Peptides and peptidomimetics that behave as low molecular weight gelators
Claudia Tomasini* and Nicola Castellucci
Dipartimento di Chimica “G. Ciamician”, Alma Mater Studiorum Università di Bologna, Via Selmi 2, I-40126 Bologna, Italy. E-mail: claudia.tomasini@unibo.it; Fax: +39-0512099456
First published on 2nd October 2012
Abstract
Gelators may be divided into chemical gels and physical gels: the internal structure of chemical gels is made of chemical bonds, while physical gels are characterized by dynamic cross-links that are constantly created and broken. The gelator present in physical gels may be an inorganic or an organic compound, the latter having a molecular weight of ≤500 amu. These compounds are generally called “low molecular weight gelators” (LMWGs). In this tutorial review we want to focus our attention on short peptides or peptidomimetics that behave as LMWGs. Peptidomimetics are small protein-like molecules designed to mimic natural peptides. To efficiently design a peptidomimetic, local constraints must be introduced into the skeleton, to induce the formation of preferred secondary structures.
 Claudia Tomasini | Claudia Tomasini received her chemistry degree cum laude in 1982 and her PhD in Organic Chemistry in 1986 at the Università di Bologna (Italy). In 1987–1988 she was a postdoctoral associate with Professor Jack E. Baldwin at the University of Oxford. In 1990 she joined as an assistant professor in the Chemistry Department at the Università di Bologna, where she became an associate professor in 1998. Her research interests include: synthesis of unusual amino acids, preparation and conformational studies of pseudopeptide foldamers, analysis of the behaviour of synthetic oligomers in solution and in the solid state for the formation of supramolecular materials. |
 Nicola Castellucci | Nicola Castellucci was born in Urbino (Italy) in 1984. In December 2008, he received his master in Advanced Chemical Methodologies from the University of Bologna under the direction of Prof. C. Trombini on “New Methods in Organocatalysis”. In January 2009, he obtained a fellowship in Bologna working in the field of the synthesis of new pseudopeptides under the supervision of Prof. Claudia Tomasini. In 2010, he started the PhD in the same group. |
1. Introduction
Dorothy Jordan Lloyd (1 May 1889 to 21 November 1946) was an early protein scientist who studied the interactions of water with proteins, particularly gelatin. She was the first to propose that the structure of globular proteins was maintained by hydrogen bonds. She accepted an invitation from R. H. Pickard in 1921 to join the newly formed British Leather Manufacturers' Research Association. In 1927, she succeeded Pickard as director, and was, until her death, the only woman leading such an association for industrial research. She was also the vice-president of the Royal Institute of Chemistry (1943–1946) and a member of the Chemical Council. Besides many contributions to scientific journals, Dorothy Jordan Lloyd was the author of The Chemistry of the Proteins and proposed in 1926 this definition for “gel”: “only one rule seems to hold for all gels and that is that they must be built up from two components, one which is a liquid at the temperature under consideration and the other which, the gelling substance proper, often spoken of as the gelator, is a solid. The gel itself has the mechanical properties of a solid, i.e. it can maintain its form under stress of its own weight and under any mechanical stress it shows the phenomenon of strain”.1 Before Jordan Lloyd, a definition of gel was given in 1861 by Thomas Graham: “while the rigidity of the crystalline structure shuts out external expressions, the softness of the gelatinous colloid partakes of fluidity and enables the colloid to become a medium for liquid diffusion, like water itself.”2What is a general definition for gel? The succinct definition of Jordon Lloyd is still useful: “if it looks like a gel it must be a gel”. A gel is a solid, in the sense that it has its own shape and stands its own weight and is easily identified by the simple “inversion test” (turn a pot of gel upside down and it is able to support its own weight without falling out onto your shoes!), but actually it is made of about 99% of a liquid and of 1% of a solid (called “gelator”). The generally accepted definition is that advanced by Flory in 1974: “a gel has a continuous structure that is permanent on the analytical time scale and is solid-like in its rheological behavior”.3
The first gelator that has been studied is gelatin, a mixture of peptides and proteins produced by partial hydrolysis of collagen extracted from the skin, boiled crushed bones, connective tissues, organs and some intestines of animals. It melts to a liquid when heated and solidifies when cooled again. Together with water, it forms a semi-solid colloid gel. Gelatin is commonly used for pharmaceutical and medical applications because of its biodegradability and biocompatibility in physiological environments.4
Several kinds of gels are currently in use for medical applications, due to their high water content and mechanical similarity to natural tissues.5 They may be divided into chemical gels and physical gels (Fig. 1): the internal structure of chemical gels is made of chemical (covalent) bonds,6 while physical gels are characterized by dynamic cross-links that are constantly created and broken, changing their state between solid and liquid under influence of environmental factors. The gelator present in physical gels may be an inorganic or an organic compound, the latter having a molecular weight of ≤500 amu. These compounds are generally called “low molecular weight gelators” and we will simply refer to them as LMWGs.
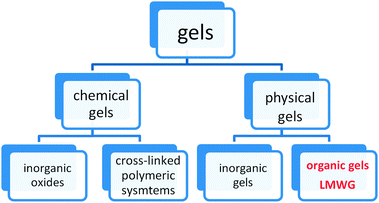 | ||
| Fig. 1 General classification of gels. | ||
The mechanism through which LMWGs operate depends on a hierarchical self-assembly process which occurs through the following sequence of steps: (i) multiple non-covalent interactions (e.g. hydrogen bonds, π–π interactions, van der Waals interactions) between molecular-scale building blocks allow them to self-assemble into supramolecular polymers referred to as fibrils; (ii) the fibrils often then assemble into nanoscale bundles, referred to as fibres; (iii) the fibres tangle and interact with one another to form a self-supporting, sample-spanning ‘solid-like’ network, which underpins the macroscopic gel. Gel-phase materials are typically prepared by heating and cooling a gelator–solvent mixture, with heating or ultrasound irradiation being used to solubilise the gelator and the gel fibres subsequently.
Some other useful definitions are:
- hydrogel: a gel made of a LMWG and water;
- organogel: a gel made of a LMWG and any kind of organic solvent;
- xerogel: a solid formed after evaporation of the solvent in a hydrogel or an organogel by drying with unhindered shrinkage. Xerogels usually retain high porosity (15–50%) and enormous surface area (150–900 m2 g−1), along with very small pore size (1–10 nm);
- aerogel: a solid formed when solvent removal occurs under supercritical conditions; the network does not shrink and a highly porous, low-density material is produced. Aerogels are materials with exceptional properties including very low density, high specific surface areas, and excellent thermal insulation properties.
Finally, a clear distinction should be made between genuine gels, which consist of solid-like fibre networks, and the highly viscous solutions of cylindrical micellar aggregates of amphiphilic molecules. The latter may have a gel-like aspect because of their high viscosity and viscoelasticity but they differ fundamentally from a gel in that they exist at temperatures above the Krafft temperature,7 and in that they can flow, albeit slowly. This distinction is important because cylindrical micelles seem to form, regardless of the presence of stereogenic centres in the amphiphile, and most of the studied micellar cylinders are made of achiral molecules.8
Although only recently systematic studies on LMWG have been undertaken, this research field has quickly blossomed and several reviews have been published in the last few years.9–14 Unlike polymeric hydrogels, supramolecular hydrogels can change their pore sizes as a result of the reassembly of LMWGs during the shrinkage/swelling processes: this property of self-adjusting pore sizes might render supramolecular hydrogels as “smart” matrices for controlled drug release.15
In this tutorial review we want to focus our attention on short peptides or peptidomimetics that behave as LMWGs.
Peptidomimetics are small protein-like molecules designed to mimic natural peptides. It is possible to design these molecules in such a way that they show the same biological effects as their peptide role models but with enhanced properties like a higher proteolytic stability, higher bioavailability and also often with improved selectivity or potency.16
2. General requirements of a LMWG
LMWGs are a large variety of molecules that share the ability to make gels.17 We can make the following broad statements about a typical gelator molecule:(i) the molecule must be partly soluble and partly insoluble in the solvent;
(ii) the molecule must have the potential to form weak intermolecular interactions, such as hydrogen bonds, electrostatic interactions, π–π stacking interactions, hydrophobic forces, non-specific van der Waals forces, and chiral dipole–dipole interactions;
(iii) the non-covalent interactions should be directional, leading to the assembly of anisotropic nanoscale fibres;18
(iv) for this reason, often LMWGs are chiral molecules.
Most LMWGs contain more than one stereogenic centre, so they can exist under several stereoisomers, that usually show completely different gelation behaviors. The spatial arrangement of different diastereoisomers strongly affects their possibility to form weak interactions, such as hydrogen bonds, π–π stacking interactions, van der Waals interactions. Thus changing the configuration of a single stereocentre may have dramatic effects on the solubility properties, the range of solvents that can be gelled, the stiffness of the gels it forms, its critical gelation concentration, and the temperature at which its gels melt.
Both peptides and peptidomimetics tend to form secondary structures, such as helices or sheets, stabilized mainly by intramolecular or intermolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds.19 Helices are stabilized by intramolecular hydrogen bonds, while sheets may be stabilized both by intermolecular or intramolecular hydrogen bonds (Fig. 2), depending on the molecular structure and the chain length. We can gather that self-organized oligomers that form helices are bad candidates as LMWGs, in contrast, oligomers that tend to have as preferred secondary structure, a β-sheet structure, are good candidates to form fibres, thus to behave as gelators.
C hydrogen bonds.19 Helices are stabilized by intramolecular hydrogen bonds, while sheets may be stabilized both by intermolecular or intramolecular hydrogen bonds (Fig. 2), depending on the molecular structure and the chain length. We can gather that self-organized oligomers that form helices are bad candidates as LMWGs, in contrast, oligomers that tend to have as preferred secondary structure, a β-sheet structure, are good candidates to form fibres, thus to behave as gelators.
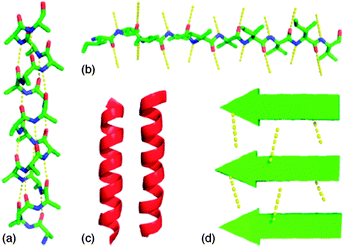 | ||
| Fig. 2 α-Helices, β-strands, and higher order structures. (a) An α-helix with the hydrogen-bonding pattern parallel to the helix axis; (b) β-strand with illustrative hydrogen bonds orthogonal to the direction of the chain; (c) α-helices forming higher order structures, the hydrophobic faces (shaded in dark red) coming together to sequester the hydrophobic residues from the solvent; (d) β-sheet assembly is stabilized by the formation of intermolecular hydrogen bonds. Reproduced from ref. 19 with kind permission of Wiley VHC. | ||
Another important feature for the formation of hydrogels from LMWG is the control of the pH. Indeed the formation of fibrils that are prodromal to the gel formation may be deeply influenced by the pH. For instance Pochan, Schneider and coworkers described the design and characterization of a 20 amino acid peptide (MAX1) that is capable of undergoing triggered hydrogelation.20 The primary sequence of MAX1 contains 20 amino acids, eight of which are lysine residues and nine of which are valine residues. When dissolved in aqueous solutions of low ionic strength at pH 9 and temperatures below ∼20 °C, the peptide remains unfolded. When MAX1 is dissolved in pH 9 buffer of low ionic strength, it is capable of undergoing temperature-induced folding and self-assembly due to the formation of a β-sheet structure (Fig. 3). A similar behavior may be found for LMWG, containing amino or/and carboxy groups.
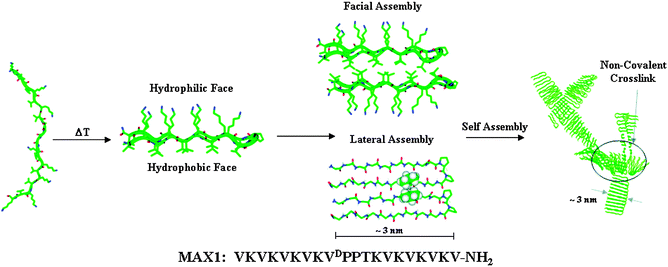 | ||
| Fig. 3 Folding and self-assembly mechanism of MAX1 leading to hydrogel formation. Increasing the temperature of a solution of unfolded peptide at pH 9 results in the formation of an amphiphilic β-hairpin structure that subsequently undergoes lateral and facial self-assembly to form a fibrillar supramolecular structure. Non-covalent cross-linking of the fibrils results in the formation of a macroscopic hydrogel. The sequence of MAX1 is shown below. Reproduced from ref. 20 with kind permission of The American Chemical Society. | ||
3. Peptide LMWG
Peptide-based scaffolds are interesting candidates for hydrogelation and organogelation because they can self-assemble using various noncovalent interactions, including hydrogen bonding, electrostatic, or π–π interactions. These interactions lead to the formation of organized supramolecular structures that can entrap and immobilize many solvent molecules under appropriate conditions. Moreover, they are easy to manufacture in large quantities, and they can also be easily modified chemically and biologically.3.1 L-Lysine based LMWG
Although Nε-lauroyl-Nα-stearylaminocarbonyl-L-lysine ethyl ester was first reported as an L-lysine based organogelator,21 several other derivatives of lysine that are efficient organogelators and/or hydrogelators have been developed in the recent years (Fig. 4). L-Lysine based gelators are prepared through easy synthetic procedures, and some classes of gelators are synthesized by the introduction of various functional groups. A recent review by Suzuki and Hanabusa describes the synthesis of organogelators, hydrogelators and amphiphilic gelators based on L-lysine and their gelation properties,22 so these derivatives will not be further described here.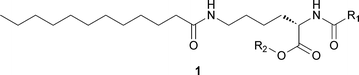 | ||
| Fig. 4 Chemical structure of a generic lysine-based LMWG. | ||
3.2 Fmoc-based LMWG
Several authors have reported dipeptides that are good gelators, when they contain an aromatic protecting group, such as fluorenyl-9-methoxycarbonyl (Fmoc). Indeed the self-assembly of Fmoc–dipeptides is driven by hydrogen bonding and π–π interactions, between π electrons in the aromatic fluorenyl rings.Fmoc-Phe-Phe (Fig. 5) is an important low molecular weight hydrogelator.23 Gazit and coworkers discovered that dissolving Fmoc-Phe-Phe peptide at a higher concentration in aqueous solution resulted in the formation of a rigid material with macroscopic characteristics of a gel.24 Although the hydrogel contained less than 1% peptide material, it kept its 3D spacious volume exceptionally well. This self-assembled gel was stable across a broad range of temperatures, over a wide pH range.
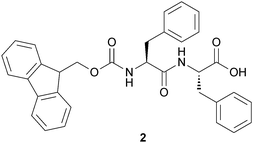 | ||
| Fig. 5 Chemical structure of Fmoc-Phe-Phe. | ||
Gelation can be induced by either lowering the pH of an aqueous solution of Fmoc-Phe-Phe or by the addition of water to a solution of Fmoc-Phe-Phe in a solvent such as DMSO. Despite the volume of literature on Fmoc-Phe-Phe, the mechanical properties reported for the gels vary significantly over four orders of magnitude and the origins of this variability are unclear.25 Ulijn and coworkers construct a model based on the data obtained comprising a new nanocylindrical molecular architecture based on π–π interlocked β-sheets.26 They applied a number of spectroscopic techniques to Fmoc-Phe-Phe to confirm the proposed model. Fig. 6 shows the model structure of Fmoc-Phe-Phe peptides that was created using the Amber forcefield.
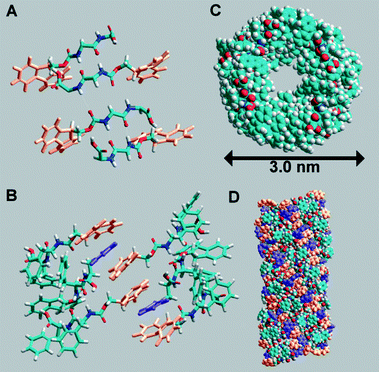 | ||
| Fig. 6 Fmoc-Phe-Phe peptides arranged in an anti-parallel β-sheet pattern (A), interlocking of Fmoc groups from alternate β-sheets to create π-stacked pairs with interleaved phenyl rings (B). Top view (C) of the structure and side view (D). Reproduced from ref. 26 with kind permission of Wiley VCH. | ||
The Ulijn group developed also a Phe-Phe-based hydrogel with the incorporation of bio-recognition function by coassembling the Fmoc-Phe-Phe and Fmoc-RGD (arginine–glycine–aspartate) peptides. Such hydrogels may be used as biomimetic fibrous scaffolds for the 3D-culture of anchorage-dependent cells,27 or may provide gels with tunable chemical and mechanical properties and may be utilized for in vitro cell culture.28 The introduction of chemical functionality into Fmoc-peptide gels may be achieved by the addition of Fmoc-protected amino acids with varying side groups. Cell culture analysis showed that the resulting scaffolds (2) and (3a–c) (Fig. 7) differ significantly in their compatibility with cell culture of bovine chondrocytes, mouse 3T3 fibroblasts and human dermal fibroblasts.
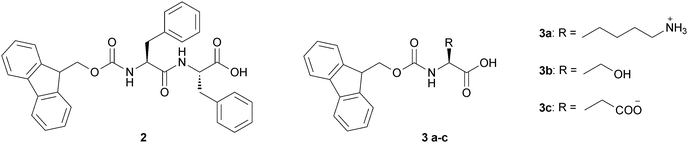 | ||
| Fig. 7 Chemical structure of Fmoc-based LMWGs (2) and (3a–c). | ||
Xu and coworkers reported a new type of supramolecular hydrogels, based on Fmoc-D-Ala-D-Ala (4),29 which exhibits gel–sol transition upon binding to its ligand (i.e., vancomycin) via a ligand–receptor interaction that can disturb the delicate balance between hydrophobic interactions and hydrogen bonds and induce a gel–sol transition. Fmoc-D-Ala-D-Ala formed hydrogels with high efficiency (one molecule of 4 can gel 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 H2O molecules).
000 H2O molecules).
Furthermore they synthesized a series of Fmoc-dipeptides (Fig. 8) for the purpose of developing a new class of low-molecular weight hydrogelators that are readily available and may serve as new biomaterials.
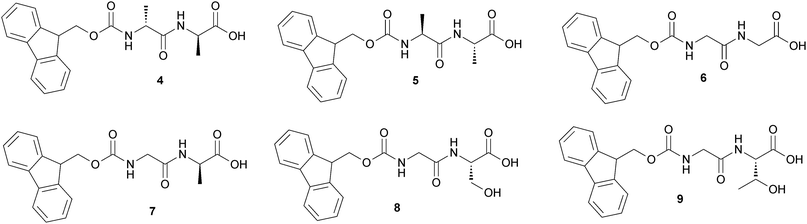 | ||
| Fig. 8 Chemical structure of Fmoc-based LMWGs (4–9). | ||
The enantiomers (4) and (5) show excellent ability to gel water at pH = 3, forming hydrogels with the gelator concentration of 4 mM. Substituting the two Ala in (4) with Gly affords (6), which exhibits behavior of gelation similar to those of (4) and (5) except at higher concentration ([6] = 11 mM). Compounds (7) and (8) form hydrogels at higher gelator concentrations and different pH and the pH response of the gel is completely reversible. Upon heating, the matrix formed by the gelators collapses into a precipitate, and water molecules are expelled. While cooling back to room temperature, the shrunken gel swells to form gel again.
The enzymatic formation of supramolecular hydrogels, using enzymes (phosphatase, thermolysin, β-lactamase or phosphatase/kinase) and Fmoc-protected amino acids or dipeptides has been reported by the same group.30 This approach provides a new strategy to detect the presence of enzymes, screen enzyme inhibitors, assist biomineralization, assay the types of bacteria, and aid the development of smart drug delivery systems, will lead to the further development of new materials for biomedical applications and improve understanding of molecular self-assembly in water.
3.3 Pyrene-based LMWG
Banerjee and coworkers have reported the organogel formed by N-terminally pyrene-conjugated oligopeptide, Py-Phe-Phe-Ala-OMe (Py = pyrene), in various organic solvents (Fig. 9).31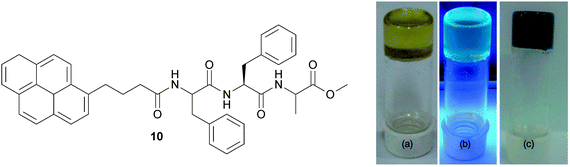 | ||
| Fig. 9 (left) Chemical structure of the pyrene-conjugated gelator peptide (10). (right) Photographs of organogel in OCDB (a) under exposure to daylight, (b) under exposure to UV light (excitation at 365 nm), and (c) graphene-containing hybrid organogel under exposure to daylight. Reproduced from ref. 31 with kind permission of Wiley VCH. | ||
The gelation behaviors of Py-Phe-Phe-Ala-OMe in different organic solvents have been tested. It forms transparent stable, supramolecular organogels in benzene, toluene, xylene, chlorobenzene, o-dichlorobenzene (ODCB), bromobenzene, chloroform, and dichloromethane. Furthermore it forms a pyrene blue-emitting transparent fluorescent organogel in ODCB. This fluorescent organogel in ODCB is capable of incorporating unfunctionalized and non-oxidized graphene nanosheets into the organogel system by utilizing non-covalent π–π stacking interactions to form a stable hybrid organogel system.
3.4 Naphthalene-based LMWG
Fluorene or pyrene groups are suspected to be carcinogenic,32 so hydrogelator dipeptides containing these moieties lack biocompatibility. As the strength of a gel is improved by hydrophobic interactions primarily provided by an aromatic moiety, Xu and coworkers introduced a new type of hydrogelator whose aromatic motif is a naphthyl group (Nap) that is a quite common fragment in clinically approved drug molecules.33They found that some of the conjugates of naphthalene and dipeptides (Fig. 10) gel water with particularly high efficiency (minimum gelation concentration = 0.07 wt%) and a cell cytotoxicity assay also indicates that these hydrogelators are biocompatible.
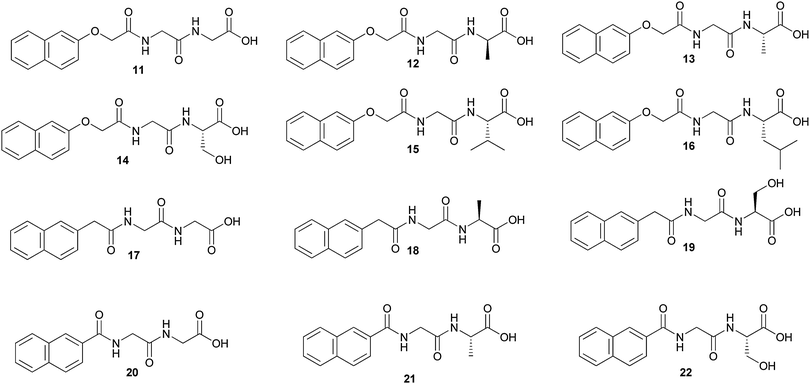 | ||
| Fig. 10 Chemical structure of naphthyl-based LMWGs (11–22). | ||
Comparing the abilities of gelation of (11), (12) and (13), the methyl group on the Ala residue in the enantiomers (12) and (13) decreases the flexibility of the dipeptides moiety, reduces the conformational entropy of the molecules, and favours more stable hydrogels. For compound (14), the hydroxylmethyl group causes steric hindrance, disfavors intermolecular interactions, and leads to less stable hydrogels. Compounds (15) and (16) fail to form hydrogels under similar conditions, likely due to the steric congestion resulted from the relatively large and hydrophobic side chains. The rest of the compounds (17–22) fail to form hydrogels, suggesting that the OCH2 linker acts as a possible acceptor of a hydrogen bond.
Xu and coworkers report also the systematic examination of the biostability of three unnatural amino acid-based hydrogelators that share structural similarities with the small-molecule hydrogelator Nap-Phe-Phe (23).34 The three molecules are Nap-D-Phe-D-Phe (24), racemic Nap-β3-HPhg-β3-HPhg (25) and Nap-L-fPhe-L-fPhe (26) (Fig. 11).
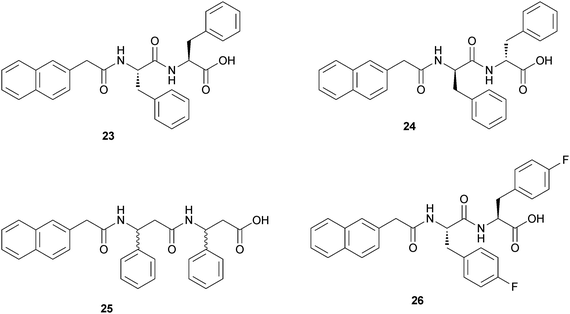 | ||
| Fig. 11 Chemical structures of Nap-L-Phe-L-Phe-OH (23) and of the three hydrogelators that share structural similarities with it. | ||
In vitro, (24) and (25) showed excellent biostability against proteinase K, a powerful enzyme hydrolase for a broad spectrum of peptides. Using radioactive tracers, a hydrogel formed by (24) exhibited good controlled release characteristics in vivo. No clinical, hematological or biochemical (including blood glucose) toxicity was observed, and there were no local or systemic bacterial, viral, or fungal infections in the mice treated over 42 days.
Adams and coworkers have utilized the hydrolysis of glucono-δ-lactone (GdL) to gluconic acid as a means of adjusting the pH in a Br-Nap-Ala-Val solution (Fig. 12) allowing the specific targeting of the final pH, because the gelation is pH-dependent.35 The slow GdL hydrolysis (as compared to fast dissolution) allows the gelation process to be followed with time. In addition, the final pH achieved can be targeted by adjusting the amount of GdL added.
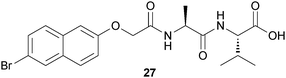 | ||
| Fig. 12 Chemical structure of naphthyl-based LMWG (27). | ||
The final pH after peptide dissolution was found to be 10.7. As the dipeptide derivative was found to be stable in aqueous solution at high pH for extended periods of time, the solution was added to a specific amount of GdL to induce assembly. The kinetics of the pH change and the final pH of the solution were found to correlate with the amount of GdL used. Combining all of these data, it is clear that the kinetics of self-assembly of the dipeptide derivative and hence hydrogelation is determined by the kinetics of pH adjustment.
Jamart-Grégoire and coworkers prepared several amino acids containing the naphthaloyl moiety as an N-protecting group and reported the results of a systematic study of the organogelation behavior of the amino acid derivatives in a wide range of solvents.36 Only the Phe and Leu derivatives shown in Fig. 13 are able to gelify some of the tested solvents, while Ala, Val, Ile and Trp derivatives (not shown) are insoluble or precipitate under the same conditions. Most of the gelified solvents are aromatic; this might suggest a primary importance of the π–π stacking interactions between the gelator and the solvent. The only exceptions to this trend are carbon tetrachloride and tetrachloroethylene. Compound 28b is able to gelify carbon tetrachloride, whereas gelator 28a form gels in both. The gelification of carbon tetrachloride is scarcely useful, due to its high toxicity. Indeed exposure to high concentrations of carbon tetrachloride or chronic exposure can affect the central nervous system, degenerate the liver and kidneys and could result in cancer.
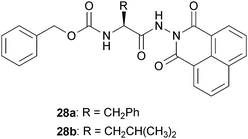 | ||
| Fig. 13 Chemical structure of naphthyl-based LMWGs (28a–b). | ||
3.5 Pyridine-based LMWG
A series of pyridine-2,6-dicarboxamide derivatives containing two α-amino acid pendant groups was prepared and characterized by Chow and Wang.37 Three of the synthesized compounds obtained from this series (Fig. 14), all having aromatic amino acid side chains, were found to be excellent organogelators toward aromatic solvents, alcoholic solvents, and carbon tetrachloride. Among the many amino acid derivatives, those containing aromatic side chain such as 29 and 30 were the best organogelators. The remaining compounds were either too soluble or too insoluble in the solvent system. It was found that the intra-molecular hydrogen bonds between the pyridine dicarboxamide N–H hydrogens and the pyridine N atom were the key structural elements for gel formation. This series of compounds represented one of the rare examples where both inter- and intra-molecular hydrogen bonds were needed for effective gel formation.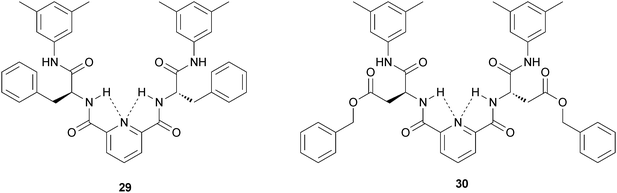 | ||
| Fig. 14 Chemical structure of pyridine-based LMWGs (29) and (30). | ||
3.6 Oligopeptide LMWG
Some interesting gelators are constituted of a few unprotected amino acids. As the natural amino acid pool consists of 20 members with different physical properties including polar, non-polar, acidic, basic and aromatic groups, these amino acids can be combined in numerous ways to produce a wide variety of building blocks with different physical properties.For β-sheet rich fibres, the self-assembly is driven by strong hydrogen bonding interactions along the fibre axes. These interactions are highly directional and link each peptide in the β-sheet to the previous and the next one in the chain creating a very stable structure. As a consequence, in this system the position of the polar group does not play a key role in the self-assembly mechanism of the peptide. In contrast, in the case of Ala based peptides, the self-assembly of the α-helices into fibres occurs through hydrophobic interactions. The position of the hydrophobic “patches” along the α-helix plays a key role in the ability of the peptide to self-assemble into long fibres. The strength of the inter-peptide interactions holding the fibre together is weaker resulting in these systems being more sensitive to charged surfaces and polar group positions. As a result, additional cooperative interactions to stabilize the structure are needed, which results in a higher peptide aggregation order (i.e. a higher number of peptides per fibre repeat unit).
Manchineella and Govindaraju reported the synthesis of the two cyclo-(Gly-L-Lys) ε-amino derivatives 31 and 32 (Fig. 15) and their behavior as LMWGs.38 The diketopiperazine 31 forms organogels in hexane, carbon tetrachloride, toluene and chloroform at minimal gelation concentrations of 5.5 wt%, 4.4 wt%, 2.6 wt% and 3.2 wt% respectively. The organogel (toluene) and the hydrogel were obtained using the diketopiperazine 32 at minimal gelation concentrations of 3.2 wt% and 2.5 wt% respectively.
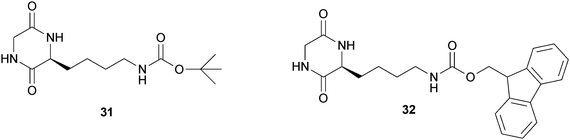 | ||
| Fig. 15 Chemical structure of cyclo-(Gly-Lys) ε-amino derivatives. | ||
Haldar and coworkers synthesized the pentapeptide Boc-Leu-Val-Aib-Phe-Aib-OMe that contains hydrophobic residues and self-assembles to form nonporous materials in the solid state.39 It forms a clear solution at elevated temperatures in aromatic solvents and microcrystals are precipitated out of the solution on cooling. It is suitable for selective gelation from an organic solvent–water mixture. In a typical procedure, 1 mL of xylene and 1 mL of water were shaken in a sample tube and 80 mg of the peptide was added. After cooling at room temperature the organic phase gelated entrapping the water inside the tube.
The same group reported the terminally protected tripeptide Boc-Val-Phe-Phe-OMe that forms sonication induced organogels.40 The peptide forms a very weak gel only in 1,2-dichlorobenzene after heating, cooling and ageing for 3 days. But ultrasound energy induces instant fibril formation and gelation in a wide range of organic solvents starting from hexane, cyclohexane, petrol, kerosene to benzene, toluene, xylene and ethanol.
Banerjee and coworkers have developed some oligopeptides that may behave as organogelators or hydrogelators, with a fine tuning of the number and the position of the amino acids.
They first studied the role of the protecting groups of the tripeptide Ala-Val-Ala in the formation of organogels. A series of eight synthetic self-assembling terminally blocked tripeptides have been studied for gelation and are reported in Fig. 16.41 Some of them form gels in various aromatic solvents including benzene, toluene, xylene, and chlorobenzene and the protecting groups play an important role in the formation of organogels. If the C-terminal protecting group has been changed from methyl ester to ethyl ester, the gelation property does not change significantly (keeping the N-terminal protecting group same), while the change of the protecting group from ethyl ester to isopropyl ester completely abolishes the gelation property. Similarly, keeping the identical C-terminal protecting group (methyl ester), the substitution of N-terminal protection tert-butyloxycarbonyl (Boc-) to benzyloxycarbonyl (Cbz-) does change the gelation property insignificantly, while the change from Boc- to pivaloyl (Piv-) or acetyl (Ac-) group completely eliminates the gelation property.
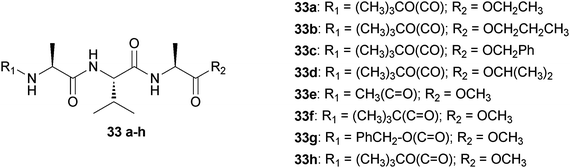 | ||
| Fig. 16 Chemical structures of peptides (33a–h). | ||
The same group studied a terminally protected self-assembling pentapeptide Boc-Leu-Val-Phe-Phe-Ala-OMe (34) that forms thermoreversible transparent gels in various organic solvents including benzene, toluene, m-xylene and 1,2-dichlorobenzene.42 A series of its variants have been synthesized in order to study the role of adjacently located Phe residues and the protecting groups for gelation in different organic solvents (Fig. 17). Replacement of any of the Phe residues of the Phe-Phe segment with any other hydrophobic α-amino acid residue drastically changes the gel forming properties indicating that both Phe residues have an important role in gel formation. Wide angle X-ray scattering (WAXS) studies of the peptide 34–benzene gel indicate that π–π interaction is responsible for gel formation and it reveals the necessity of the Phe residues in gel formation. Transmission electron microscopy (TEM) of the gels reveals a nanofibrillar morphology, which is obtained from the self-assembled gelators in the gel phase.
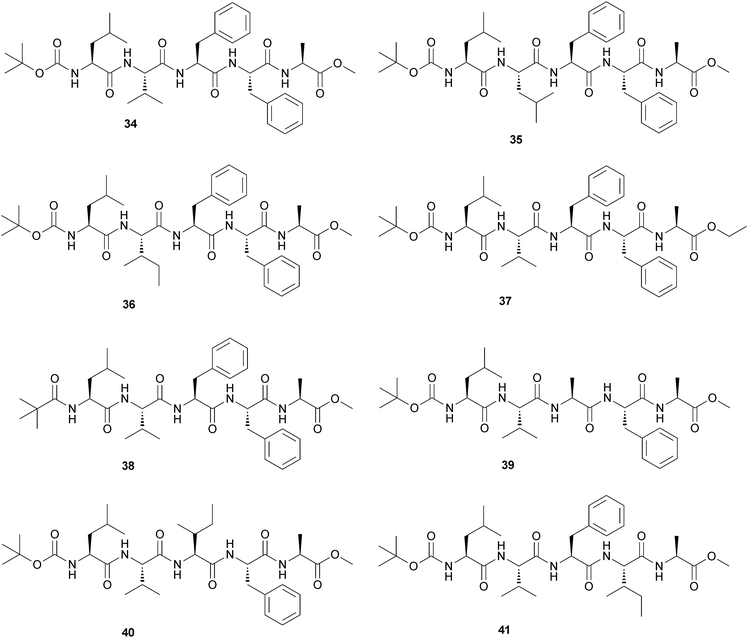 | ||
| Fig. 17 Schematic representation of peptides (34–41). | ||
Finally the same group reported two synthetic oligopeptide-based, thermoreversible, pH-sensitive hydrogels.43 Two peptides, Gly-Ala-Ile-Leu (42) and Gly-Phe-Ile-Leu (43), without having any protecting group, can form hydrogels at physiological pH (Fig. 18). They are stable in the pH range 6–8.5. 42 can form a hydrogel at high concentration, and its minimum gelation concentration was 17% (w/v). When they replaced the Ala residue with Phe, they obtained the more efficient gelator (43) that forms a thermoreversible pH-sensitive hydrogel with a minimum gelation concentration of 2.65% (w/v). These two hydrogels have been utilized for entrapment and slow release of an anticancer drug doxorubicin at physiological pH, promising their future application as a drug delivery vehicle.
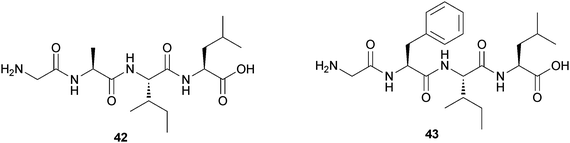 | ||
| Fig. 18 Chemical structure of hydrogelators (42) and (43). | ||
The different effect of the use of Ala or Phe on the gelator propensities of oligopeptides has been studied also by Saiani and coworkers.44 They compared the self-assembly and gelation properties of the four octapeptides reported in Fig. 19: two contain four Ala moieties and the other two contain four Phe moieties.
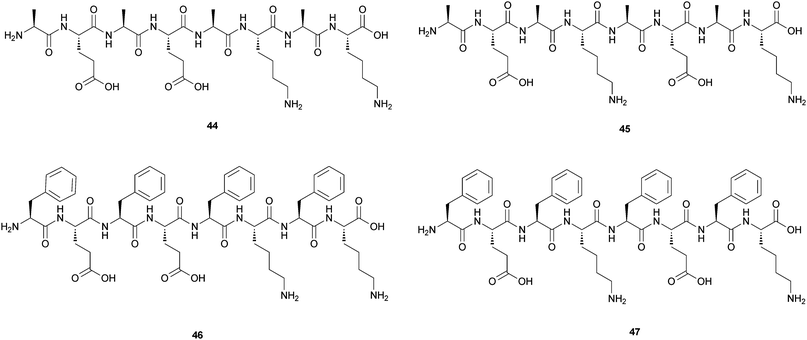 | ||
| Fig. 19 Chemical structures of the four octapeptides containing Ala, Phe, Lys and Glu (44–47). | ||
They are based on four different amino acids: Ala, Phe, Lys and Glu and have formula: (Ala-Glu)2-(Ala-Lys)244, (Ala-Glu-Ala-Lys)245, (Phe-Glu)2-(Phe-Lys)246 and (Phe-Glu-Phe-Lys)247. No self-assembly in solution was observed for 45, while 44 was found to self-assemble forming thick, rigid fibres with a diameter of ∼6 nm. These fibres were composed of two fibrils aggregating side by side to form “pearl-necklace” morphologies. No gelation was observed for 44 in the concentration range investigated (0 to 100 mg mL−1). In contrast, both 46 and 47 found to self-assemble in solution and to form hydrogels at an initial concentration of ∼8 mg mL−1.
3.7 Two-components peptide LMWG
It is possible to induce gelation by mixing compounds that tend to self-organize in organic solution, as different geometric and structural requirements are important in the self-organisation of mixtures of peptide gelators.Smith and coworkers demonstrated that a class of dendritic gelators self-assembles into gels as a consequence of intermolecular hydrogen bond interactions between the CONH groups in the peptide head groups.45 This family of gelators is ideal for probing the behaviour of mixtures, because they have a range of different variable structural features: (i) molecular “size”, (ii) molecular “shape” and (iii) molecular “chirality”, because gelators can be constructed using D-Lys, rather than L-Lys (Fig. 20).
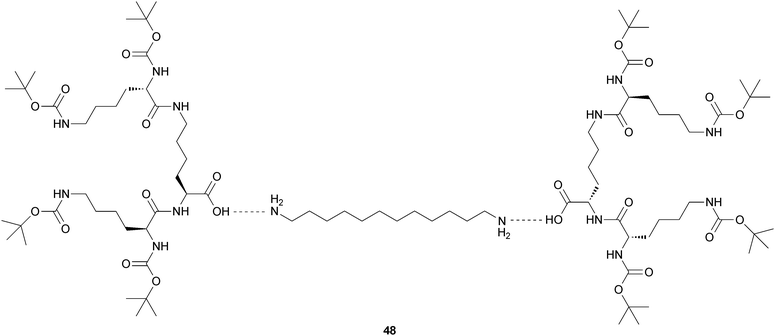 | ||
| Fig. 20 Two-components system in which dendritic head groups can exchange, hence giving rise to a disruptive chirality effect when a mixture of head groups based on L- and D-Lys is employed. | ||
The authors could demonstrate that of the three parameters investigated, mixtures with different “size” and “chirality” can self-organise their molecular building blocks, while mixtures with different “shape” disrupt another self-assembly. Interestingly, the “size” and “chirality” parameters correspond to the molecular information programmed into the peptide head group and the hydrogen-bond interactions between these head groups enable the self-assembly.
Another interesting example of two component gelators has been reported by Shinkai and coworkers.46 They developed a new class of organic organogelators (49, 50 and 51) based on 2-anthracenecarboxylic acid (2Ac), attached noncovalently with the gelator counterpart containing a 3,4,5-tris(n-dodecyloxy)-benzoylamide backbone (Fig. 21). Among the three gelators, two (50 and 51) are chiral containing D-Ala or L-2-phenylglycine moieties, respectively. They can act as efficient gelators of organic solvents with varying polarity depending upon the gelator systems, while 49 even gelates chiral solvents. The photoirradiation of the gel samples produces photocyclodimers having different degrees of stereoselectivity for different systems.
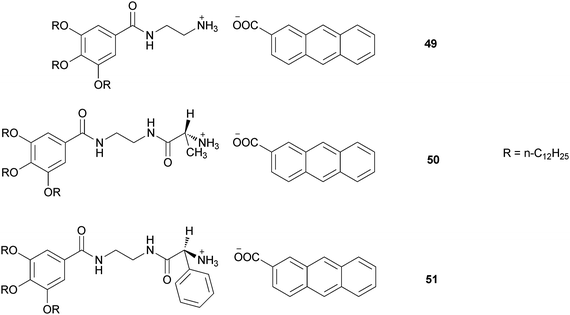 | ||
| Fig. 21 Chemical structure of the binary gelators (49), (50) and (51). | ||
4. Peptidomimetic LMWG
With this short overview among the peptide gelators, we have shown that several different structures are able to behave as hydrogelators and/or organogelators. They are chiral molecules and contain groups able to form hydrogen bonds and groups that form hydrostatic interaction, such as aromatic rings or aliphatic long chains. Now we will show peptidomimetic compounds that are good gelators and share with peptide LMWG the same characteristics.4.1 Cyclobutane derivative LMWG
Ortuño and coworkers prepared two chiral synthetic β-dipeptides that are efficient organogelators.47 One contains two trans-cyclobutane residues and the other one a trans- and a cis-cyclobutane fragment (Fig. 22). | ||
| Fig. 22 Chemical structure of cyclobutane derivative gelators (52 and 53). | ||
Gels of both β-dipeptides (52 and 53) were easily obtained from 40 mM solutions in toluene and in 1:1 mixtures of dichloromethane/pentane or ethyl acetate/pentane. By using NMR techniques, it was possible to keep under control the temperature at which the gelation process occur, by checking the chemical shift of the NH proton signal. Two main gelation processes were clearly distinguishable: one cooling from 298 to 269 K, the other one when cooling from 269 to 250 K. Here an abrupt decay in the NH protons signal area was noticeable, which clearly indicated that a sol–gel transition arose.
The same group had previously demonstrated that also the β-tetrapeptide (54) (Fig. 23) readily forms organogels in selected solvents.48 It is made up of homochiral cyclobutane residues and displays conformational bias in solution prompted by the formation of intramolecular hydrogen bonds. This compound self-assembles to produce nanosized fibrils and forms an organogel in 3:2 ethyl acetate/hexane and 3:1 acetone/hexane mixtures.
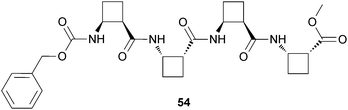 | ||
| Fig. 23 Chemical structure of β-tetrapeptide gelator (54). | ||
4.2 C3-peptidomimetic LMWG
Feringa and coworkers developed three 1,3,5-trisamide-cyclohexane-based gelators (55–57) that are derivatised with hydrophobic amino acids (Fig. 24) to shield the amide groups from competitive interactions with water and have thereby enforced the anisotropic self-assembly of the gelator molecules in water due to the concurrent action of hydrogen bonding and hydrophobic effects.49 Compounds (55–57) all form thermoreversible gels in water at very low concentrations. The very low critical gelation concentrations and the high gel–sol phase transition temperatures clearly indicate that their self-assembly is driven by strong intermolecular interactions. | ||
| Fig. 24 Chemical structure of 1,3,5-trisamide–cyclohexane-based gelators (55–57). | ||
The compatibility of hydrogel formation by (55–57) with various types of surfactants (sodium dodecyl sulfate SDS, cetyltrimethylammonium CTAB, n-octyl-β-D-glucopyranoside OG) was investigated by dissolving (55–57) in surfactant solutions below and above the critical micelle concentration (cmc) and by subsequently examining the samples. Gels of 55 are formed in combination with nonionic (OG) or anionic (SDS) surfactants either by temperature-induced gelation or by the lowering of the pH from 7 to 3.5. The gelation behavior of the nonionic gelators 56 and 57 is even more tolerant toward the presence of surfactants, and transparent gels are obtained in the presence of SDS, CTAB, or OG below and well above the cmc of the surfactants.
Later on, the same group reported a class of C3-symmetric amino acid based LMWG that allowed a systematic and broad structural variation.50 A schematic representation of the structure of the C3-symmetric compounds is shown in Fig. 25. The C3-symmetric compounds are built up in sections from the centre towards the periphery. The first central section is formed by a C3-symmetric rigid core. The C3-symmetry of this core reduces the occurrence of polymorphism and allows a better control over the intermolecular and fibre/solvent interactions. The second section consists of hydrogen-bonding units, in general ureas or amides. The application of different types of these units enables variation in the interaction strength. The units have to be connected to the core in an orientation that enables simultaneous, self-complementary, intermolecular hydrogen bonding of the three units, allowing one-dimensional aggregation and hence gelation to occur.
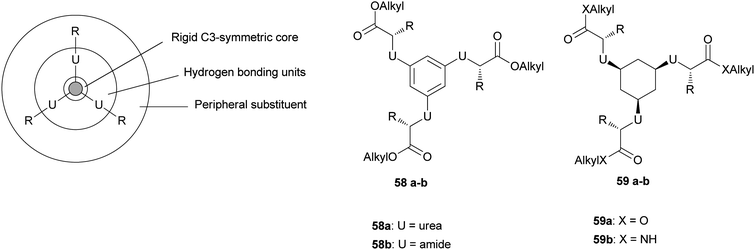 | ||
| Fig. 25 (left) Schematic design of the C3-symmetric compounds. (right) General structure of the different classes of compounds. | ||
An excellent core and a hydrogen-bonding-unit candidate that fulfils these criteria is the cis,cis-1,3,5-cyclohexanetricarboxamide scaffold (59). In this structure (Fig. 25, right), the amide groups are orientated perpendicularly to the mean plane of the molecule and exhibit a parallel alignment with respect to each other. This provides the strong uni-axial intermolecular interactions affording one-dimensional columnar self-assembly. A comparable scaffold is the 1,3,5-benzenetricarboxyamide core, which has also been reported to have a hydrogen-bonded columnar type of packing.
Structural variation yielded a number of compounds, the aggregation behaviour and resulting aggregates and gels were studied by FTIR spectroscopy, dropping ball measurements, differential scanning calorimetry and transmission electron microscopy. These studies showed that the nature of the core unit, the type of hydrogen-bonding units and the applied amino acids have a strong influence on the interactions, resulting in large differences in aggregation properties, thermal stability and morphology between the various compounds. Both gelation studies and FTIR spectroscopy showed that the cyclohexane core was superior to the benzene core.
Yamanaka and Fujii developed a C3-symmetrical tris-urea low molecular weight gelator,51 in three steps starting from phloroglucin, by nucleophilic aromatic substitution of phloroglucin and 4-fluoronitrobenzene, reduction of nitro groups and reaction of the resulting primary amines with an excess amount of octylisocyanate. The desired tris-urea 60 (Fig. 26) was obtained as a white solid and used for gelation experiments in four chloroalkanes: CH2Cl2, CHCl3, 1,1,2-trichloroethane, and 1,1,2,2-tetrachloroethane, under various conditions. Thermal treatment was effective in gelating 1,1,2-trichloroethane. Mixtures of 60 and CHCl3 or 1,1,2,2-tetrachloroethane gave gels after ultrasound irradiation.
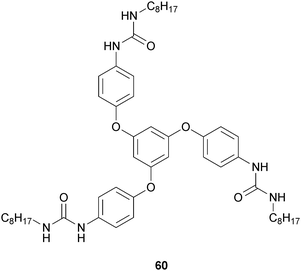 | ||
| Fig. 26 Chemical structure of C3-symmetrical tris-urea low molecular weight gelator (60). | ||
It is known that some low molecular weight compounds form supramolecular gels by adding metal salts,52 and the effect of metal salts on the gelation of 60 and chloroalkane solvents was investigated. The addition of CuBr2 was effective for the gelation of 60 in 1,1,2-trichloroethane and afforded a pale brown semitransparent gel after thermal dissolution and cooling. CH2Cl2 changed into a gel in the presence of equimolar 60 and BiCl3 after sonication. Spherical particles with rough surfaces were found by SEM observation of CHCl3 gel prepared from ultrasound irradiation of 60 and Y(NO3)3 (Fig. 27).
 | ||
| Fig. 27 SEM images of gels (xerogels) prepared by ultrasound irradiation: (a) CHCl3 gel of 60, (b) 1,1,2,2-tetrachloroethane gel of 60, (c) 1,1,2,2-tetrachloroethane gel of 60 and CuBr2, (d) CHCl3 gel of 60 and Y(NO3)3. Reproduced from ref. 52 with kind permission of the American Chemical Society. | ||
Meijer and coworkers reported on various C3-symmetrical columnar supramolecular architectures relying on hydrogen bonding in combination with π–π stacking. Twelve C3-symmetrical molecules have been investigated, six of which contain three central amide functionalities and six of which contain three central urea groups.53 Peripheral groups of the disks are “small”, “medium”, or “large”, half of them being achiral and the other half being chiral. In all of the cases, elongated, helical stacks are formed in apolar solution, except for the “medium” amide disks. Among all these compounds, only the “small” achiral urea disk (61) (Fig. 28) efficiently forms an organogel in toluene and this outcome confirms the directionality of the interactions.
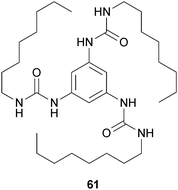 | ||
| Fig. 28 Chemical structure of C3-symmetrical organogelator (61). | ||
4.3 Peptide amphiphilic gels
Peptide amphiphiles (PAs) are a class of molecules consisting of a hydrophobic nonpeptidic tail covalently conjugated to a peptide sequence. These molecules consist of three segments (Fig. 29): | ||
| Fig. 29 Molecular structure of the investigated peptide amphiphile. The molecule is composed of three segments, the alkyl tail, the β-sheet forming peptide sequence, and the charged peptide headgroup. | ||
a hydrophobic sequence, which is commonly an alkyl tail that drives aggregation through hydrophobic collapse;
a β-sheet-forming peptide that promotes nanofibre formation;
a peptide segment that contains ionizable side chains and often an amino acid sequence of interest for biological signaling.
Stupp and coworkers have developed bioactive scaffolds composed of peptide amphiphiles that self-assemble into nanofibre networks.54 The PA molecules self-assemble into cylindrical nanofibres when the strong electrostatic repulsion between molecules is either neutralized by changing pH or screened by adding ions. At certain concentrations and in the presence of sufficient electrostatic screening, these PA molecules can form a three-dimensional network that is observed macroscopically as a self-supporting hydrogel. The authors found that systematic variations in the β-sheet sequences of peptide amphiphiles are very effective in tailoring the stiffness of gels formed by their nanofibre networks. Increasing both the number and fraction of Val residues are especially effective at raising mechanical stiffness, whereas Ala tends to reduce it. On the other hand, supramolecular design of softer gels can be achieved with sequences that promote twisting of β-sheets given their ability to weaken their peripheral hydrogen bonds.
The same authors recently reported that noodle-like strings of arbitrary length could be formed by manually drawing the heated aqueous peptide amphiphile solution into a salty medium from a pipette (Fig. 30).55 This observation suggested that macroscopic alignment extending over centimetres was achieved. Using the same methods, the unheated solutions did not form mechanically stable string gels or show any birefringence. Scanning electron microscopy (SEM) indicated that strings formed from heated peptide amphiphile solutions contained extraordinarily long arrays of aligned nanofibre bundles, while unheated peptide amphiphile solutions formed matrices of randomly entangled nanofibres.
 | ||
| Fig. 30 (a) and (b) A peptide amphiphile solution coloured with trypan blue injected into phosphate-buffered saline after heat treatment. (c) A knot made with peptide amphiphile string. Reproduced from ref. 55 with kind permission of Nature Publishing Group. | ||
A particular kind of amphiphilic peptides are bolaamphiphiles (two-headed amphiphiles). They are named after the “bola”, which is a South American weapon made of two balls connected by a string. As with conventional amphiphiles, the chemical functionality of the headgroups and linking group can be varied to change the aggregation properties. Some groups have reported the design and synthesis of hydrogelators and/or organogelators that have this skeleton.
Escuder, Miravet and coworkers reported a family of bolaamphiphilic gelators (Fig. 31).56 Compound (63) is known to be an “ambidextrous” gelator, because it is capable of forming gels in organic solvents and water, but it is a hydrogelator with moderate efficiency in terms of minimum gelator concentration.
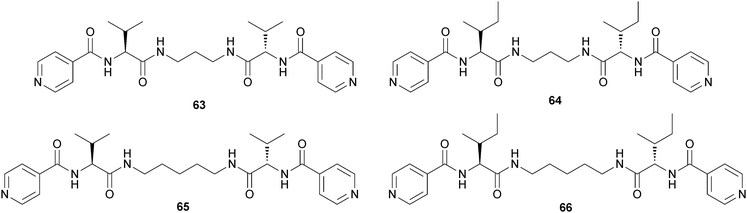 | ||
| Fig. 31 Chemical structures of bolamphiphilic gelators (63–66). Compound (63) is an “ambidextrous” gelator. | ||
Considering that the entropy change associated with the hydrophobic effect is the driving force for the aggregation, the authors prepared compounds (64–66), where they introduced groups with enhanced hydrophobicity into the molecule to boost the gelation properties as a result of an improved favorable entropic term. It was found that compounds (63–64) form hydrogels that were made of fibrillar networks and that improvement of the gelation efficiency was easily achieved by increasing the hydrophobic character of the gelators.
Das and coworkers reported the self-assembly of peptide based bolaamphiphiles upon sonication.57 Peptide-appended bolaamphiphiles (67) and (68) were capable of forming stable, transparent, self-supporting hydrogels upon sonication at room temperature and a final pH of 5.4, whereas bolaamphiphile (69) was unable to form a gel under similar conditions (Fig. 32). The self-assembly process occurs mainly through hydrogen bonding interactions and π-stacking interactions. In the gel phase, these peptide bolaamphiphile molecules adopt long, twisted nanofibrillar network structures. Finally, the hydrogel of the Tyr rich peptide bolaamphiphile molecule (67) is used as a nanoreactor for in situ synthesis of Pt nanoparticles that are used as a catalyst in the hydrogenation reaction of p-nitroaniline to p-phenylenediamine.
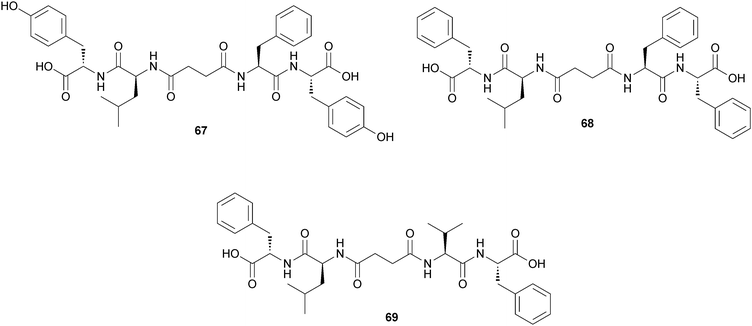 | ||
| Fig. 32 Chemical structure of bolaamphiphilic gelators (67–69). | ||
Our group has recently reported the synthesis of the compounds 70a–d and 71a–d that contain the Phe-D-Oxd [D-Oxd = (4R,5S)-4-carboxy-5-methyl oxazolidin-2-one] moiety (Fig. 33). These compounds may behave as efficient organogelators or hydrogelators, simply transforming the benzyl ester into the corresponding free acid.58 We have also replaced the D-Oxd moiety with a D-Pro moiety, to check if the presence of the Oxd moiety was essential for the formation of gels. In contrast to the D-Oxd-containing compounds (70a–b and 71a–b), no gel was ever formed with any of the D-Pro-containing molecules (70c–d and 71c–d). This outcome suggests that the Phe-D-Oxd moiety may be defined as a “privileged scaffold” for the formation of gels.
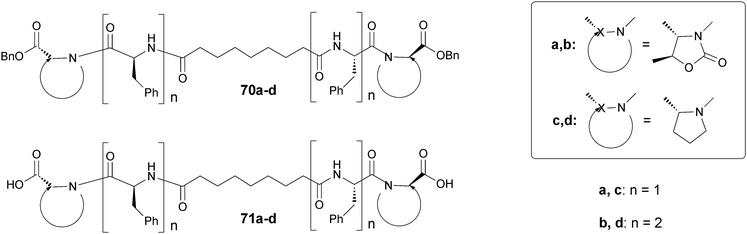 | ||
| Fig. 33 Chemical structure of bolaamphiphilic pseudopeptides (70a–d) and (71a–d). | ||
Then we reported the synthesis of a small library of stereoisomeric pseudopeptides (72a–h) that are able to make gels in different solvents and in the presence of several metal ions.59 Four benzyl esters and four carboxylic acids, all containing a moiety of azelaic acid (a long chain dicarboxylic acid) coupled with four different pseudopeptide moieties sharing the same skeleton were synthesized (Fig. 34).
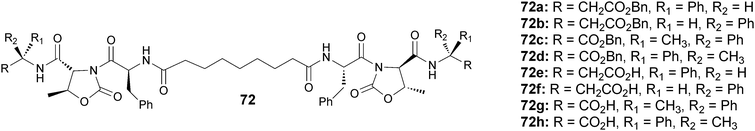 | ||
| Fig. 34 Chemical structure of bolaamphiphilic pseudopeptides (72a–h). | ||
The tendency of these pseudopeptides to form gels was evaluated by using the inversion test of 10 mM solutions of pure compounds and of stoichiometric mixtures of pseudopeptides and metal ions. The formation of gel containing Zn(II) or Cu(II) ions gave good results in terms of incorporation of the metal ions, while the presence of Cu(I), Al(III) and Mg(II) gave less satisfactory results.
Tovar and coworkers reported some new hydrogel “noodles” that are robust enough to be manipulated into complex shapes such as knots and spirals.60 They found that the two bolaamphiphilic peptides (73) and (74) (Fig. 35) form strong and rigid self-supporting hydrogels under physiological conditions upon sonication. The self-assembly process occurs mainly through hydrogen bonding interactions and π-stacking interactions. In the gel phase, these peptide bolaamphiphile molecules adopt long, twisted nanofibrillar network structures.
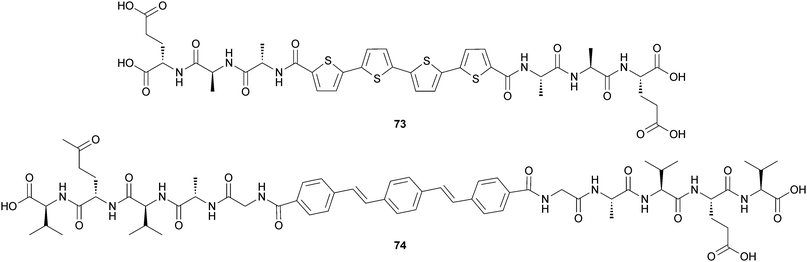 | ||
| Fig. 35 Chemical structure of bolaamphiphilic peptides (73) and (74). | ||
This noodle formation technique leads to the macroscopic alignment of peptide motifs bearing π-conjugated functionality and results in materials displaying unique photophysical and anisotropic electrical responses as evidenced by the alignment of two representative π-conjugated peptide nanostructures (Fig. 36). The alignment of these π-electron oligomers within the noodle macrostructures leads to global directionality among the internal π-stacked chromophores. They also found that marked photophysical differences are present between the hydrogels derived from randomly oriented nanostructures and those generated via this noodle producing method.
 | ||
| Fig. 36 Representative SEM images of aligned structures composed of 73 (a) and 74 (c) constructed through the noodle preparation technique (white arrows indicate direction in which the peptide solutions were dispensed into the assembly solution). Optical birefringence of macroscale peptide noodles composed of 72 (b) and 73 (d) as seen under crossed polarizers. Reproduced from ref. 60 with kind permission of Wiley VCH. | ||
5. Conclusions
In this review we have shown a general survey on a particular kind of LMWGs: small peptides and peptidomimetics that behave as gelators. Among them, some are organogelators and others are hydrogelators, depending on their structure.Some general requirements are needed for the formation of a good gelator: (i) it should not be too soluble or too insoluble; (ii) it should contain a hydrophilic head, as a peptide bond or a urea bond; (iii) it should contain some hydrophobic moieties, usually aromatic rings or occasionally long aliphatic chains; (iv) it should be a chiral compound.
Most of them can gelify water and/or organic solvents and form robust gels in fairly low concentration, compared to most of the polymer gels. This is due to some characteristics of the analyzed structures. Self-organized oligomers that form helices are bad candidates as LMWGs; in contrast, oligomers that tend to fold in a β-sheet structure, are good candidates to form fibres, thus to behave as gelators.
Several peptide gelators contain a Phe moiety: some authors have shown that upon replacing the Phe group with an Ala group the gelation does not occur. Also the presence of an Asp or a Lys moiety favours the formation of gels.
Peptidomimetics are small protein-like molecules designed to mimic natural peptides. To design peptidomimetic gelators some requirements are needed: local constrains must be introduced into the skeleton, to induce the formation of β-sheet structures. Thus the introduction of a constrain (a cyclobutane or an oxazolidin-2-one ring) or the preparation of C3-symmetric compounds produces efficient gelators. Furthermore, outstanding results have been obtained with amphiphilic or bolaamphiphilic gelators. These molecules consist of three segments: (i) a hydrophobic sequence, which is commonly an alkyl tail, that drives aggregation through hydrophobic collapse; (ii) a β-sheet-forming peptide that promotes nanofibre formation; (iii) a peptide segment that contains ionizable side chains and often an amino acid sequence of interest for biological signaling.
References
- D. Jordan Lloyd, in Colloid Chemistry, ed. J. Alexander, The Chemical Catalog Co, New York, 1926, vol. 1, pp. 767–782 Search PubMed
.
- T. Graham, Philos. Trans. R. Soc. London, 1861, 151, 183–224 CrossRef
- P. J. Flory, Faraday Discuss. Chem. Soc., 1974, 57, 7–18 RSC
- S. Sakai, K. Hirose, K. Taguchi, Y. Ogushi and K. Kawakami, Biomaterials, 2009, 30, 3371–3377 CrossRef CAS
- D. J. Overstreet, D. Dutta, S. E. Stabenfeldt and B. L. Vernon, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 881–903 CrossRef CAS
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS
- H. Kunleda and K. Shinoda, J. Phys. Chem., 1976, 80, 2468–2470 CrossRef
- A. Brizard, R. Oda and I. Huc, Top. Curr. Chem., 2005, 256, 167–218 CrossRef CAS
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC
- Molecular Gels. Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Printed in the Netherlands, 2006, pp. 1–13 Search PubMed
- D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18–33 RSC
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024–2027 RSC
- J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC
- S.-L. Zhou, S. Matsumoto, H.-D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem.–Eur. J., 2005, 11, 1130–1136 CrossRef CAS
- A. Grauer and B. König, Eur. J. Org. Chem., 2009, 5099–5111 CrossRef CAS
- D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS
- L. Boyle and D. N. Woolfson, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. J. W. Steed and P. A. Gale, Wiley VHC, 2012, pp. 1639–1664 Search PubMed
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS
- E. S. Amis and V. K. Lamer, Science, 1939, 90, 90 CAS
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2009, 38, 967–975 RSC
- X. Yan, P. Zhua and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS
- J. Raeburn, G. Pont, L. Chen, Y. Cesbron, R. Lèvy and D. J. Adams, Soft Matter, 2012, 8, 1168–1174 RSC
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS
- V. Jayawarna, S. M. Richardson, A. R. Hirst, N. W. Hodson, A. Saian, J. E. Gough and R. V. Ulijn, Acta Biomater., 2009, 5, 934–943 CrossRef CAS
- Y. Zhang, H. W. Gu, Z. M. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS
- B. Adhikari, J. Nanda and A. Banerjee, Chem.–Eur. J., 2011, 17, 11488–11496 CrossRef CAS
- J. Wilson, D. F. V. Lewis and T. J. B. Gray, Chem. Res. Toxicol., 1993, 6, 535–541 CrossRef
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, J. Mater. Chem., 2007, 17, 850–854 RSC
- G. Liang, Z. Yang, R. Zhang, L. Li, Y. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419–8422 CrossRef CAS
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS
- P. Curcio, F. Allix, G. Pickaert and B. Jamart-Grégoire, Chem.–Eur. J., 2011, 17, 13603–13612 CrossRef CAS
- H.-F. Chow and G.-X. Wang, Tetrahedron, 2007, 63, 7407–7418 CrossRef CAS
- S. Manchineella and T. Govindaraju, RSC Adv., 2012, 2, 5539–5542 RSC
- S. Maity, P. Jana and D. Haldar, CrystEngComm, 2011, 13, 3064–3071 RSC
- S. Maity, P. Kumar and D. Haldar, Soft Matter, 2011, 7, 5239–5245 RSC
- A. K. Das, P. P. Bose, M. G. B. Drew and A. Banerjee, Tetrahedron, 2007, 63, 7432–7442 CrossRef CAS
- A. Banerjee, G. Palui and A. Banerjee, Soft Matter, 2008, 4, 1430–1437 RSC
- J. Naskar, G. Palui and A. Banerjee, J. Phys. Chem. B, 2009, 113, 11787–11792 CrossRef CAS
- A. Saiani, A. Mohammed, H. Frielinghaus, R. Collins, N. Hodson, C. M. Kielty, M. J. Sherratt and A. F. Miller, Soft Matter, 2009, 5, 193–202 RSC
- R. Hirst, B. Huang, V. Castelletto, I. W. Hamley and D. K. Smith, Chem.–Eur. J., 2007, 13, 2180–2188 CrossRef
- A. Dawn, N. Fujita, S. Haraguchi, K. Sada, S.-i. Tamaru and S. Shinkai, Org. Biomol. Chem., 2009, 7, 4378–4385 CAS
- E. Gorrea, P. Nolis, E. Torres, E. Da Silva, D. B. Amabilino, V. Branchadell and R. M. Ortuño, Chem.–Eur. J., 2011, 17, 4588–4597 CrossRef CAS
- F. Rùa, S. Boussert, T. Parella, I. Dìez-Pérez, V. Branchadell, E. Giralt and R. M. Ortuño, Org. Lett., 2007, 9, 3643–3645 CrossRef
- A. Heeres, C. van der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS
- M. de Loos, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron, 2007, 63, 7285–7301 CrossRef CAS
- M. Yamanaka and H. Fujii, J. Org. Chem., 2009, 74, 5390–5394 CrossRef CAS
- P. Terech, G. Gebel and R. Ramasseul, Langmuir, 1996, 12, 4321–4323 CrossRef CAS
- J. J. van Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS
- M. A. Greenfield, J. R. Hoffman, M. Olvera de la Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. Olvera de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS
- V. J. Nebot, J. Armengol, J. Smets, S. Fernàndez Prieto, B. Escuder and J. F. Miravet, Chem.–Eur. J., 2012, 18, 4063–4072 CrossRef CAS
- I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301–5308 RSC
- N. Castellucci, G. Angelici, G. Falini, M. Monari and C. Tomasini, Eur. J. Org. Chem., 2011, 3082–3088 CrossRef CAS
- N. Castellucci, G. Falini, G. Angelici and C. Tomasini, Amino Acids, 2011, 41, 609–620 CrossRef CAS
- D. Wall, S. R. Diegelmann, S. Zhang, T. J. Dawidczyk, W. L. Wilson, H. E. Katz, H.-Q. Mao and J. D. Tovar, Adv. Mater., 2011, 23, 5009–5014 CrossRef
| This journal is © The Royal Society of Chemistry 2013 |