., 2003, 2, 511 CrossRef CAS.
- S. Hüfner, Photoelectron Spectroscopy, Springer, Berlin, 1995 Search PubMed.
- M. P. Seah and W. A. Dench, Surf. Interface Anal., 1979, 1, 2 CrossRef CAS.
- H. Siegbahn and K. Siegbahn, J. Electron Spectrosc. Relat. Phenom., 1973, 2, 319 CrossRef CAS.
- H. Siegbahn, J. Phys. Chem., 1985, 89, 897 CrossRef CAS.
- R. W. Joyner and M. W. Roberts, Surf. Sci., 1979, 87, 501 CrossRef CAS.
- H.-J. Ruppender, M. Grunze, C. W. Kong and M. Wilmers, Surf. Interface Anal., 1990, 15, 245 CrossRef CAS.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872 CrossRef CAS.
- H. Bluhm, M. Hävecker, A. Knop-Gericke, D. Teschner, E. Kleimenov, V. I. Bukhtiyarov, D. F. Ogletree, M. Salmeron and R. Schlögl, J. Phys. Chem. B, 2004, 108, 14340 CrossRef CAS.
- D. F. Ogletree, H. Bluhm, E. L. D. Hebenstreit and M. Salmeron, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601, 151 CrossRef.
- M. E. Grass, P. G. Karlsson, F. Aksoy, M. Lundqvist, B. Wannberg, B. S. Mun, Z. Hussain and Z. Liu, Rev. Sci. Instrum., 2010, 81, 053106 CrossRef.
- F. Aksoy, M. E. Grass, S. H. Joo, N. Jabeen, Y. P. Hong, Z. Hussain, B. S. Mun and Z. Liu, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 645, 260 CrossRef CAS.
- S. Kaya, H. Ogasawara, L.-Å. Näslund, J.-O. Forsell, H. S. Casalonguea, D. J. Miller and A. Nilsson, Catal. Today, 2013, 205, 105 CrossRef.
- J. Schnadt, J. Knudsen, J. N. Andresen, H. Siegbahn, A. Pietzsch, F. Hennies, N. Johansson, N. Mårtensson, G. Öhrwall, S. Bahr, S. Mähl and O. Schaff, J. Synchrotron Radiat., 2012, 19, 701 CrossRef CAS.
- R. Toyoshima, M. Yoshida, Y. Monya, Y. Kousa, K. Suzuki, H. Abe, B. S. Mun, K. Mase, K. Amemeiya and H. Kondoh, J. Phys. Chem. C, 2012, 116, 18691 CAS ; Supporting Information: http://pubs.acs.org/doi/suppl/10.1021/jp301636u/suppl_file/jp301636u_si_001.pdf.
- Specs Surface Nano Analysis GmbH, Berlin, Germany. http://www.specs.de/cms/front_content.php?idcat=269.
- V. G. Scienta, Uppsala, Sweden. http://www.vgscienta.com/productlist.aspx?MID=397.
- Omicron NanoTechnology GmbH, Taunusstein, Germany. http://www.omicron.de/en/products/ea-125-/instrument-concept.
- M. A. Kelly, M. L. Shek, P. Pianetta, T. M. Gür and M. R. Beasley, J. Vac. Sci. Technol., A, 2001, 19, 2127 CAS.
- J. Pantförder, S. Pöllmann, J. F. Zhu, D. Borgmann, R. Denecke and H.-P. Steinrück, Rev. Sci. Instrum., 2005, 76, 014102 CrossRef.
- F. Mangolini, J. Åhlund, G. E. Wabiszewski, V. P. Adiga, P. Egberts, F. Streller, K. Backlund, P. G. Karlsson, B. Wannberg and R. W. Carpick, Rev. Sci. Instrum., 2012, 83, 093112 CrossRef CAS.
- A. Jürgensen, N. Esser and R. Hergenröder, Surf. Interface Anal., 2012, 44, 1100 CrossRef.
- H. Bluhm, M. Hävecker, A. Knop-Gericke, M. Kiskinova, R. Schlögl and M. Salmeron, MRS Bull., 2007, 32, 1022 CrossRef CAS.
- M. Salmeron and R. Schlögl, Surf. Sci. Rep., 2008, 63, 169 CrossRef CAS.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. V. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, High-pressure X-ray photoelectron spectroscopy: a tool to investigate heterogeneous catalytic processes, in Advances in Catalysis, ed. B. C. Gates and H. Knözinger, Burlington, Academic Press, 2009, vol. 52, pp. 213–272 Search PubMed.
- H. Bluhm, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 71 CrossRef CAS.
- D. E. Starr, H. Bluhm, Z. Liu, A. Knop-Gericke and M. Hävecker, Application of Ambient Pressure X-ray Photoelectron Spectroscopy for the In-situ Investigation of Catalytically Relevant Processes, in In-situ Characterization of Heterogeneous Catalysts, ed. J. A. Rodriguez, J. C. Hanson and P. J. Chupas, John Wiley & Sons, Inc., NY, 2013, pp. 315–344 Search PubMed.
- A. Shavorskiy and H. Bluhm, Ambient pressure X-ray photoelectron spectroscopy, in Handbook of Heterogeneous Catalysts for Clean Technology – Design, Analysis and Application, ed. K. Wilson and A. Lee, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
- J. A. Whaley, A. H. McDaniel, F. El Gabaly, R. L. Farrow, M. E. Grass, Z. Hussain, Z. Liu, M. A. Linne, H. Bluhm and K. F. McCarty, Rev. Sci. Instrum., 2010, 81, 086104 CrossRef.
- R. Westerström, M. E. Messing, S. Blomberg, A. Hellman, H. Grönbeck, J. Gustafson, N. M. Martin, O. Balmes, R. van Rijn, J. N. Andersen, K. Deppert, H. Bluhm, Z. Liu, M. E. Grass, M. Hävecker and E. Lundgren, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115440 CrossRef.
- R. Toyoshima, M. Yoshida, Y. Monya, K. Suzuki, B. S. Mun, K. Amemeiya, K. Mase and H. Kondoh, J. Phys. Chem. Lett., 2012, 3, 3182 CrossRef CAS.
- S. Blomberg, M. J. Hoffmann, J. Gustafson, N. M. Martin, V. R. Fernandes, A. Borg, Z. Liu, R. Chang, S. Matera, K. Reuter and E. Lundgren, Phys. Rev. Lett., 2013, 110, 117601 CrossRef.
- Z. Zhu, D. R. Butcher, B. Mao, Z. Liu, M. Salmeron and G. A. Somorjai, J. Phys. Chem. C, 2013, 117, 2799 CAS.
- O. Balmes, A. Resta, D. Wermeille, R. Felici, M. E. Messing, K. Deppert, Z. Liu, M. E. Grass, H. Bluhm, R. Van Rjin, J. W. M. Frenken, R. Westerström, S. E. J. Blomberg, J. N. Andersen, J. Gustafson and E. Lundgren, Phys. Chem. Chem. Phys., 2012, 14, 4796 RSC.
- G. Ketteler, D. F. Ogletree, H. Bluhm, H. Liu, E. L. D. Hebenstreit and M. Salmeron, J. Am. Chem. Soc., 2005, 127, 18269 CrossRef CAS.
- D. Teschner, A. Pestryakov, E. Kleimenov, M. Hävecker, H. Bluhm, H. Sauer, A. Knop-Gericke and R. Schlögl, J. Catal., 2005, 230, 186 CrossRef CAS.
- D. Teschner, A. Pestryakov, E. Kleimenov, M. Hävecker, H. Bluhm, H. Sauer, A. Knop-Gericke and R. Schlögl, J. Catal., 2005, 230, 195 CrossRef CAS.
- J. Pantförder, S. Pöllmann, J. F. Zhu, D. Borgmann, R. Denecke and H.-P. Steinrück, Rev. Sci. Instrum., 2005, 76, 014102 CrossRef.
- V. V. Kaichev, I. P. Prosvirin, V. I. Bukhtiyarov, H. Unterhalt, G. Rupprechter and H.-J. Freund, J. Phys. Chem. B, 2003, 107, 3522 CrossRef CAS.
- V. V. Kaichev, A. V. Miller, I. P. Prosvirin and V. I. Bukhtiyarov, Surf. Sci., 2012, 606, 420 CrossRef CAS.
- H. Gabasch, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, D. Zemlyanov, B. Aszalos-Kiss, K. Hayek and B. Klötzer, J. Catal., 2006, 242, 340 CrossRef CAS.
- D. Zemlyanov, B. Aszalos-Kiss, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, H. Gabasch, W. Unterberger, K. Hayek and B. Klötzer, Surf. Sci., 2006, 600, 983 CrossRef CAS.
- H. Gabasch, W. Unterberger, K. Hayek, B. Klötzer, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, J. Han, F. H. Ribeiro, B. Aszalos-Kiss, T. Curtin and D. Zemlyanov, Surf. Sci., 2006, 600, 2980 CrossRef CAS.
- D. Teschner, E. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, H. Sauer, A. Knop-Gericke, R. Schlögl, M. Chamam, A. Wootsch, A. S. Canning, J. J. Gamman, S. D. Jackson, J. McGregor and L. F. Gladden, J. Catal., 2006, 242, 26 CrossRef CAS.
- H. Gabasch, K. Hayek, B. Klötzer, W. Unterberger, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, B. Aszalos-Kiss and D. Zemlyanov, J. Phys. Chem. C, 2007, 111, 7957 CAS.
- R. Toyoshima, M. Yoshida, Y. Monya, Y. Kousa, K. Suzuki, H. Abe, B. S. Mun, K. Mase, K. Amemeiya and H. Kondoh, J. Phys. Chem. C, 2012, 116, 18691 CAS.
- A. Haghofer, K. Föttinger, F. Girgsdies, D. Teschner, A. Knop-Gericke, R. Schlögl and G. Rupprechter, J. Catal., 2012, 286, 13 CrossRef CAS.
- C. Rameshan, C. Weilach, W. Stadlmayr, S. Penner, H. Lorenz, M. Hävecker, R. Blume, T. Rocha, D. Teschner, A. Knop-Gericke, R. Schlögl, D. Zemlyanov, N. Memmel, G. Rupprechter and B. Klötzer, J. Catal., 2010, 276, 113 CrossRef.
- D. Teschner, J. Borsodi, Z. Kis, L. Szentmiklósi, Z. Révay, A. Knop-Gericke, R. Schlögl, D. Torres and Ph. Sautet, J. Phys. Chem. C, 2010, 114, 2293 CAS.
- D. Teschner, Z. Révay, J. Borsodi, M. Hävecker, A. Knop-Gericke, R. Schlögl, D. Milroy, S. D. Jackson, D. Torres and Ph. Sautet, Angew. Chem., Int. Ed., 2008, 47, 9274 CrossRef CAS.
- E. M. Vass, M. Hävecker, S. Zafeiratos, D. Teschner, A. Knop-Gericke and R. Schlögl, J. Phys.: Condens. Matter, 2008, 20, 184016 CrossRef.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86 CrossRef CAS.
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. Knop-Gericke, Z. Paál and R. Schlögl, J. Catal., 2006, 237, 17 CrossRef CAS.
- F. Tao, S. Dag, L.-W. Wang, Z. Liu, D. R. Butcher, H. Bluhm, M. Salmeron and G. A. Somorjai, Science, 2010, 327, 850 CrossRef CAS.
- Z. Zhu, F. F. Tao, F. Zheng, R. Chang, Y. Li, L. Heinke, Z. Liu, M. Salmeron and G. A. Somorjai, Nano Lett., 2012, 12, 1491 CrossRef CAS.
- S. Günther, A. Scheibe, H. Bluhm, M. Hävecker, E. Kleimenov, A. Knop-Gericke, R. Schlögl and R. Imbihl, J. Phys. Chem. C, 2008, 112, 15382 Search PubMed.
- J. Y. Chung, F. Aksoy, M. E. Grass, H. Kondoh, P. Ross, Z. Liu and B. S. Mun, Surf. Sci., 2009, 603, L35 CrossRef CAS.
- D. R. Butcher, M. E. Grass, Z. Zeng, F. Aksoy, H. Bluhm, G. A. Somorjai, W.-X. Li, B. S. Mun and Z. Liu, J. Am. Chem. Soc., 2011, 133, 20319 CrossRef CAS.
- D. J. Miller, H. Öberg, S. Kaya, H. S. Casalongue, D. Friebel, T. Anniyev, H. Ogasawara, H. Bluhm, L. G. M. Pettersson and A. Nilsson, Phys. Rev. Lett., 2011, 107, 195502 CrossRef CAS.
- R. B. Getman, W. F. Schneider, A. D. Smeltz, W. N. Delgass and F. H. Ribeiro, Phys. Rev. Lett., 2009, 102, 076101 CrossRef CAS.
- T. Shimada, B. S. Mun, I. F. Nakai, A. Banno, H. Abe, Y. Iwasawa, T. Ohta and H. Kondoh, J. Phys. Chem. C, 2010, 114, 17030 CAS.
- Z. Zhu, D. R. Butcher, B. Mao, Z. Liu, M. Salmeron and G. A. Somorjai, J. Phys. Chem. C, 2013, 117, 2799 CAS.
- A. Virnovskaia, S. Jørgensen, J. Hafizovic, Ø. Prytz, E. Kleimenov, M. Hävecker, H. Bluhm, A. Knop-Gericke, R. Schlögl and U. Olsbye, Surf. Sci., 2007, 601, 30 CrossRef CAS.
- L. R. Merte, J. Knudsen, F. M. Eichhorn, S. Porsgaard, H. Zeuthen, L. C. Grabow, E. Lægsgaard, H. Bluhm, M. Salmeron, M. Mavrikakis and F. Besenbacher, J. Am. Chem. Soc., 2011, 133, 10692 CrossRef CAS.
- D. Teschner, A. Wootsch, O. Pozdnyakova-Tellinger, J. Kröhnert, E. M. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, P. C. Jentoft, A. Knop-Gericke and R. Schlögl, J. Catal., 2007, 249, 318 CrossRef CAS.
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. Knop-Gericke, Z. Paál and R. Schlögl, J. Catal., 2006, 237, 1 CrossRef CAS.
- S. Porsgaard, L. R. Merte, L. K. Ono, F. Behafarid, J. Matos, S. Helveg, M. Salmeron, B. R. Cuenya and F. Besenbacher, ACS Nano, 2012, 6, 10743 CAS.
- S. Schäfer, S. A. Wyrzgol, R. Caterino, A. Jentys, S. J. Schoell, M. Hävecker, A. Knop-Gericke, J. A. Lercher, I. D. Sharp and M. Stutzmann, J. Am. Chem. Soc., 2012, 134, 12528 CrossRef.
- P. Jiang, S. Porsgaard, F. Borondics, M. Köber, A. Caballero, H. Bluhm, F. Besenbacher and M. Salmeron, J. Am. Chem. Soc., 2010, 132, 2858 CrossRef CAS.
- E. A. Willneff, S. Braun, D. Rosenthal, H. Bluhm, M. Hävecker, E. Kleimenov, A. Knop-Gericke, R. Schlögl and S. L. M. Schroeder, J. Am. Chem. Soc., 2006, 128, 12052 CrossRef CAS.
- T. Herranz, X. Y. Deng, A. Cabot, P. Alivisatos, Z. Liu, G. Soler-Illia and M. Salmeron, Catal. Today, 2009, 143, 158 CrossRef CAS.
- T. Herranz, X. Deng, A. Cabot, Z. Liu and M. Salmeron, J. Catal., 2011, 283, 119 CrossRef CAS.
- S. Porsgaard, P. Jiang, S. Borondics, S. Wendt, H. Bluhm, F. Besenbacher and M. Salmeron, Angew. Chem., Int. Ed., 2011, 50, 2266 CAS.
- A. V. Bukhtiyarov, R. I. Kvon, A. V. Nartova, I. P. Prosvirin and V. I. Bukhtiyarov, Surf. Sci., 2012, 606, 559 CrossRef CAS.
- V. I. Bukhtiyarov, A. I. Nizovskii, H. Bluhm, M. Hävecker, E. Kleimenov, A. Knop-Gericke and R. Schlögl, J. Catal., 2006, 238, 260 CrossRef CAS.
- R. W. Joyner and M. W. Roberts, Chem. Phys. Lett., 1979, 60, 459 CrossRef CAS.
- A. I. Boronin, V. I. Bukhtiyarov, A. L. Vishnevskii, G. K. Boreskov and V. I. Savchenko, Surf. Sci., 1988, 201, 195 CrossRef CAS.
- T. C. R. Rocha, A. Oestereich, D. V. Demidov, M. Hävecker, S. Zafeiratos, G. Weinberg, V. I. Bukhtiyarov, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2012, 14, 4554 RSC.
- D. V. Demidov, I. P. Prosvirin, A. M. Sorokin, T. Rocha, A. Knop-Gericke and V. I. Bukhtiyarov, Kinet. Catal., 2011, 52, 855 CrossRef CAS.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224 CrossRef CAS.
- M. E. Glass, D. Butcher, Y. Zhang, Y. Li, H. Bluhm, K. Bratlie, T. Zhang, J. Y. Park and G. A. Somorjai, Angew. Chem., Int. Ed., 2008, 47, 8893 CrossRef.
- F. Aksoy, M. E. Grass, S. H. Joo, N. Jabeen, Y. P. Hong, Z. Hussain, B. S. Mun and Z. Liu, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 645, 260 CrossRef CAS.
- F. G. Requejo, E. L. D. Hebenstreit, D. F. Ogletree and M. Salmeron, J. Catal., 2004, 226, 83 CrossRef CAS.
- R. W. Joyner, M. W. Roberts and S. P. Singh-Boparai, Surf. Sci. Lett., 1981, 104, L199 CrossRef CAS.
- R. Blume, M. Hävecker, S. Zafeiratos, D. Teschner, A. Knop-Gericke, R. Schlögl, P. Dudin, A. Barinov and M. Kiskinova, Catal. Today, 2007, 124, 71 CrossRef CAS.
- R. Blume, M. Hävecker, S. Zafeiratos, D. Teschner, E. Kleimenov, A. Knop-Gericke, R. Schlögl, A. Barinov, P. Dudin and M. Kiskinova, J. Catal., 2006, 239, 354 CrossRef CAS.
- D. E. Starr and H. Bluhm, Surf. Sci., 2013, 608, 241 CrossRef CAS.
- R. Blume, M. Hävecker, S. Zafeiratos, D. Teschner, E. Vass, P. Schnörch, A. Knop-Gericke, R. Schlögl, S. Lizzit, P. Dudin, A. Barinov and M. Kiskinova, Phys. Chem. Chem. Phys., 2007, 9, 3648 RSC.
- K. Qadir, S. H. Joo, B. S. Mun, D. R. Butcher, J. R. Renzas, F. Aksoy, Z. Liu, G. A. Somorjai and J. Y. Park, Nano Lett., 2012, 12, 5761 CrossRef CAS.
- H. J. Ruppender, M. Grunze, C. W. Kong and M. Wilmers, Surf. Interface Anal., 1990, 15, 245 CrossRef CAS.
- B. C. Bayer, M. Fouquet, R. Blume, C. T. Wirth, R. S. Weatherup, K. Ogata, A. Knop-Gericke, R. Schlögl, S. Hofmann and J. Robertson, J. Phys. Chem. C, 2012, 116, 1107 CAS.
- K. Wang, Z. Liu, T. H. Cruz, M. Salmeron and H. Liang, J. Phys. Chem. A, 2010, 114, 2489 CrossRef CAS.
- A. Caballero, J. P. Holgado, V. M. Gonzalez-delaCruz, S. E. Habas, T. Herranz and M. Salmeron, Chem. Commun., 2010, 46, 1097 RSC.
- V. V. Kaichev, A. Yu. Gladky, I. P. Prosvirin, A. A. Saraev, M. Hävecker, A. Knop-Gericke, R. Schlögl and V. I. Bukhtiyarov, Surf. Sci., 2013, 609, 113 CrossRef CAS.
- V. Papaefthimiou, T. Dintzer, V. Dupuis, A. Tamion, F. Tournus, A. Hillion, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl and S. Zafeiratos, ACS Nano, 2011, 5, 2182 CrossRef CAS.
- S. Zafeiratos, T. Dintzer, D. Teschner, R. Blume, M. Hävecker, A. Knop-Gericke and R. Schlögl, J. Catal., 2010, 269, 309 CrossRef CAS.
- S. Alayoglu, S. K. Beaumont, F. Zheng, V. V. Pushkarev, H. Zheng, V. Iablokov, Z. Liu, J. Guo, N. Kruse and G. A. Somorjai, Top. Catal., 2011, 54, 778 CrossRef CAS.
- X. Deng, A. Verdaguer, T. Herranz, Ch. D. Weiss, H. Bluhm and M. Salmeron, Langmuir, 2008, 24, 9474 CrossRef CAS.
- D. M. Littrell and B. J. Tatarchuk, J. Vac. Sci. Technol., A, 1986, 4, 1608 CAS.
- V. I. Bukhtiyarov, I. P. Prosvirin, E. P. Tikhomirov, V. V. Kaichev, A. M. Sorokin and V. V. Evstigneev, React. Kinet. Catal. Lett., 2003, 79, 181 CrossRef CAS.
- K. Andersson, G. Ketteler, H. Bluhm, S. Yamamoto, H. Ogasawara, L. G. M. Pettersson, M. Salmeron and A. Nilsson, J. Am. Chem. Soc., 2008, 130, 2793 CrossRef CAS.
- K. Andersson, G. Ketteler, H. Bluhm, S. Yamamoto, H. Ogasawara, L. G. M. Pettersson, M. Salmeron and A. Nilsson, J. Phys. Chem. C, 2007, 111, 14493 CAS.
- S. Günther, L. Zhou, M. Hävecker, A. Knop-Gericke, E. Kleimenov, R. Schlögl and R. Imbihl, J. Chem. Phys., 2006, 125, 114709 CrossRef.
- S. Yamamoto, K. Andersson, H. Bluhm, G. Ketteler, D. E. Starr, Th. Schiros, H. Ogasawara, L. G. M. Pettersson, M. Salmeron and A. Nilsson, J. Phys. Chem. C, 2007, 111, 7848 CAS.
- J. P. Baxter, M. Grunze and C. W. Kong, J. Vac. Sci. Technol., A, 1988, 6, 1123 CAS.
- K. Mudiyanselage, S. D. Senanayake, L. Feria, S. Kundu, A. E. Baber, J. Graciani, A. B. Vidal, S. Agnoli, J. Evans, R. Chang, S. Axnanda, Z. Liu, J. F. Sanz, P. Liu, J. A. Rodriguez and D. J. Stacchiola, Angew. Chem., Int. Ed., 2013, 52 DOI:10.1002/anie.201210077.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893 CrossRef CAS.
- R. Würz, M. Rusu, Th. Schedel-Niedrig, M. Ch. Lux-Steiner, H. Bluhm, M. Hävecker, E. Kleimenov, A. Knop-Gericke and R. Schlögl, Surf. Sci., 2005, 580, 80 CrossRef.
- M. A. Kelly, M. L. Shek, P. Pianetta, T. M. Gür and M. R. Beasley, J. Vac. Sci. Technol., A, 2001, 19, 2127 CAS.
- Y. Enta, B. S. Mun, M. Rossi, J. P. N. Ross, Z. Hussain, C. S. Fadley, K.-S. Lee and S.-K. Kim, Appl. Phys. Lett., 2008, 92, 012110 CrossRef.
- M. Rossi, B. S. Mun, Y. Enta, C. S. Fadley, K.-S. Lee, S.-K. Kim, H.-J. Shin, Z. Hussain and J. P. N. Ross, J. Appl. Phys., 2008, 103, 044104 CrossRef.
- B. C. Bayer, C. Zhang, R. Blume, F. Yan, M. Fouquet, C. T. Wirth, R. S. Weatherup, L. Lin, C. Baehtz, R. A. Oliver, A. Knop-Gericke, R. Schlögl, S. Hofmann and J. Robertson, J. Appl. Phys., 2011, 109, 114314 CrossRef.
- B. C. Bayer, S. Hofmann, C. Castellarin-Cudia, R. Blume, C. Baehtz, S. Esconjauregui, C. T. Wirth, R. A. Oliver, C. Ducati, A. Knop-Gericke, R. Schlögl, A. Goldoni, C. Cepek and J. Robertson, J. Phys. Chem., 2011, 115, 4359 CrossRef CAS.
- A. Rinaldi, J.-P. Tessonnier, M. E. Schuster, R. Blume, F. Girgsdies, Q. Zhang, T. Jacob, S. B. Abd Hamid, D. S. Su and R. Schlögl, Angew. Chem., Int. Ed., 2011, 50, 3313 CrossRef CAS.
- B. Frank, A. Rinaldi, R. Blume, R. Schlögl and D. S. Su, Chem. Mater., 2010, 22, 4462 CrossRef CAS.
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlögl and D. S. Su, Science, 2008, 322, 5898 Search PubMed.
- J. Zhang, D. S. Su, R. Blume, R. Schlögl, R. Wang, X. Yang and A. Gajović, Angew. Chem., Int. Ed., 2010, 49, 8640 CrossRef CAS.
- St. Hofmann, R. Blume, Ch. T. Wirth, M. Cantoro, R. Sharma, C. Ducati, M. Hävecker, S. Zafeiratos, P. Schnörch, A. Oestereich, D. Teschner, M. Albrecht, A. Knop-Gericke, R. Schlögl and J. Robertson, J. Phys. Chem. C, 2009, 113, 1648 CAS.
- C. Zhang, F. Yan, B. C. Bayer, R. Blume, M. H. van der Veen, R. Xie, G. Zhong, B. Chen, A. Knop-Gericke, R. Schlögl, B. D. Capraro, S. Hofmann and J. Robertson, J. Appl. Phys., 2012, 111, 064310 CrossRef.
- B. C. Bayer, S. Hofmann, C. Castellarin-Cudia, R. Blume, C. Baehtz, S. Esconjauregui, C. T. Wirth, R. A. Oliver, C. Ducati, A. Knop-Gericke, R. Schlögl, A. Goldoni, C. Cepek and J. Robertson, J. Phys. Chem. C, 2011, 115, 4359 CAS.
- G. Ketteler, P. Ashby, B. S. Mun, I. Ratera, H. Bluhm, B. Kasemo and M. Salmeron, J. Phys.: Condens. Matter, 2008, 20, 184024 CrossRef.
- A. Shavorskiy, F. Aksoy, M. Grass, Z. Liu, H. Bluhm and G. Held, J. Am. Chem. Soc., 2011, 133, 6659 CrossRef CAS.
- A. Shavorskiy, T. Eralp, K. Schulte, H. Bluhm and G. Held, Surf. Sci., 2013, 607, 10 CrossRef CAS.
- G. Ketteler, S. Yamamoto, H. Bluhm, K. Andersson, D. E. Starr, D. F. Ogletree, H. Ogasawara, A. Nilsson and M. Salmeron, J. Phys. Chem. C, 2007, 111, 8278 CAS.
- J. Haubrich, R. G. Quiller, L. Benz, Z. Liu and C. M. Friend, Langmuir, 2010, 26, 2445 CrossRef CAS.
- R. Jribi, E. Barthel, H. Bluhm, M. Grunze, P. Koelsch, D. Verreault and E. Sondergård, J. Phys. Chem. C, 2009, 113, 8273 CAS.
- O. Rosseler, M. Sleiman, V. N. Montesinos, A. Shavorskiy, V. Keller, N. Keller, M. I. Litter, H. Bluhm, M. Salmeron and H. Destaillats, J. Phys. Chem. Lett., 2013, 4, 536 CrossRef CAS.
- X. Deng, T. Herranz, Ch. Weis, H. Bluhm and M. Salmeron, J. Phys. Chem. C, 2008, 112, 9668 CAS.
- P. Jiang, D. Prendergast, F. Borondics, S. Porsgaard, L. Giovanetti, E. Pach, J. Newberg, H. Bluhm, F. Besenbacher and M. Salmeron, J. Chem. Phys., 2013, 138, 024704 CrossRef.
- J. T. Newberg, D. E. Starr, S. Porsgaard, S. Yamamoto, S. Kaya, E. R. Mysak, T. Kendelewicz, M. Salmeron, G. E. Brown, Jr., A. Nilsson and H. Bluhm, Surf. Sci., 2011, 605, 89 CrossRef CAS.
- J. T. Newberg, D. E. Starr, S. Porsgaard, S. Yamamoto, S. Kaya, E. R. Mysak, T. Kendelewicz, M. Salmeron, G. E. Brown, Jr., A. Nilsson and H. Bluhm, J. Phys. Chem. C, 2011, 115, 12864 CAS.
- Y. Gassenbauer, R. Schafranek, A. Klein, S. Zafeiratos, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 245312 CrossRef.
- E. Kleimenov, H. Bluhm, M. Hävecker, A. Knop-Gericke, A. Pestryakov, D. Teschner, J. A. Lopez-Sanchez, J. K. Bartley, G. J. Hutchings and R. Schlögl, Surf. Sci., 2005, 575, 181 CrossRef CAS.
- M. Hävecker, R. W. Mayer, A. Knop-Gericke, H. Bluhm, E. Kleimenov, A. Liskowsi, D. Su, R. Follath, F. G. Requejo, D. F. Ogletree, M. Salmeron, J. A. Lopez-Sanchez, J. K. Bartley, G. J. Hutchings and R. Schlögl, J. Phys. Chem. B, 2003, 107, 4587 CrossRef.
- M. Eichelbaum, M. Hävecker, Ch. Heine, A. Karpov, C.-K. Dobner, F. Rosowski, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2012, 51, 6246 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski, Y.-L. Lee, D. Morgan, H. M. Christen, H. Bluhm and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 6081 CAS.
- J. L. Hueso, D. Martinez-Martinez, A. Caballero, A. R. Gonzalez-Elipe, B. S. Mun and M. Salmeron, Catal. Commun., 2009, 10, 1898 CrossRef CAS.
- M. Hävecker, S. Wrabetz, J. Kröhnert, L.-I. Csepei, R. Naumann d'Alnoncourt, Y. V. Kolen'ko, F. Girgsdies, R. Schlögl and A. Trunschke, J. Catal., 2012, 285, 48 CrossRef.
- A. C. Sanfiz, T. W. Hansen, D. Teschner, P. Schnörch, F. Girgsdies, A. Trunschke, R. Schlögl, M. H. Looi and S. B. Abd Hamid, J. Phys. Chem. C, 2010, 114, 1912 CAS.
- Y.-C. Lu, E. J. Crumlin, G. M. Veith, J. R. Harding, E. Mutoro, L. Baggetto, N. J. Dudney, Z. Liu and Y. Shao-Horn, Sci. Rep., 2012, 2, 715 Search PubMed.
- W. C. Chueh, A. H. McDaniel, M. E. Grass, Y. Hao, N. Jabeen, Z. Liu, S. M. Haile, K. F. McCarty, H. Bluhm and F. El Gabaly, Chem. Mater., 2012, 24, 1876 CrossRef CAS.
- C. Wen, Y. Zhu, Y. Ye, S. Zhang, F. Cheng, Y. Liu, P. Wang and F. Tao, ACS Nano, 2012, 6, 9305 CrossRef CAS.
- S. Yamamoto, T. Kendelewicz, J. T. Newberg, G. Ketteler, D. E. Starr, E. R. Mysak, K. Andersson, H. Ogasawara, H. Bluhm, M. Salmeron, G. E. Brown, Jr. and A. Nilsson, J. Phys. Chem. C, 2010, 114, 2256 CAS.
- E. de Smit, M. M. van Schooneveld, F. Cinquini, H. Bluhm, Ph. Sautet, F. M. F. de Groot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2011, 50, 1584 CrossRef CAS.
- X. Y. Deng, J. Lee, C. J. Wang, C. Matranga, F. Aksoy and Z. Liu, Langmuir, 2011, 27, 2146 CrossRef CAS.
- X. Y. Deng, J. Lee, C. J. Wang, C. Matranga, F. Aksoy and Z. Liu, J. Phys. Chem. C, 2010, 114, 22619 CAS.
- E. de Smit, F. M. F. de Groot, R. Blume, M. Hävecker, A. Knop-Gericke and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 667 RSC.
- T. Kendelewicz, S. Kaya, J. T. Newberg, H. Bluhm, N. Mulakaluri, W. Moritz, M. Scheffler, A. Nilsson, R. Pentcheva and G. E. Brown, Jr., J. Phys. Chem. C, 2013, 117, 2719 CAS.
- E. R. Mysak, D. E. Starr, K. R. Wilson and H. Bluhm, Rev. Sci. Instrum., 2010, 81, 016106 CrossRef.
- A. Verdaguer, Ch. D. Weis, G. Oncins, G. Ketteler, H. Bluhm and M. Salmeron, Langmuir, 2007, 23, 9699 CrossRef CAS.
- A. Mura, I. Hedishemi, Z. Liu, T. Hosoi, H. Watanaba and K. Arima, J. Phys. Chem. C, 2013, 117, 165 CAS.
- M. Krisch, R. D'Auria, M. A. Brown, D. J. Tobias, J. C. Hemminger, M. Ammann, D. E. Starr and H. Bluhm, J. Phys. Chem. C, 2007, 111, 13497 CAS.
- M. A. Brown, Z. Liu, P. D. Ashby, A. Mehta, R. L. Grimm and J. C. Hemminger, J. Phys. Chem. C, 2008, 112, 18287 CAS.
- M. A. Brown, P. D. Ashby, M. J. Krisch, Z. Liu, B. S. Mun, R. G. Green, J. B. Giorgi and J. C. Hemminger, J. Phys. Chem. C, 2010, 114, 14093 CAS.
- S. Ghosal, J. C. Hemminger, H. Bluhm, B. S. Mun, E. L. D. Hebenstreit, G. Ketteler, D. F. Ogletree, F. G. Requejo and M. Salmeron, Science, 2005, 307, 563 CrossRef CAS.
- K. Arima, P. Jiang, X. Deng, H. Bluhm and M. Salmeron, J. Phys. Chem. C, 2010, 114, 14900 CAS.
- A. Krepelová, T. Huthwelker, H. Bluhm and M. Ammann, ChemPhysChem, 2010, 11, 3859 CrossRef.
- A. Verdaguer, J. J. Segura, J. Fraxedas, H. Bluhm and M. Salmeron, J. Phys. Chem. C, 2008, 112, 16898 CAS.
- M. D. Baer, I.-F. W. Kuo, H. Bluhm and S. Ghosal, J. Phys. Chem. B, 2009, 113, 15843 CrossRef CAS.
- S. Ghosal, M. A. Brown, M. Krisch, H. Bluhm, M. Salmeron, P. Jungwirth and J. C. Hemminger, J. Phys. Chem. A, 2008, 112, 12378 CrossRef CAS.
- S. Kaya, S. Yamamoto, J. T. Newberg, H. Bluhm, H. Ogasawara, T. Kendelewicz, G. E. Brown Jr., L. G. M. Pettersson and A. Nilsson, Sci. Rep., 2013, 3, 1074 CrossRef.
- M. H. Cheng, K. M. Callahan, A. M. Margarella, D. J. Tobias, J. C. Hemminger, H. Bluhm and M. J. Krisch, J. Phys. Chem. C, 2012, 116, 4545 CAS.
- H. Bluhm, D. F. Ogletree, Ch. Fadley, Z. Hussain and M. Salmeron, J. Phys.: Condens. Matter, 2002, 14, L227 CrossRef CAS.
- A. Krepelová, J. T. Newberg, T. Huthwelker, H. Bluhm and M. Ammann, Phys. Chem. Chem. Phys., 2010, 12, 8870 RSC.
- D. E. Starr, D. Pan, J. T. Newberg, M. Ammann, E. G. Wang, A. Michaelidis and H. Bluhm, Phys. Chem. Chem. Phys., 2011, 13, 19988 RSC.
- A. Krepelová, Th. Bartels-Rausch, M. A. Brown, H. Bluhm and M. Ammann, J. Phys. Chem. A, 2013, 117, 401 CrossRef.
- C. Zhang, M. E. Grass, A. H. McDaniel, S. C. DeCaluwe, F. El Gabali, Z. Liu, K. F. McCarty, R. L. Farrow, M. A. Linne, Z. Hussain, G. S. Jackson, H. Bluhm and B. W. Eichhorn, Nat. Mater., 2010, 9, 944 CrossRef CAS.
- S. C. DeCaluwe, M. E. Grass, C. Zhang, F. El Gabaly, H. Bluhm, Z. Liu, G. S. Jackson, A. H. McDaniel, K. F. McCarty, R. L. Farrow, M. A. Linne, Z. Hussain and B. W. Eichhorn, J. Phys. Chem. C, 2010, 114, 19853 CAS.
- S. C. DeCaluwe, G. S. Jackson, R. L. Farrow, A. H. McDaniel, F. El Gabaly, K. F. McCarty, S. Nie, M. A. Linne, H. Bluhm, J. T. Newberg, Z. Liu and Z. Hussain, ECS Trans., 2009, 16, 253 CrossRef CAS.
- C. Zhang, M. E. Grass, Y. Yu, K. J. Gaskell, S. C. DeCaluwe, R. Chang, G. S. Jackson, Z. Hussain, H. Bluhm, B. W. Eichhorn and Z. Liu, ACS Catal., 2012, 2, 2297 CrossRef CAS.
- F. El Gabaly, A. H. McDaniel, M. E. Grass, Z. Liu, H. Bluhm, M. A. Linne, Z. Hussain, R. L. Farrow and K. F. McCarty, Phys. Chem. Chem. Phys., 2010, 12, 12138 RSC.
- F. El Gabaly, A. H. McDaniel, M. E. Grass, W. Chueh, H. Bluhm, Z. Liu and K. F. McCarty, Chem. Commun., 2012, 48, 8338 RSC.
- V. Papaefthimiou, M. Shishkin, D. K. Niakolas, M. Athanasiou, Y. T. Law, R. Arrigo, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, T. Ziegler, S. G. Neophytides and S. Zafeiratos, Adv. Energy Mater., 2013 DOI:10.1002/aenm.201200727.
- P. S. Miedema, M. M. van Schooneveld, R. Bogerd, T. C. R. Rocha, M. Hävecker, A. Knop-Gericke and F. M. F. de Groot, J. Phys. Chem. C, 2011, 115, 25422 CAS.
- F. Tao, M. E. Grass, Y. W. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932 CrossRef CAS.
- F. Tao, M. E. Grass, Y. W. Zhang, D. R. Butcher, F. Aksoy, S. Aloni, V. Altoe, S. Alayoglu, J. R. Renzas, C. K. Tsung, Z. W. Zhu, Z. Liu, M. Salmeron and G. A. Somorjai, J. Am. Chem. Soc., 2010, 132, 8697 CrossRef CAS.
- J. R. Renzas, W. Y. Huang, Y. W. Zhang, M. E. Grass, D. T. Hoang, S. Alayoglu, D. R. Butcher, F. Tao, Z. Liu and G. A. Somorjai, Phys. Chem. Chem. Phys., 2011, 13, 2556 RSC.
- M. E. Grass, M. Park, F. Aksoy, Y. Zhang, M. Kunz, Z. Liu and B. S. Mun, Langmuir, 2010, 26, 16362 CrossRef CAS.
- S. Alayoglu, F. Tao, V. Altoe, C. Specht, Z. W. Zhu, F. Aksoy, D. R. Butcher, R. J. Renzas, Z. Liu and G. A. Somorjai, Catal. Lett., 2011, 141, 633 CrossRef CAS.
- F. Zheng, S. Alayoglu, V. V. Pushkarev, S. K. Beaumont, C. Specht, F. Aksoy, Z. Liu, J. H. Guo and G. A. Somorjai, Catal. Today, 2012, 182, 54 CrossRef CAS.
- M. D. Ong, B. W. Jacobs, J. D. Sugar, M. E. Grass, Z. Liu, G. M. Buffleben, W. M. Clift, M. E. Langham, P. Cappillino and D. B. Robinson, Chem. Mater., 2012, 24, 996 CrossRef CAS.
- D. Teschner, A. Wootsch and Z. Paál, Appl. Catal., A, 2012, 411, 31 CrossRef.
- Y. Jugnet, D. Loffreda, C. Dupont, F. Delbecq, E. Ehret, F. J. Cadete Santos Aires, B. S. Mun, F. Aksoy Akgul and Z. Liu, J. Phys. Chem. Lett., 2012, 3, 3707 CrossRef CAS.
- Z. Paál, A. Wootsch, D. Teschner, K. Lázár, I. E. Sajó, N. Győrffy, G. Weinberg, A. Knop-Gericke and R. Schlögl, Appl. Catal., A, 2011, 391, 377 CrossRef.
- C. Rameshan, W. Stadlmayr, S. Penner, H. Lorenz, N. Memmel, M. Hävecker, R. Blume, D. Teschner, T. Rocha, D. Zemlyanov, A. Knop-Gericke, R. Schlögl and B. Klötzer, Angew. Chem., Int. Ed., 2012, 51, 3002 CrossRef CAS.
- M. Friedrich, D. Teschner, A. Knop-Gericke and M. Armbrüster, J. Catal., 2012, 285, 41 CrossRef CAS.
- V. Papaefthimiou, T. Dintzer, V. Dupuis, A. Tamion, F. Tournus, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl and S. Zafeiratos, J. Phys. Chem. Lett., 2011, 2, 900 CrossRef CAS.
- V. Papaefthimiou, T. Dintzer, M. Lebedeva, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, V. Pierren-Bohnes, E. Savinova and S. Zafeiratos, J. Phys. Chem. C, 2012, 116, 14342 CAS.
- L. Shao, W. Zhang, M. Armbrüster, D. Teschner, F. Girgsdies, B. Zhang, O. Timpe, M. Friedrich, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2011, 50, 10231 CrossRef CAS.
- K. Kovnir, M. Armbrüster, D. Teschner, T. V. Venkov, L. Szentmiklósi, F. C. Jentoft, A. Knop-Gericke, Yu. Grin and R. Schlögl, Surf. Sci., 2009, 603, 1784 CrossRef CAS.
- K. Kovnir, M. Armbrüster, D. Teschner, T. V. Venkov, F. C. Jentoft, A. Knop-Gericke, Y. Grin and R. Schlögl, Sci. Technol. Adv. Mater., 2007, 8, 420 CrossRef CAS.
- Ch. Rameshan, W. Stadlmayr, S. Penner, H. Lorenz, L. Mayr, M. Hävecker, R. Blume, T. Rocha, D. Teschner, A. Knop-Gericke, R. Schlögl, D. Zemlyanov, N. Memmel and B. Klötzer, J. Catal., 2012, 290, 126 CrossRef CAS.
- B. Bridier, J. Pérez-Ramírez, A. Knop-Gericke, R. Schlögl and D. Teschner, Chem. Sci., 2011, 2, 1379 RSC.
- S. Carenco, A. Tuxen, M. Chintapalli, E. Pach, C. Escudero, T. D. Ewers, P. Jiang, F. Borondics, G. Thornton, A. P. Alivisatos, H. Bluhm, J. Guo and M. Salmeron, J. Phys. Chem. C, 2013, 117, 6259 CAS.
- S. Zafeiratos, F. Paloukis, G. Papakonstantinou, D. Teschner, M. Hävecker, E. Vass, P. Schnörch, A. Knop-Gericke, R. Schlögl, B. Moreno, E. Chinarro, J. R. Jurado and S. G. Neophytides, Catal. Today, 2010, 157, 250 CrossRef CAS.
- B. Halevi, E. J. Peterson, A. Delariva, E. Jeroro, V. M. Lebarbier, Y. Wang, J. M. Vohs, B. Kiefer, E. Kunkes, M. Hävecker, M. Behrens, R. Schlögl and A. K. Datye, J. Phys. Chem. C, 2010, 114, 17181 CAS.
- B. Halevi, E. J. Peterson, A. Roy, A. DeLariva, E. Jeroro, F. Gao, Y. Wang, J. M. Vohs, B. Kiefer, E. Kunkes, M. Hävecker, M. Behrens, R. Schlögl and A. K. Datye, J. Catal., 2012, 291, 44 CrossRef CAS.
- C. Rameshan, W. Stadlmayr, C. Weilach, S. Penner, H. Lorenz, M. Hävecker, R. Blume, T. Rocha, D. Teschner, A. Knop-Gericke, R. Schlögl, N. Memmel, D. Zemlyanov, G. Rupprechter and B. Klötzer, Angew. Chem., Int. Ed., 2010, 49, 3224 CrossRef CAS.
- C. Rameshan, H. Lorenz, L. Mayr, S. Penner, D. Zemlyanov, R. Arrigo, M. Hävecker, R. Blume, A. Knop-Gericke, R. Schlögl and B. Klötzer, J. Catal., 2012, 295, 186 CrossRef CAS.
- S. Piccinin, S. Zafeiratos, C. Stampfl, T. W. Hansen, M. Hävecker, D. Teschner, V. I. Bukhtiyarov, F. Girgsdies, A. Knop-Gericke, R. Schlögl and M. Scheffler, Phys. Rev. Lett., 2010, 104, 035503 CrossRef.
- F. Zheng, S. Alayoglu, V. V. Pushkarev, S. K. Beaumont, C. Specht, F. Aksoy, Z. Liu, J. Guo and G. A. Somorjai, Catal. Today, 2012, 182, 54 CrossRef CAS.
- G. Abrasonis, S. Wintz, M. O. Liedke, F. Aksoy, M. Krause, K. Kuepper, D. Banerjee, Z. Liu and S. Gemming, J. Phys. Chem. C, 2012, 116, 14401 CAS.
- A. Toghan, R. Arrigo, A. Knop-Gericke and R. Imbihl, J. Catal., 2012, 296, 99 CrossRef CAS.
- M. Friedrich, D. Teschner, A. Knop-Gericke and M. Armbrüster, J. Phys. Chem. C, 2012, 116, 14930 CAS.
- Y. Zhu, S. Zhang, Y. Ye, X. Zhang, L. Wang, W. Zhu, F. Cheng and F. Tao, ACS Catal., 2012, 2, 2403 CrossRef CAS.
- H. Pfnür and D. Menzel, J. Chem. Phys., 1983, 79, 2400 CrossRef.
- J. C. Fuggle, T. E. Madey, M. Steinkilberg and D. Menzel, Surf. Sci., 1975, 52, 521 CrossRef CAS.
- F. M. Hoffmann, J. Chem. Phys., 1989, 90, 2816 CrossRef CAS.
- F. M. Hoffmann and J. L. Robbins, J. Electron Spectrosc. Relat. Phenom., 1987, 45, 421 CrossRef CAS.
- N. Schumacher, A. Boisen, S. Dahl, A. A. Gokhale, S. Kandoi, L. C. Grabow, J. A. Dumesic, M. Mavrikakis and I. Chorkendorff, J. Catal., 2005, 229, 265 CrossRef CAS.
- J. Yoshihara and C. T. Campbell, J. Catal., 1996, 161, 776 CrossRef CAS.
- K. Andersson, A. Gómez, C. Glover, D. Nordlund, H. Öström, T. Schiros, O. Takahashi, H. Ogasawara, L. G. M. Pettersson and A. Nilsson, Surf. Sci., 2005, 585, L183 CrossRef CAS.
- T. Yamada, S. Tamamori, H. Okuyama and T. Aruga, Phys. Rev. Lett., 2006, 96, 036105 CrossRef CAS.
- T. Schiros, S. Haq, H. Ogasawara, O. Takahashi, H. Öström, K. Andersson, L. G. M. Pettersson, A. Hodgson and A. Nilsson, Chem. Phys. Lett., 2006, 429, 415 CrossRef CAS.
- J. Ren and S. Meng, J. Am. Chem. Soc., 2006, 128, 9282 CrossRef CAS.
- J. Nakamura, J. M. Campbell and C. T. Campbell, J. Chem. Soc., Faraday Trans., 1990, 86, 2725 RSC.
- K. Bange, D. E. Grider, T. E. Madey and J. K. Sass, Surf. Sci., 1984, 136, 38 CrossRef.
- A. Spitzer and H. Lüth, Surf. Sci., 1982, 120, 376 CrossRef CAS.
- A. Spitzer and H. Lüth, Surf. Sci., 1985, 160, 353 CrossRef CAS.
- J. Kubota, J. Kondo, K. Domen and C. Hirose, Surf. Sci., 1993, 295, 169 CrossRef CAS.
- M. Polak, Surf. Sci., 1994, 321, 249 CrossRef CAS.
- D. Kolovos-Vellianitis and J. Küppers, J. Phys. Chem. B, 2003, 107, 2559 CrossRef CAS.
- Ch. Ammon, A. Bayer, H. P. Steinrück and G. Held, Chem. Phys. Lett., 2003, 377, 163 CrossRef CAS.
- W. D. Clendening, J. A. Rodriguez, J. M. Campbell and C. T. Campbell, Surf. Sci., 1989, 216, 429 CrossRef CAS.
- S. Hagstrom, H. B. Lyon and G. A. Somorjai, Phys. Rev. Lett., 1965, 15, 491 CrossRef CAS.
- L. L. Kesmodel, L. H. Dubois and G. A. Somorjai, Chem. Phys. Lett., 1987, 56, 267 CrossRef.
- O. Björnehom, A. Nilsson, H. Tillborg, P. Bennich, A. Sandell, B. Hernnäs, C. Puglia and N. Mårtensson, Surf. Sci., 1994, 315, L983 CrossRef.
- B. L. M. Hendriksen and J. W. M. Frenken, Phys. Rev. Lett., 2002, 89, 046101 CrossRef CAS.
- B. K. Hodnett, Heterogeneous catalytic oxidation reaction: Fundamental and technological aspects of the selective and total oxidation of organic compounds, John Wiley & Sons LTD, 2000 Search PubMed.
- Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany, 2008 Search PubMed.
- J. Schnadt, J. Knudsen, X. L. Hu, A. Michaelides, R. T. Vang, K. Reuter, Z. Li, E. Laegsgaard, M. Scheffler and F. Besenbacher, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 075424 CrossRef.
- M. Rocca, L. Savio, L. Vattuone, U. Burghaus, V. Palomba, N. Novelli, F. B. de Mongeot, U. Valbusa, R. Gunnella, G. Comelli, A. Baraldi, S. Lizzit and G. Paolucci, Phys. Rev. B, 2000, 61, 213 CrossRef CAS.
- X. Bao, M. Muhler, T. Schedel-Niedrig and R. Schlögl, Phys. Rev. B, 1996, 54, 2249 CrossRef CAS.
- D. N. Futuba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987 CrossRef.
- K. B. K. Teo, E. Minoux, L. Hudanski, F. Peauger, J.-P. Schell, P. Legagneux, D. Dieumegardt, G. A. Amaratunga and W. I. Milne, Nature, 2005, 437, 968 CrossRef CAS.
- F. Kreupl, A. P. Graham, G. S. Duesberg, W. Steinhoegl, M. Liebau, E. Unger and W. Hoenlein, Microelectron. Eng., 2002, 64, 399 CrossRef CAS.
- K. Hata, D. N. Futaba, K. Mizuno, T. Namai, M. Yumura and S. Iijima, Science, 2004, 306, 1362 CrossRef CAS.
- C. Mattevi, C. T. Wirth, S. Hofmann, R. Blume, M. Cantoro, C. Ducati, C. Cepek, A. Knop-Gericke, S. Milne, C. Castellarin-Cudia, S. Dolafi, A. Goldoni, R. Schlögl and J. J. Robertson, J. Phys. Chem. C, 2008, 112, 12207 CAS.
- T. De los Arcos, F. Vonau, M. G. Garnier, V. Thommen, H.-G. Boyen, P. Oelhafen, M. Dueggelin, D. Mathis and R. Guggenheim, Appl. Phys. Lett., 2002, 80, 2383 CrossRef CAS.
- E. G. Colgan, J. P. Gambino and Q. Z. Hong, Mater. Sci. Eng., R, 1996, 16, 43 CrossRef.
- C. Zhang, F. Yan, C. S. Allen, B. C. Bayer, S. Hofmann, B. J. Hickey, D. Cott, G. Zhong and J. J. Robertson, J. Appl. Phys., 2010, 108, 024311 CrossRef.
- S. B. Adler, Chem. Rev., 2004, 104, 4791 CrossRef CAS.
- K. Kobayashi, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601, 32 CrossRef CAS.
- G. E. Brown, Jr., V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Grätzel, G. Maciel, M. I. McCarthy, K. H. Nealson, D. A. Sverjensky, M. F. Toney and J. M. Zachara, Chem. Rev., 1999, 99, 77 CrossRef.
- D. Emfietzogloua and H. Nikjoob, Radiat. Res., 2007, 167, 110 CrossRef.
- S. Matsuyama, H. Mimura, H. Yumoto, H. Hara, K. Yamamura, Y. Sano, K. Endo, Y. Mori, Y. Nishino, K. Tamasaku, M. Yabashi, T. Ishikawa and K. Yamauchi, Proc. SPIE 5918, Laser-Generated, Synchrotron, and Other Laboratory X-Ray and EUV Sources, Optics, and Applications II, p. 591804, 2005.
- W. Chao, P. Fischer, T. Tyliszczak, S. Rekawa, E. Anderson and P. Naulleau, Opt. Express, 2012, 23, 9777 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |