 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInverse electron demand Diels–Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme
Astrid-Caroline
Knall
* and
Christian
Slugovc
Institute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9/5, A-8010 Graz, Austria. E-mail: a.knall@tugraz.at; Web: http://ictm.tugraz.at Fax: +43 316 1032284
First published on 8th April 2013
Abstract
Inverse electron demand Diels–Alder reactions (iEDDA) between 1,2,4,5-tetrazines and olefins have emerged into a state-of-the art concept for the conjugation of biomolecules. Now, this reaction is also increasingly being applied in polymer science and materials science. The orthogonality of this exciting reaction to other well-established click chemistry schemes, its high reaction speed and its biocompatibility are key features of iEDDA making it a powerful alternative to existing ligation chemistries. The intention of this tutorial review is to introduce the reader to the fundamentals of inverse electron demand Diels–Alder additions and to answer the question whether iEDDA chemistry is living up to the criteria for a “click” reaction and can serve as a basis for future applications in post-synthetic modification of materials.
 Astrid-Caroline Knall | Astrid-Caroline Knall studied Industrial Chemistry at the Graz University of Technology in Austria. Her dissertation (under the supervision of Franz Stelzer) focused on the synthesis of Europium coordination compounds and their application in water vapour sensors. She then moved to Borealis Polyolefine GmbH where her main research areas were reactive modification and additivation of polyolefins. In 2011, she returned to Graz University of Technology as a postdoctoral research fellow where she is now working at the Institute for Chemistry and Technology of Materials. Her research interests include tetrazines as building blocks for functional (stimuli-responsive, fluorescent and photosensitive) polymers. |
 Christian Slugovc | Christian Slugovc studied Chemistry at the Vienna University of Technology and received his PhD in 1998. After a postdoc-year at the CSIC in Sevilla, working on C–H activation with iridium complexes, he joined Graz University of Technology in 2002 where he worked on ring opening metathesis polymerisation for his habilitation which he finished in 2007. Since then he has been an Associate Professor at the Institute for Chemistry and Technology of Materials at Graz University of Technology. His research interests comprise the application of olefin metathesis in macromolecular and materials chemistry, research on olefin metathesis catalysts, organometallic phosphorescent complexes and porous polymers. |
Key learning points(1) Electron-rich dienophiles and electron-poor dienes (e.g. tetrazines) react in an inverse electron demand Diels–Alder reaction (iEDDA).(2) iEDDA fulfils Sharpless' criteria for a “click” reaction while offering advantages over already established “click” chemistry schemes. (3) Tetrazines and suitable dienophiles are readily available with a variety of chemical functionalities. (4) Both reaction partners can be modified to balance parameters like stability, reactivity and availability of reagents. (5) iEDDA can be performed simultaneously with other “click” reactions without the need for protecting groups. |
1. Introduction
Soon after its introduction by Sharpless et al. in 2001,1 the concept of “click chemistry” has evoked increasing interest in the scientific community. Initiated by the archetypal “click” reaction – copper-catalysed azide–alkyne Huisgen 1,3-dipolar cycloaddition (CuAAC) – a variety of reactions has been identified to be suitable for the conjugation of molecules using different ligation concepts.2,3 Among these, the inverse electron demand Diels–Alder addition (iEDDA, Scheme 1) of 1,2,4,5-tetrazines and olefins, occasionally referred to as Carboni–Lindsey4 reaction, has increasingly gained importance in the last few years. Other than iEDDA,5 SPIEDAC6,7 (strain-promoted inverse electron demand Diels–Alder cycloaddition), DAinv8 (Diels–Alder with inverse electron demand) or ihDA/rDA9 (inverse electron demand hetero Diels–Alder–retro-Diels–Alder) are used as abbreviations. Initially applied for the synthesis of pyridazines,4,9 iEDDA was introduced as a potential click chemistry scheme almost simultaneously by two groups in 2008.10,11 Since then, it has gathered a remarkable amount of attention, especially in the life sciences, where challenging problems could be solved12 and therefore already became an established method in the fields of bioorthogonal and metal-free click chemistry.3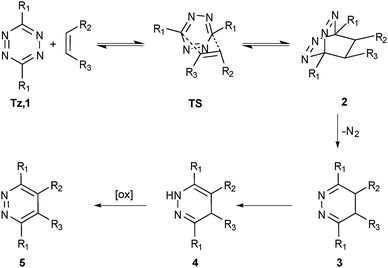 | ||
| Scheme 1 iEDDA reaction scheme. | ||
The question to be answered is now whether iEDDA fully lives up to the criteria of a “click reaction”1 and is a suitable tool also for other tasks, like the connection of building blocks to achieve new functional materials. Equimolarity, high yields, generation of inoffensive by-products and stable products are desirable properties to avoid tedious purification steps, especially when large scale is of interest. Furthermore, chemoselectivity, a wide scope, a fast timescale and readily available stable starting materials would be beneficial for a successful, modular “click” approach.
In this tutorial review, we first aim to introduce the reader to the basics of iEDDA. That is, we discuss the mechanism of the inverse electron demand Diels–Alder addition–retro Diels–Alder reaction cascade in more detail and present suitable reaction partners. Then, synthetic approaches toward tetrazines and methods to characterise iEDDA reactions in terms of reaction speed are presented. Secondly, we demonstrate that the criteria mentioned above are fulfilled and highlight three key assets of iEDDA: fast kinetics, bioorthogonality and the opportunity to perform mutually orthogonal click chemistries. For this purpose recent examples from different fields are used.
2. iEDDA basics
2.1 Mechanism
Tetrazines (Scheme 1, Tz, 1) and suitable dienophiles (e.g.6–13, Fig. 1) react in an inverse electron demand hetero-Diels–Alder (ihDA)–retro-Diels–Alder (rDA) reaction cascade9 to form dihydropyridazines which might be further oxidised to the corresponding pyridazines. The first step in this sequence is a Diels–Alder [4+2] cycloaddition with inverse electron demand resulting in a highly strained bicyclic adduct (2).![(a) Dienophiles14trans-bicyclo[6.1.0]nonene (6), trans-cyclooctene (TCO, 7), methylcyclopropene (8), bicyclo[6.1.0]nonyne (9), cyclooctyne (10), norbornene (11), cyclopentene (12), styrene (13) and (b) tetrazines5,13,15,16 (e.g. 3-(benzylamino)-tetrazine (14) or 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (15)) used in iEDDA reactions and their (estimated) relative reactivities.](/image/article/2013/CS/c3cs60049a/c3cs60049a-f1.gif) | ||
| Fig. 1 (a) Dienophiles14trans-bicyclo[6.1.0]nonene (6), trans-cyclooctene (TCO, 7), methylcyclopropene (8), bicyclo[6.1.0]nonyne (9), cyclooctyne (10), norbornene (11), cyclopentene (12), styrene (13) and (b) tetrazines5,13,15,16 (e.g. 3-(benzylamino)-tetrazine (14) or 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (15)) used in iEDDA reactions and their (estimated) relative reactivities. | ||
Due to this concerted reaction mechanism, the reaction is characterised by second-order kinetics, the rate determining step being the initial [4+2] cycloaddition between Tz as diene and the olefin as dienophile.5 The initially formed, highly strained bicyclic adduct 2 is then rapidly converted in a retro-Diels–Alder step (upon release of nitrogen) to the corresponding 4,5-dihydropyridazine 3. A subsequent 1,3-prototropic isomerisation leads to the corresponding 1,4-dihydro-isomer 4. This process was reported to take place via aminal formation as confirmed by 1H-NMR.13
Concise information on isomerisation from 3 to 4 as well as on the final oxidation step in terms of a systematic study is still missing. This may be the case due to two reasons: firstly, the initial [4+2] cycloaddition was identified to be the rate determining step and thus further reaction steps were neglected. Secondly, if iEDDA is used as a conjugation tool, it is not of prime importance whether the coupling sites are present in their oxidised form, unless the formed pyridazines are used for further studies e.g. as coordinating sites. Depending on the type of added alkene, intermediate 4 is quite stable, meaning that also after allowing some time, the NH signal peaking up at 8.5–9 ppm is still present in 1H-NMR spectra of iEDDA reactions and oxidants are needed to obtain the pyridazine product 5. Examples of such oxidising agents are nitrous gases, isoamyl nitrite, hydrogen peroxide or DDQ (2,3-dichloro-5,6-dicyanobenzoquinone). If alkynes are used as dienophiles, 5 is formed without this final oxidation step. Surprisingly, this is not linked to a higher reactivity of alkynes towards tetrazines which is the actual opposite to azide–alkyne click chemistry.
According to frontier orbital theory,9 Diels–Alder reactions can be divided into three types (Fig. 2): in neutral Diels–Alder additions ΔE between the HOMO and LUMO of both reactants is similar, normal Diels–Alder additions show a reduced HOMOdiene–LUMOdienophile separation (compared to the HOMOdienophile–LUMOdiene separation) which dominates the reactivity and finally, inverse electron demand Diels–Alder reactions are governed by the HOMOdienophile–LUMOdiene gap.17 Therefore, as a general rule of thumb, electron-withdrawing substituents at 3- and 6-positions of the tetrazine lower the LUMO of the diene and thus accelerate iEDDA while electron-donating substituents have a negative effect on reaction rates.9 Furthermore, dienophiles with electron-rich substituents (raising the HOMO of the dienophile) are preferred. Concomitantly, steric effects need to be considered. Sterically demanding substituents on both reaction partners raise the distortion energy18 needed to attain the aforementioned transition state TS (Fig. 1), whereas a high degree of ring strain of the dienophile facilitates iEDDA reactions by reducing the activation energy (caused by a raised HOMOdienophile and reduction of the distortion energy).
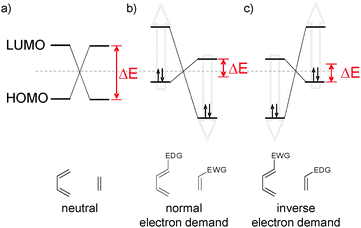 | ||
| Fig. 2 Frontier orbital model of (a) neutral, (b) normal electron demand and (c) inverse electron demand Diels–Alder additions (EDG = electron-donating group, EWG = electron-withdrawing group).9,17 | ||
Studies on the iEDDA addition rates of substituted styrenes (13) as substrates and py-Tz (15, Fig. 1) led to the determination of Hammett parameters which were found to be reduced in polar solvents.19 Generally, the dependence of reaction rates on solvent polarity was found to be small.17 Furthermore, a drastic increase of reaction rates was observed, when water was added to the reaction mixtures, as observed in other publications.10,20 This is caused by a stabilising interaction between water and the activated complex TS and an enhancement of hydrophobic interactions between the two reactants, which is also the case for Diels–Alder reactions with “normal” electron demand. These results further support a concerted [4+2]-cycloaddition. A slight acceleration of reaction rates at lower pH due to protonation of the pyridine rings (and consequently, increased electron-deficiency of the diene) was observed, while the ionic strength did not seem to influence the reaction rate, which suggests a simple kind of Lewis acid catalysis. Less attention has been, so far, devoted to the final oxidation step, which is occasionally accelerated due to elimination of reaction products (amines, alcohols, cyclopentadiene)9 from the corresponding dihydropyridazines. “Self-oxidation” by unreacted tetrazine was observed when styrenes (13) were used as dienophiles.14
According to the literature, the Alder–Stein principle (the relative stereochemistry of dienes and dienophiles is conserved), and also Alder's endo-rule (the endo-adduct is the kinetically preferred product), should apply,14 however, upon oxidation/rearomatisation, this stereochemical information is lost. LC-MS (liquid chromatography-mass spectrometry) chromatograms after iEDDA of BAT (14, Fig. 1) and a monosubstituted norbornene, however, showed a mixture of four dihydropyridazine products11,16 with the same base peak. Therefore, both endo- and exo-addition products are formed, each with the two respective possible orientations, which indicates a limited stereoselectivity.11
2.2 iEDDA precursors
Previous reviews on tetrazine chemistry treated iEDDA rather as a side aspect and concentrated on their strong electron-deficiency, their ability to be reversibly reduced and their occasional fluorescence.9,21 A lot of applications for tetrazine-based materials, for example, in the field of addressable sensors, displays, high-energy materials (used in propellants, explosives and pyrotechnics), in coordination chemistry, in the synthesis of heterocycles9 and in total synthesis already exist, however, another new and exciting application of tetrazines is their incorporation as electron-deficient comonomers in alternating donor–acceptor copolymers used for photovoltaics.22This has led to synthesis routes for a considerable amount of different asymmetric and symmetric tetrazines with various functionalities.21 While the aforementioned application of tetrazines in high-energy compounds and their high reactivity in [4+2] cycloadditions suggest a limited stability of tetrazines, many representatives, such as the aryl-substituted derivatives presented in Fig. 1, are stable under ambient conditions and also safe to handle, unless they bear substituents with high nitrogen content. However, it is generally advisable to perform tetrazine synthesis in a well-ventilated hood. Tetrazine syntheses are to date mostly performed on a small scale, also owing to the prices and availability of their nitrile precursors.
A straightforward method to prepare tetrazines is a two-step procedure starting from aromatic nitriles (or their analogues, such as nitrile imines or aldehydes, Scheme 2a) using hydrazine or formamidine acetate (for asymmetrically substituted tetrazines, Scheme 2b) as reagents.21 Depending on the structure of the nitrile starting material, anhydrous hydrazine or hydrazine hydrate may be used; common (co)solvents are THF or ethanol. Sometimes, sulphur5 or acids are added as catalysts. A recently developed transition metal-catalysed microwave synthesis protocol enabled the conversion of aliphatic nitriles (Scheme 2a) to tetrazines.23 In the second step, the resulting dihydrotetrazine is oxidised (for this purpose, DDQ,24 isoamyl nitrite22 or HONO14 are often used depending on the substrate structure). Occasionally, this oxidation step is already accomplished in the presence of oxygen (as in the synthesis of unsubstituted tetrazine).14
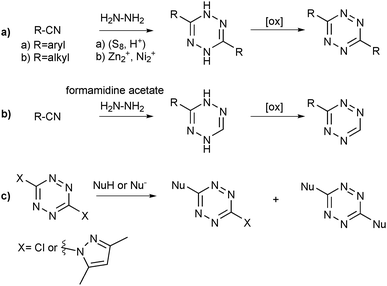 | ||
| Scheme 2 Synthesis strategies for tetrazines.21,23 | ||
This nitrile-based route has the drawback that attempts to prepare asymmetrically functionalised tetrazines will lead to a statistical mixture of products (a theoretical maximum of 50% yield can be achieved for the target compound when none of the three products is preferably formed) which have to be purified, mostly involving chromatographic methods. This and side reactions (e.g. formation of the corresponding 1,2,4-triazoles or of thiadiazoles in the presence of sulphur)21 both may reduce the effective yield. However, the synthesis of symmetrical bisaryltetrazines like pyTz proceeds smoothly, owing to their good crystallising behaviour which facilitates purification. Compared to this, the preparation of asymmetrically substituted tetrazines using formamidine acetate (Scheme 2b) has the advantage that firstly, only a single product is expected which is monofunctionalised and secondly, more reactive monosubstituted tetrazines are obtained.
Alternatively, 1,2,4,5-tetrazine-3,6-dicarboxylic acid can be obtained via dimerisation of ethyldiazoacetate in the presence of a base, while nucleophilic substitution of 1,2,4,5-substituted tetrazines (Scheme 2c) allows the preparation of differently functionalised tetrazines.9,21
Most tetrazines are characterised by their bright colours which are typically ranging from different shades of pink to red. Upon successful Diels–Alder reaction, the pink color vanishes (corresponding to a decrease of the characteristic absorption band at around 540 nm) which can be used as a means of reaction control, but also for the determination of reaction rates under pseudo first-order conditions using UV/Vis spectroscopy5,13,15,16 while for ultrafast iEDDA kinetics, stopped-flow approaches were used.43 Researchers used this technique to evaluate the hydrolytic stability of tetrazines,15 too, which is relevant when iEDDA reactions are performed in water, buffer solutions or even in in vivo approaches. Here, a general relationship between increased reactivity and reduced hydrolytic stability can be concluded, however, not all tetrazines presented in Fig. 1 have been compared in this regard. Furthermore, occasional exceptions from this correlation occur (for example, BAT (14) is less prone to hydrolysis than pyTz (15) but due to reduced sterical hindrance, more reactive than expected).15
Alternatively, reaction progress can be monitored via1H-NMR,14,24 infrared spectroscopy or high-performance liquid chromatography (in combination with mass spectrometry11 or UV/Vis detectors). The formed “click” products may be characterised, in addition to these techniques, with more sophisticated methods, such as MALDI-TOF-MS (matrix-assisted laser desorption ionisation time-of-flight mass spectrometry)20,24 or SEC (size exclusion chromatography).24
3. iEDDA features and applications
A previous review article covers advances in biomedical applications of iEDDA click chemistry for multistep labeling of live cell surface antigens, intracellular imaging of small molecules and in vivo imaging applications as well as modification of cells with nanomaterials for clinical diagnostics.12 Therefore, this section concentrates on the underlying chemistry to give an impression that features of iEDDA are responsible for its success in life sciences and could also be of interest for other fields. Herein, we focus on the three most impressive features of iEDDA: fast reaction rates, bioorthogonality and mutual orthogonality with other click reactions.Basically, two main strategies have been followed. On the one hand, iEDDA was used to construct molecular probes which were then applied in labeling experiments or, on the other hand, for the conjugation of tetrazines and alkenes or alkynes directly in vitro or in vivo (termed pretargeted labeling). While in the former concept, especially the equimolar ratio and the high reaction speed are of interest, the second strategy takes benefit of iEDDA's versatility, bioorthogonality, robustness and selectivity. Therefore, scientists were enabled, in either of these approaches, to solve challenging tasks, which will be used to illustrate the capability of this rather new conjugation chemistry.
3.1 Fast kinetics
All click chemistry schemes involving azides have in common that tuning of reaction rates via modification of the azide reaction partner is virtually impossible (albeit aryl azides tend to be less reactive than their alkyl counterparts25). In CuAAC reactions, rates are influenced by employing different solvents, copper species or the use of specific ligands which accelerate CuAAC, for example via stabilisation of copper(I) in aqueous solution. Modification of the alkyne reaction partner using electron-withdrawing substituents or strained systems (strain-promoted azide–alkyne click chemistry, SPAAC) led to significant improvements in terms of reaction rates so that copper catalysts could be omitted.2,3,7 Similarly, increasing the electron density of the phosphine substituents in Staudinger ligation experiments raised reaction rates, but at the cost of increased phosphine oxidation as an unwanted side reaction.26 Demanding applications in the field of chemical biology, however, require even higher reaction rates which could be offered by iEDDA chemistry (Table 1).| Reaction | Reactants | Rate [M−1 s−1] | Reference |
|---|---|---|---|
a Difluorocyclooctyne.
b Biarylazacyclooctynone.
c Measured in CD3CN.
d In H2O–MeOH (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), 21 °C.
e H2O–dioxane (90:10), 25 °C. :5), 21 °C.
e H2O–dioxane (90:10), 25 °C.
|
|||
| CuAAC | Terminal alkyne + azide | 10−2–10−4 | 20 |
| Staudinger–Bertozzi ligation | Azide + phosphine | 2.2 × 10−3c |
26 |
| Electron deficient AAC | DIFOa + azide | 8 × 10−2c |
26 |
| SPAAC | BARACb + azide | 1c | 26 |
| iEDDA | Norbornene + tetrazine | 1–10d | 16 |
| iEDDA | 6 + tetrazine | 3.8 × 105e |
8 |
Moreover, both iEDDA reaction partners can be used to modulate the reaction speed while the total reactivity range spans nine orders of magnitude, depending on the nature of the two reactants and furthermore, reaction conditions.7,14 For example, speeding up the reaction by replacement of the initially used norbornenes 11 by more reactive trans-cyclooctenes 7 (conveniently obtained via photoisomerisation of the corresponding cis-isomers)20 allowed the use of less reactive but more stable tetrazines (with respect to hydrolysis as well as nucleophiles present in biological systems), thus opening up a plethora of bioorthogonal applications. Further chemical modification, for example, enhancement of ring strain of 713 or 95 by a condensed cyclopropanyl ring resulted in even faster iEDDA reactions. To date, the fastest iEDDA example, using trans-bicyclo[6.1.0]nonene (6) as dienophile, proceeds at an impressive rate of up to 380000 M−1 s−1,8 which is more than five orders of magnitude higher than SPAAC.7
Attempts to prepare Tz derivatives with 18F were less successful, one reason being that diaryltetrazines were found to be not stable under commonly used mild nucleophilic fluorination conditions.27
iEDDA click chemistry was also applied for the synthesis of radiometalated probes (64Cu,3089Zr30 or 111In31). In these approaches tetrazine-derivatised chelators (like 19, Fig. 4) or desferrioxamine (DFO) derivatives were used to prepare radiolabeled antibodies30 or to conjugate the radiolabels to tumor cells pretargeted with TCO-bearing antibodies directly in vivo.31
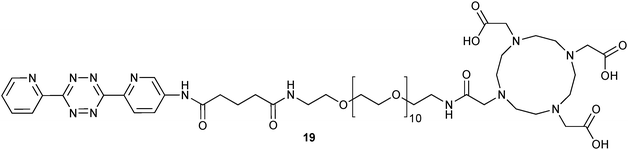 | ||
| Fig. 4 DOTA (1,4,7,10-tetraazacyclo-dodecane-1,4,7,10-tetraacetic acid) derivatised tetrazine (19) for 111In radiometalated “chasers”. | ||
3.2 Bioorthogonality
The development of bioorthogonal reactions was driven by biologists and chemists striving for techniques to study biomolecules in their natural environment.3,26 Initially, inherent functionalities were used to connect biomolecules to labels and the resulting bioconjugates were applied to monitor uptake, conversion or excretion of relevant substances, e.g. metabolites. Examples of these inherent functionalities are the amino groups in lysine or the thiol groups in cysteine or in polypeptides, as well as the 5′ and 3′ termini of DNA. In addition to conversions of these functionalities like condensation of amines with aldehydes, click chemistry emerged as a powerful tool for bioorthogonal chemistry. Another breakthrough in this field was achieved by genetic encoding, that is, the incorporation of non-natural amino acids in proteins via genetic engineering, which can be then converted using different conjugation techniques.6,16,20 For bioorthogonal reactions, a prerequisite is that the conjugation chemistry uses functionalities which are absent from natural biomolecules, and furthermore, the structural perturbation imposed by the newly introduced functional group(s) must be minimal so that the target molecule's natural bioactivity is not altered.3 Furthermore, a high reaction rate is desirable as labeling experiments are often performed at very low concentration levels.This excludes, for example, thiol–ene reactions: radical thiol–ene reactions would lead to unspecific labeling, whereas Michael thiol–ene acceptors are subject to cross-reactions with several nucleophiles present in typical in vitro or in vivo applications. Another prerequisite is the presence of thiol groups in the targeted molecules, which is, for example, not the case in phospholipids or DNA.
A major advantage of iEDDA ligations (over CuAAC) is the lack of need of copper catalysts. In general, metal-free click reactions are of high interest for life science applications2,3 given the cytotoxic nature of copper(I) already at low concentration levels.3,26 Thereby, a limiting factor of most copper-free click reactions is their moderate reaction speed which leads to reduced coupling efficiencies and, consequently, to unsatisfying signal-to-noise ratios in molecular imaging.
Cross-reactivity of tetrazines with nucleophiles like thiols, as suggested occasionally,3 cannot be fully excluded, however, this seems not to be a problem, at least in the short timescale required for iEDDA reactions,13 and can be further improved by the choice of a less reactive tetrazine. This trade-off between reactivity and stability15 can be compensated by a highly reactive dienophile (e.g. TCO or derivatives of 6). Moreover, trans-configured double bonds, as prevalent in cell membranes, are, for example, only sluggishly converted.12 Consequently, iEDDA can be regarded as bioorthogonal which will be further illustrated by the examples presented in the next sections.
Another application was the conjugation of nanoparticles to pretargeted cells using iEDDA, where tetrazine33,34 or alkene-derivatised35 cappers served as conjugation handles. These systems were then conjugated to small molecule antibiotics (for the detection of gram-positive bacteria)33 or antibodies (for diagnostic sensing and pathogen detection).34 Furthermore, this technique was used to label cancer cells with fluorescent35 quantum dots. Several consecutive iEDDA conjugation steps to accumulate a higher amount of nanoparticles further amplified the corresponding signals. This higher labeling density, especially compared to avidin–biotin affinity techniques, could be achieved due to the smaller size of Tz and conjugation partners.34
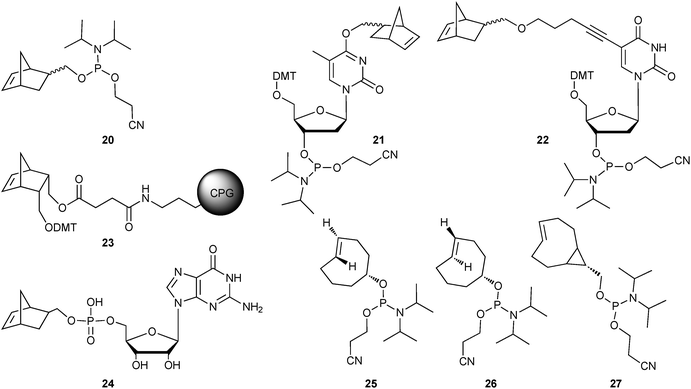 | ||
| Fig. 5 Incorporation of iEDDA dienophiles into DNA and RNA. Norbornene-containing building blocks for PCR (polymerase chain reaction) and solid phase synthesis (20–24; DMT = 4,4′-dimethoxytrityl; CPG = controlled pore glass)36,37 and TCO-derivatised phosphoramidites (25–27).8 | ||
Genetic encoding utilises stop codons which normally direct termination of protein synthesis (for example, the “amber” UAG codon). This “stop” codon can be suppressed while a non-natural amino acid is encoded onto the complementary tRNA instead.16 Examples of such non-natural amino acids, which allow for introduction of clickable groups into specific sites in the final protein, are given in Fig. 6. Using this approach, a 4-aminophenylalanine-derived tetrazine (32)20 as well as non-natural lysine derivatives bearing TCO (28), cyclooctyne (29) or norbornenes (30, 31) have been successfully incorporated in proteins and subsequent iEDDA derivatisation was performed. This concept was further combined with fluorogenic probes and used for the imaging of bacteria6 and cancer cells.16
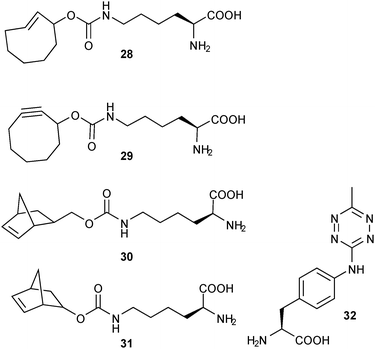 | ||
| Fig. 6 Non-natural amino acids 28,629,630,631,6,16 and 3220 used for genetic encoding of tetrazines and olefins. | ||
3.3. Mutual orthogonality with other click reactions
One of the most appealing features of iEDDA click chemistry is its mutual orthogonality with azide–alkyne click chemistry, thiol–ene click chemistry and, potentially, further click reactions. In contrast to previous attempts where protecting groups, partial conversion of “clickable” groups,38 or different reactivities (while using one click chemistry scheme)39 were applied, orthogonal click chemistry allows these conjugations to take place next to each other, ideally in an “one-pot approach”. Chemists from diverse fields are attracted by the concept of a multiple “clickable” scaffold to connect complex functionalities in a facile fashion. Accordingly, an increasing number of publications in the field of chemical biology, drug discovery,40 activity profiling,41 macromolecular chemistry2,7 and materials science is showing up in recent literature.The very first example of orthogonal click chemistry was already presented in one of the pioneering iEDDA publications by the Fox group,10 where a dually clickable scaffold (Scheme 3, 33) was used to connect thioredoxin (Trx, a 11.7 kDa protein containing a single disulfide) via Michael addition to the maleimide followed by iEDDA addition of py-Tz to the TCO double bond. The authors used the electron-deficient nature of the tetrazines to selectively attack the strained, electron-rich double bond while leaving the maleimide unaffected. At the same time, the thiol–ene reaction favoured the electron-deficient maleimide double bond under Michael conditions. This allows us to use the inherently reactive thiol groups to attach more complex functionalities by applying 33 in combination with iEDDA click chemistry.
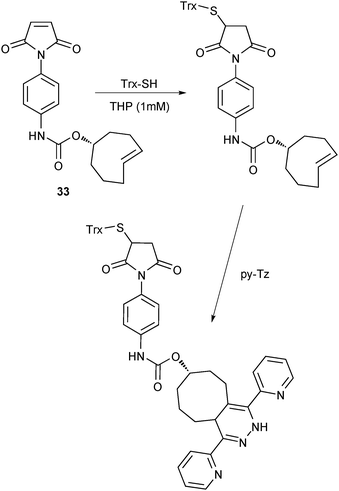 | ||
| Scheme 3 Mutual orthogonality of iEDDA and Michael thiol–ene chemistry (THP = tri(3-hydroxypropyl)phosphine).10 | ||
Later approaches involving tetrazines and alkenes combined norbornenes for iEDDA and terminal alkynes for CuAAC using DNA8 or block copolymers42 as backbone. The respective “cross-reactions” (1,3-dipolar addition of azides to the norbornene double bond38 and iEDDA between tetrazines and terminal alkynes) have been occasionally observed, yet under very harsh reaction conditions. The next, logical step was to extend this approach to a combination with copper-free click chemistry which was used for the simultaneous labelling of two different types of cells.6 This was achieved by genetic engineering to encode non-natural amino acids. The thus obtained cells expressed proteins containing an iEDDA and SPAAC acceptor, respectively, and could be selectively labelled using fluorogenic agents. However, cross-reactivity between the cyclooctyne derivative 29 used in this work and tetrazines was noted as drawback which could be avoided by adding firstly an azide-bearing, then a tetrazine-bearing profluorescent labelling agent.6 Cyclooctynes (see Fig. 7) are prone to undergo both iEDDA and SPAAC, therefore, these two reactions cannot be performed simultaneously and the azide–alkyne click reaction, being the slower one (Table 1), has to be performed prior to the iEDDA reaction. This could be circumvented by the design of new cycloalkynes which are virtually unreactive towards tetrazines but still possess a pronounced reactivity in SPAAC reactions.18 These compounds are characterised by fused benzene rings or methyl groups at the alpha-position to the alkyne. Furthermore, terminal alkynes, which are highly reactive in CuAAC reactions, only react with tetrazines, unless electron-donating substituents are present or relatively harsh reaction conditions are applied.
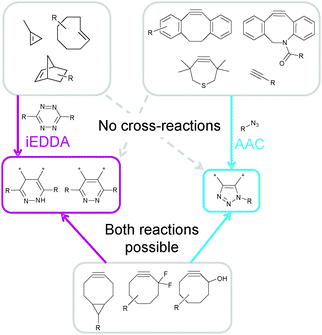 | ||
| Fig. 7 Suitable pairs for mutually orthogonal azide–alkyne/iEDDA click chemistry. | ||
The combination of iEDDA dienophiles (TCO43 and methylcyclopropene,44 respectively) and dibenzocyclooctyne as the SPAAC acceptor finally allowed a copper-free, mutually orthogonal and bioorthogonal protocol. DFT (density functional theory) calculations revealed that the introduction of the two fused benzene rings in dibenzocyclooctyne derivatives raised the distortion energy necessary to attain the first transition state TS in iEDDA reactions,18 so that the reaction rate for tetrazines and dibenzocyclooctyne was drastically reduced. Additionally, two compounds, methylcyclopropene and 3,3,6,6-tetramethylthiacycloheptyne, were predicted to be a mutually orthogonal couple for SPAAC and iEDDA, respectively.
Not much later, attempts to an activity-profiling system with orthogonally clickable probes (using an iEDDA reaction, a Staudinger–Bertozzi ligation and a CuAAC or SPAAC) were presented.41 While the authors were able to perform the three “click” reactions in parallel, practical reasons (background fluorescence due to CuSO4 and a poor signal-to-noise ratio when SPAAC was applied) finally led to a multistep protocol.
The first examples of iEDDA applications in materials science comprise the derivatisation of MOFs (metal–organic frameworks)46 or utilisation of the inherent electron-rich double bonds of multi-walled carbon nanotubes47 using tetrazines. iEDDA was also applied for the immobilisation of carbohydrate arrays for high-throughput screening of carbohydrate–protein interactions.48
Only very recently, the iEDDA reaction has also found application in polymer science. These first macromolecular applications of iEDDA concentrate on norbornenes as the alkene reaction partner, in spite of its comparably modest reactivity, due to the easy availability and stability of norbornenes compared to trans-cyclooctenes. Because mostly organic solvents are employed, more reactive tetrazines, which would undergo hydrolysis in aqueous solution10 (like py-Tz) can be selected and still, reaction rates typically exceed those of comparable CuAAC approaches (cf.Table 1).
Still originating from the field of life science, the first examples of iEDDA conjugation of polymers comprised dextrans with different molecular weight which were attached to a tetrazine labelling agent to modify its pharmacokinetics (which was then used for in vivo PET and fluorescence imaging of tumour epitopes)29 and post-modification of a polymeric imidazole ligand (34, Fig. 8) with a norbornoyl active ester for the preparation of norbornene-coated water-soluble quantum dots which were then conjugated to pretargeted cells bearing TCO-modified antibodies.35 Furthermore, a polyamide with pendent Reppe anhydride (35) and pentenoyl (36) side chains was derivatised by two consecutive iEDDA click chemistry steps due to the different reactivities of the respective tetrazines and alkenes.39
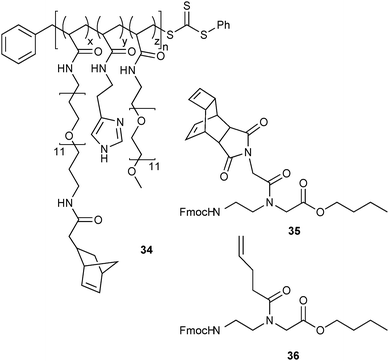 | ||
| Fig. 8 Norbornyl-modified random copolymer (34)35 and monomers (35, 36) for consecutive iEDDA transformations.39 | ||
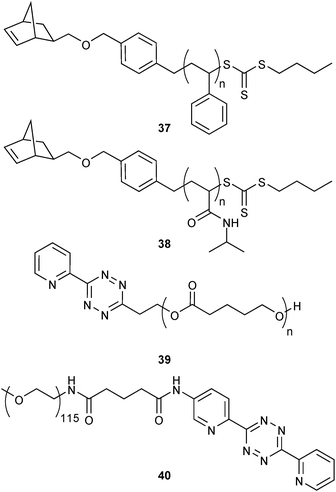
Thereby, the attention of polymer scientists was drawn toward iEDDA as a potential conjugation tool. Initially, the orthogonality of iEDDA and different polymerisation techniques was exploited. For example, iEDDA was found to be compatible with the trithiocarbonate group in RAFT (reversible addition–fragmentation chain transfer polymerisation) chain transfer agents24 or acidic catalysts for ring-opening polymerisation24,49 and could be therefore applied for the end-functionalisation of different norbornyl-terminated (37, 38) or tetrazine-terminated (39, 40) semi-telechelic polymers (Fig. 9). Herein, the authors demonstrated for the first time the suitability of iEDDA to act as a versatile conjugation tool in polymer chemistry. The fast and quantitative nature of iEDDA together with the irreversible “click” junction allows preparation of block copolymers while the discoloration of the initially pink tetrazine-containing materials can be used as a straightforward proof for successful conversion. Due to the high efficiency of iEDDA, none of the conjugation partners has to be applied in excess which facilitates product purification. Size-exclusion chromatography confirmed that the narrow molecular weight distribution of the constituent homopolymers could be conserved.
In analogy to this, later on, a norbornene initiator for the preparation of polylactide chains was presented. The resulting short polymer chains (41, Fig. 10) were then again used for end-functionalisation with py-Tz and preparation of block copolymers with 40. In the same publication, poly(lactides) with norbornene side chains (42) were derivatised with py-Tz using polymer-analog click chemistry.49 In addition to GPC (gel permeation chromatography), successful “click” modification could be proved, in both cases, by MALDI-TOF MS.
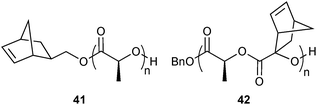 | ||
| Fig. 10 Semi-telechelic (41) and side-chain functionalised (42) poly(lactides) for iEDDA post-polymerisation functionalisation.49 | ||
The compatibility of iEDDA and RAFT was further utilised for orthogonal, one-pot labelling of micelles42 combining mutually orthogonal iEDDA and CuAAC. Taking benefit of the living nature of RAFT, norbornene side chains were introduced into the hydrophobic part of a block copolymer and subsequently, TIPS-protected terminal alkynes were copolymerised to obtain a second, hydrophilic block. After deprotection of the alkynes and self-assembly of the resulting copolymers (Fig. 11, 43) in H2O, the resulting micelles could be selectively decorated in the core (using py-Tz to perform iEDDA) or the shell (performing CuAAC with a profluorescent coumarin azide) and moreover, both reactions could be performed in parallel. However, in this case, a reduction of CuAAC reactivity was noted (also, when the iEDDA reaction was performed first), most likely due to the coordinating nature of the tetrazines. Therefore, the CuAAC reaction had to be performed prior to tetrazine addition, which then resulted in complete functionalisation of both clickable groups.
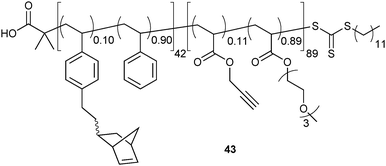 | ||
| Fig. 11 Orthogonally clickable amphiphilic block copolymers.42 | ||
A later paper from the same group uses poly(carbonates) with norbornene side chains as versatile conjugation handles which were used as acceptors for radical thiol–ene, 1,3-dipolar cycloaddition with azides at elevated temperature and iEDDA click chemistry. Also, when the first two “click” reactions were driven to partial conversion, performing the three reactions sequentially led to multi-functionalised copolymers.38
While these first results illustrate the scope and versatility of norbornenes as functional units in macromolecular iEDDA click chemistry concepts, they also give a first idea of its potential limitations. That is, firstly, potential side reactions which have to be considered, especially during polymerisation (which is also the case for unprotected terminal alkynes). A suitable polymerisation method (which is not compromised by iEDDA and vice versa) has to be identified unless a facile post-polymerisation modification is envisageable (e.g. termination agents bearing dienes or dienophiles). For example, polymerisation techniques like ROMP (ring-opening metathesis polymerisation), which rely on the release of ring strain, are excluded when iEDDA dienophiles shall be incorporated in a polymer chain. Secondly, reaction partners, especially in orthogonal approaches, need to be selected carefully to avoid side reactions and also to avoid interference with the material's intended functionality. For example, while cytotoxicity might not be an issue, copper might have, nonetheless, negative effects on material properties, for example, polymer solubility or nanoparticle fluorescence.35 Therefore, a copper-free, orthogonal approach still is the method of choice. However, one should not forget that new functionalities are introduced which will, especially in the case of polymer-analog transformations, affect the final material properties, which could, however offer additional benefits.
4. Summary and outlook
Going back to the initially posed question, iEDDA is undoubtedly fulfilling the criteria for a “click reaction”. The examples presented in this tutorial review demonstrate that the iEDDA reaction is fast enough to be applied for the conjugation of 18F-labeled PET tracers while the chemoselectivity of the reaction allows it to be performed in bioorthogonal in vivo setups. This guarantees site-specific binding while avoiding side reactions, which could be demonstrated by modification of DNA, biocatalytic incorporation and, even more impressively, genetic encoding of tetrazine or alkene-derivatised non-natural amino acids. Accomplishment of these highly challenging tasks proves the wide scope and modularity of iEDDA.These features make iEDDA click chemistry an interesting alternative, also for the modification of materials, especially polymers, not only because of its equimolarity and high yields (as demonstrated in several publications discussed in this review). Unlike in the case of thiol–ene and azide–alkyne click chemistry, both reaction partners can be modified in order to tune their reactivity. Moreover, in polymer and materials sciences, where reaction rates do not need to be as fast as for bioconjugation reactions, a “reasonable time frame” is still easily achieved when less reactive but more abundant substrates like norbornenes are used. Furthermore, due to their numerous applications in other fields, tetrazines have been prepared comprising a number of functionalities and are therefore readily available. This allows us to specifically design tetrazine and alkene reagents for a given application with respect to potential side reactions and linker groups to connect the tetrazines with the substrate/functionality of choice. Additionally, tetrazines are, especially compared to azides, easier to store and handle. Due to the extrusion of nitrogen (no harmful by-products are formed), a stable product is formed and connects the two parts which are to be conjugated.
The assets of iEDDA compared to other click chemistry schemes are obvious: in addition to its high selectivity and impressive reaction speed (in comparison to Staudinger–Bertozzi ligations, strain-promoted or electron-deficient azide–alkyne click chemistry), it also avoids the use of additional catalysts (such as in CuAAC) or initiators (such as for radical thiol–ene reactions). Upon the appropriate choice of reaction partners, hydrolysis (such as in succinimide ester chemistry) or side reactions with other nucleophiles (such as in Michael thiol–ene reactions) are avoided. Furthermore, iEDDA can be performed in parallel with several other “click” reactions, without the need for protecting groups. First corresponding results from theoretical and experimental studies indicate a mutual orthogonality not only between CuAAC or (Michael) thiol–ene reactions and iEDDA, respectively, but also between strain-promoted azide–alkyne click chemistry and iEDDA, provided that suitable reaction partners are selected. In orthogonal click approaches, the formed pyridazines could, due to their coordinating nature, change the kinetics of CuAAC click reactions.
A major current drawback is the synthetic availability of tetrazines using toxic hydrazine as starting material and the potentially explosive nature of some tetrazine precursors (which, however, also applies for azides). Nevertheless, first examples of symmetrical tetrazines as comonomers in electroactive polymers (such as in OPV applications) suggest that these can be produced in appropriate amounts for material synthesis, even when they are used as comonomers. Due to the sometimes expensive starting materials (suitably derivatised nitriles), new synthetic approaches to (especially asymmetrically substituted) tetrazines with improved yields will contribute to the success of iEDDA as a conjugation technique and make it more accessible for applications in materials science.
The first impressive examples from different fields of life sciences and materials science, however, illustrate that iEDDA will act as a powerful additional tool to already established “click” reactions for efficient post-functionalisation of materials. Researchers are provided with a big “playground” of suitable reaction partners, linker groups and reaction conditions which can be easily implemented and are ready to be applied for the synthesis of new functional conjugates. The fine-tuning of the aforementioned orthogonal approaches, new and improved routes to tetrazines and innovative applications of orthogonal concepts are expected to open up avenues to yet unforeseen material combinations with exciting properties.
Acknowledgements
Financial support for a Hertha-Firnberg position for ACK from the Austrian Science Fund (FWF): [T 578-N19] is gratefully acknowledged.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- K. Kempe, A. Krieg, C. Remzi Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176 RSC , and references therein.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC , and references therein.
- R. A. Carboni and R. V. Lindsey, J. Am. Chem. Soc., 1959, 81, 4342 CrossRef CAS.
- W. Chen, D. Wang, C. Dai, D. Hamelberg and B. Wang, Chem. Commun., 2012, 48, 1736 RSC.
- T. Plass, S. Milles, C. Koehler, J. Szymański, R. Mueller, M. Wiessler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166 CrossRef CAS.
- C.-H. Wong and S. C. Zimmermann, Chem. Commun., 2013, 49, 1679 RSC , and references therein.
- J. Schoch, M. Staudt, A. Samanta, M. Wiessler and A. Jäschke, Bioconjugate Chem., 2012, 23, 1382 CrossRef CAS.
- R. A. A. Foster and M. C. Willis, Chem. Soc. Rev., 2013, 42, 63 RSC , and references therein.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518 CrossRef CAS.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297 CrossRef CAS.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816 CrossRef CAS.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646 CrossRef CAS.
- J. Sauer, D. K. Heldmann, J. Hetzenegger, J. Krauthan, H. Sichert and J. Schuster, Eur. J. Org. Chem., 1998, 2885 CrossRef CAS , and references therein.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263 CrossRef CAS.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298 CrossRef CAS.
- J. Sauer and R. Sustmann, Angew. Chem., Int. Ed. Engl., 1980, 19, 779 CrossRef.
- Y. Liang, J. L. Mackey, S. A. Lopez, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 17904 CrossRef CAS.
- J. W. Wijnen, S. Zavarise and J. B. F. N. Engberts, J. Org. Chem., 1996, 61, 2001 CrossRef CAS.
- J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898 CrossRef CAS.
- G. Clavier and P. Audebert, Chem. Rev., 2010, 110, 3299 CrossRef CAS , and references therein.
- Z. Li and J. Ding, Macromol. Chem. Phys., 2011, 212, 2260 CrossRef CAS , and references therein.
- J. Yang, M. R. Karver, W. Li, S. Sahu and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 5222 CrossRef CAS.
- C. F. Hansell, P. Espeel, M. M. Stamenović, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828 CrossRef CAS.
- H. A. Michaels and L. Zhu, Chem.–Asian J., 2011, 6, 2825 CrossRef CAS , and references therein.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef CAS , and references therein.
- Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. D. Brown, P. S. Conti and J. M. Fox, Chem. Commun., 2010, 46, 8043 RSC.
- T. Reiner, E. J. Keliher, S. Earley, B. Marinelli and R. Weissleder, Angew. Chem., Int. Ed., 2011, 50, 1922 CrossRef CAS.
- N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4762 CrossRef CAS.
- B. M. Zeglis, P. Mohindra, G. I. Weissmann, V. Divilov, S. A. Hilderbrand, R. Weissleder and J. S. Lewis, Bioconjugate Chem., 2011, 22, 2048 CrossRef CAS.
- R. Rossin, P. R. Verkerk, S. M. van den Bosch, R. C. M. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., Int. Ed., 2010, 49, 3375 CrossRef CAS.
- D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792 CrossRef CAS.
- H. J. Chung, T. Reiner, G. Budin, C. Min, M. Liong, D. Issadore, H. Lee and R. Weissleder, ACS Nano, 2011, 5, 8834 CrossRef CAS.
- V. M. Peterson, C. M. Castro, H. Lee and R. Weissleder, ACS Nano, 2012, 6, 3506 CrossRef CAS.
- H. S. Han, N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 7838 CrossRef CAS.
- J. Schoch, M. Wiessler and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8846 CrossRef CAS.
- J. Schoch, S. Ameta and A. Jäschke, Chem. Commun., 2011, 47, 12536 RSC.
- R. J. Williams, I. A. Barker, R. K. O'Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285 CrossRef CAS.
- R. Pipkorn, M. Wiessler, W. Waldeck, P. Lorenz, U. Muehlhausen, H. Fleischhacker, M. Koch and K. Braun, Int. J. Med. Sci., 2011, 8, 387 CrossRef CAS , and references therein.
- D. M. Beal and L. H. Jones, Angew. Chem., Int. Ed., 2012, 51, 6320 CrossRef CAS.
- L. I. Willems, N. Li, B. I. Florea, M. Ruben, G. A. van der Marel and H. S. Overkleeft, Angew. Chem., Int. Ed., 2012, 51, 4431 CrossRef CAS.
- C. F. Hansell and R. K. O'Reilly, ACS Macro Lett., 2012, 1, 896 CrossRef CAS.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Angew. Chem., Int. Ed., 2012, 51, 920 CrossRef CAS , and references therein.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60 CrossRef CAS.
- C. Chen, C. A. Allen and S. M. Cohen, Inorg. Chem., 2011, 50, 10534 CrossRef CAS.
- H. Hayden, Y. K. Gun'ko and T. S. Perova, Chem. Phys. Lett., 2007, 435, 84 CrossRef CAS.
- H. G. S. Beckmann, A. Niederwieser, M. Wiessler and V. Wittmann, Chem.–Eur. J., 2012, 18, 6548 CrossRef CAS.
- I. A. Barker, D. J. Hall, C. F. Hansell, F. E. Du Prez, R. K. O'Reilly and A. P. Dove, Macromol. Rapid Commun., 2011, 32, 1362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |