Optical switches with biplanemers obtained by intramolecular photocycloaddition reactions of tethered arenes
Herbert
Meier
*a and
Derong
Cao
*b
aDepartment of Chemistry, Institute of Organic Chemistry, University of Mainz, Duesbergweg 10-14, 55099 Mainz, Germany. E-mail: hmeier@uni-mainz.de; Fax: +49 (0)6131-39 25396
bSchool of Chemistry and Chemical Engineering, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Wushan, 510641 Guangzhou, China. E-mail: drcao@scut.edu.cn; Fax: +86 20 87110245
First published on 2nd October 2012
Abstract
The dimerization of anthracene by a [4π + 4π] cycloaddition is one of the oldest and best known reactions in photochemistry. In the series of tethered bichromophoric arenes, this reaction type could be extended to anthracene–naphthalene, naphthalene–naphthalene and recently even to anthracene–benzene and naphthalene–benzene systems. Cyclophanes, which can be regarded as twofold or multiple tethered systems, are not discussed here. The cycloisomerizations are performed by irradiation at the long-wavelength absorption (λ > 270 nm), whereas shorter wavelengths (λ < 270 nm) lead to cycloreversions, which can be also achieved by a thermal route. The systems represent therefore a P- and T-type photochromism, which can be used for optical or chiroptical switches. An acceleration of the switch is possible by a singlet energy transfer (light harvesting antenna effect) in dendritic compounds. In the past 5 to 10 years many applications of these switches were studied in the context of photonic devices, sensor techniques, lithographic processes, imaging techniques, data processing and data storage.
 Herbert Meier | Herbert Meier studied chemistry and mathematics at the University of Tübingen and the Free University of Berlin, Germany, and completed in 1968 his doctoral thesis in organic chemistry with Prof. Müller in Tübingen. In 1972 he became a Docent and in 1975 an Associate Professor in Tübingen. In 1982 he accepted the chair of Prof. Horner at the University of Mainz. His research focuses on organic compounds with interesting properties for materials science. The scientific results were documented in more than 500 publications and in several guest professorships in USA, China, Russia and Croatia. |
 Derong Cao | Derong Cao studied Chemistry in Lanzhou University, China. In 1997 he finished his doctoral thesis in Organic Chemistry under the supervision of Prof. Meier at the University of Mainz, Germany and Prof. Lin at Lanzhou University. In 1997 he became an Associate Professor in Lanzhou and in 2002 a Full Professor in Guangzhou Institute of Chemistry, Chinese Academy of Sciences. Now he works in the School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou. Since 2008 he has been the head of the Department of Chemistry. His main scientific interests, documented in over 150 papers, focus on organic functional molecules and materials. |
1. Introduction
Two arene ring systems A and B (benzene, naphthalene, anthracene), which are connected by a (saturated) bridge, can undergo intramolecular [4π + 4π] photocycloadditions to so-called biplanemers (1 → 2) in which the two π systems are arranged face-to-face. Owing to the spatial and through-bond interaction of the π centres, this topology leads to special physical and chemical properties. Due to the generated 1,4-cyclohexadiene rings, both π systems are slightly bent. Although the newly formed σ bonds are extraordinarily long (up to 166 pm),1 the distance between the repelling π systems is relatively short. Hence, in addition to the reverse photochemical reaction, a thermal cycloreversion can take place (Scheme 1).![Intramolecular [4π + 4π] cycloaddition (cycloisomerization) of benzenoid aromatics and cycloreversions.](/image/article/2013/CS/c2cs35271k/c2cs35271k-s1.gif) | ||
| Scheme 1 Intramolecular [4π + 4π] cycloaddition (cycloisomerization) of benzenoid aromatics and cycloreversions. | ||
The tether – normally attached at the 1-position of a naphthalene or at the 9-position of an anthracene ring system – enhances the cycloaddition tendency and permits clean reactions at high dilution.
The bichromophoric compounds 1 can contain the following combinations of arenes:
| A–X–B |
| Benzene–X–naphthalene |
| Benzene–X–anthracene |
| Naphthalene–X–naphthalene |
| Naphthalene–X–anthracene |
| Anthracene–X–anthracene |
Whereas the anthracene–anthracene combination is long known, the benzene–naphthalene and the benzene–anthracene combinations are quite new.2,3 Until now no benzene–benzene system A–X–B, which cycloisomerized, has been known.
The face-to-face orientation of the ring systems A and B does not correspond to a populated conformer of 1. Hence, the flexibility of the tether X plays an important role. It turned out that three-atom tethers, such as CH2–CH2–CH2, CH2–O–CH2 and O–CH2–O, are very favorable. One of the two new σ bonds in 2 forms then together with the tether a five-membered ring. The so-called “rule of five” was originally proposed by Srinivasan and Carlough4 and Liu and Hammond5 for triplet sensitized internal cycloadditions. The preferred formation of fluorescing excimers in the series of α,ω-diphenylalkanes was also established for C3 chains, that is for 1,3-diphenylpropanes (Hirayama rule6). The focus of the present article lies therefore on three-atom tethers.
The photochromic system 1 ⇄ 2 represents an optical switch which can be applied in photonic devices, sensor techniques, lithographic patterning, imaging techniques and especially for data processing and data storage. In previous years many publications on applications of the reversible reaction 1 ⇄ 2 in supramolecular chemistry and materials science appeared.7–26 In principle, such a switch can be monitored by any (physical) property which is sufficiently different for 1 and 2. An easy control is possible by measuring the absorbance of 1 and 2 at a certain excitation wavelength. Ideal is a selective light emission of 1 or 2. Moreover, chiroptical switches can be constructed based on this molecular basis.8
The anthracene photodimerization was discussed in some excellent reviews by Becker27,28 and by Bouas-Laurent et al.29–31 Therefore we refer to these articles when older results are mentioned (mostly in Section 3.6) and concentrate here on results which were obtained in the previous decade.
2. Reaction routes
The cycloisomerization 1 → 2 occurs normally in the excited singlet state S1 and has to compete with fluorescence F, radiationless deactivation (internal conversion IC) and intersystem crossing. Apart from special compounds 1, the intersystem crossing or the triplet sensitized process does not yield the cycloisomers 2 at all or leads to a mixture of [2 + 4]- and [4 + 4] cycloadducts. Nevertheless, very high quantum yields of about 70% have been reported for some anthracene–anthracene biplanemers which were formed by a triplet reaction (see Section 3.6). According to the orbital symmetry conservation, the intramolecular [4π + 4π] cycloaddition is photochemically allowed. The compounds 1, in which two arene ring systems A and B are connected by a saturated tether, represent bichromophoric systems. The electronic excitation in the S1 state of 1 is located (first approximation) in the arene of lower singlet energy (S1(B) ≤ S1(A)). The cycloisomerization 1 → 2 can be regarded as cycloaddition of the excited part B and part A in the ground state. Large orbital coefficients of HOMO and LUMO in the reacting centres are important for a highly positive orbital overlap and determine the reactive sites. The cycloisomerization 1(S1) → 2 is always diabatic (DC) and includes therefore an intermediate return from the excited state to the ground state So – however, first a crossing to a higher singlet state Sn can occur.Fig. 1 reveals that there are three routes for the reverse reaction 2 → 1, namely the thermal cycloreversion (TCR), which is an exothermic process in the ground state So, a diabatic and an adiabatic photochemical cycloreversion route (DCR and ACR).
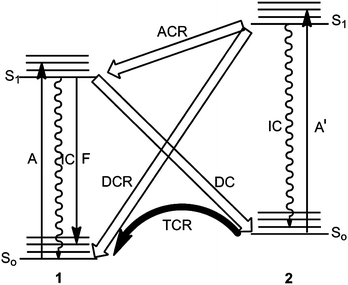 | ||
| Fig. 1 Reaction routes for the interconversion 1 ⇄ 2 on the singlet energy hypersurfaces. DC: diabatic cycloisomerization, DCR: diabatic cycloreversion, ACR: adiabatic cycloreversion, TCR: thermal cycloreversion; A,A′: absorptions, F: fluorescence (fluorescence of 2 is rather exceptional), IC: internal conversion. | ||
The thermal activation barrier depends on the individual system. In some cases, the thermal process 2 → 1 occurs already at room temperature, in some other cases temperatures of more than 150 °C are needed. The average activation energy Ea amounts to 35 ± 5 kcal mol−1.31
The light-triggered reaction 2 → 1 is normally performed by irradiation at the benzene absorption A′ (for example at λ = 254 nm). The photoreaction 1 → 2, on the other hand, is performed at λ > 270 nm by irradiation at the naphthalene or anthracene absorption. Surprisingly, a recent paper by Turro et al.32 showed that the adiabatic route ACR can amount to 90% of the whole process. A typical example is the photochemical cycloreversion of the intramolecular adduct 2a to bis(9-anthrylmethyl)ether 1a32 (Scheme 2). The ratio 1a (S1)/1a (So) was determined by quantitative fluorescence spectroscopy.
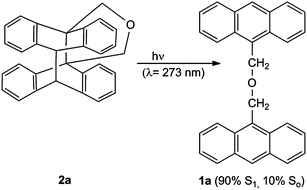 | ||
| Scheme 2 Photochemical cycloreversion: adiabatic and diabatic reaction routes. | ||
Many details of the energy hypersurfaces are unknown. In addition to the pericyclic minimum PM*, one or more minima for intramolecular excimers/exciplexes E* can exist. Four different possibilities have to be considered:
(i) A fluorescent excimer/exciplex can be detected. It can be located on the photochemical reaction route 1 → 2 and/or 2 → 1 or is formed independently.
(ii) A fluorescent excimer/exciplex cannot be recognized in the photochemical reaction route 1 → 2 or 2 → 1.
(iii) An excimer/exciplex emission of 1 can be seen, but the reaction 1 → 2 does not take place.
(iv) The most difficult case is the participation of a non-fluorescent excimer/exciplex.30,31
Altogether the generation of fluorescing intramolecular excimers/exciplexes is the most general case (i). Reaction 1a ⇄ 2a is an example of case (ii). Aromatic substituents, such as phenyl, at the 10-position of the anthracene moiety lead to case (iii).30,31
The reaction mechanism ranges from a concerted process to a stepwise process via an intermediate diradical DR. A reliable proof for the participation of diradicals is given when epidioxides are formed in the presence of oxygen (trapping of 1,8-biradicals).33
Fig. 2 visualizes intramolecular excimers/exciplexes E*, pericyclic minima PM* for a concerted reaction and diradicals DR, which may have zwitterionic character.2,3,14
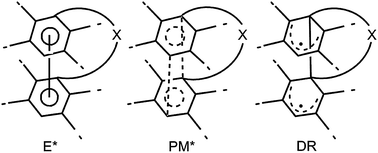 | ||
| Fig. 2 E*: intramolecular excimer/exciplex, PM*: pericyclic minimum, DR: diradical. | ||
Normally, the switching processes 1 ⇄ 2 are performed in solution; however, many topochemically controlled reactions in the solid state are known as well.
3. Biplanemers
3.1 Benzene–benzene biplanemers
Although the existence of tetracyclo[6.2.2.24,7.01,4] tetradeca-5,9,11,13-tetraene (4, n = 2) was claimed on the basis of CIDNP studies,344 or its derivatives cannot be obtained by photocycloisomerization reactions of 1,2-diphenylethanes (Scheme 3). Diphenylmethane or higher α,ω-diphenylalkanes do not show such a photoreaction either.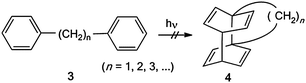 | ||
| Scheme 3 α,ω-Diphenylalkanes do not photocycloisomerize. | ||
3.2 Naphthalene–benzene biplanemers
Naphthalene–benzene biplanemers are the smallest biplanemers which can be obtained by intramolecular photocycloaddition reactions of naphthalenes tethered at the 1-position to an electron rich benzene ring system. When irradiated at λ > 270 nm, the compounds 5a,b show a quantitative cycloisomerization to 6a,b. This process is slower and less effective for 5c3,35 (Scheme 4). Irradiation of 6a–c at λ = 254 or heating to 90–100 °C causes the reverse reaction.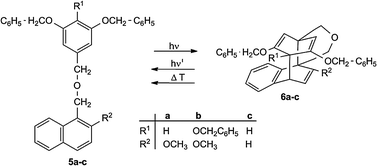 | ||
| Scheme 4 Generation and cleavage of naphthalene–benzene biplanemers 6a–c. | ||
Hydrolysis of the enol ethers 6 by formic acid generates the corresponding ketones.35Scheme 5 illustrates the formation of monoketone 7c and diketone 9c by enol ether cleavage of 6c. The dienone 8c, an isomer of 7c, shows a spontaneous, irreversible Cope rearrangement to the conjugated compound 10c. The central [4.2.2.22,5] tricycle is thereby transformed to a [6.4.0.02,7] tricycle. The biplanemers 6a–c themselves do not show such a [3,3] shift.
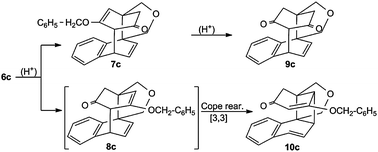 | ||
| Scheme 5 Enol ether cleavage of 6c. | ||
Rearrangements of the central skeleton can already occur directly after the protonation of 6a to three isomeric carbenium ions 11a, 12a, 13a (Scheme 6). Instead of the equilibria 12a ⇄ 12a′, 13a ⇄ 13a′ and 18a ⇄ 18a′, non-classical carbocations cannot a priori be excluded.35
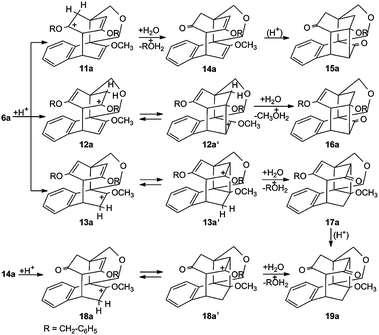 | ||
| Scheme 6 Enol ether cleavage of 6a. | ||
An excess of formic acid leads to the product distribution 15a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16a:17a:19a = 18:11:18:23. The compounds 17a and 19a, which have a central tetracyclo[6.4.0.02,7.04,11] dodecane structure, predominate slightly.35
:16a:17a:19a = 18:11:18:23. The compounds 17a and 19a, which have a central tetracyclo[6.4.0.02,7.04,11] dodecane structure, predominate slightly.35
3.3 Naphthalene–naphthalene biplanemers
Chandross and Dempster36 obtained the first intramolecular cycloaddition of a tethered bichromophoric naphthalene system 20a ⇄ 21a. De Schryver et al.37 investigated the reaction 20c → 21c, 22c. Recently Turro et al.32 studied the reverse reactions 21a–c ⇄ 20a–c and 22c ⇄ 20c (Scheme 7). A three-atom tether X–Y–X is best suited for this photocyclization, neither a (CH2)2 nor a (CH2)4 chain permits the process. In contrast to all other biplanemers discussed here, the photolysis of 20 can lead to two stereoisomers, the syn configuration 21 and the anti configuration 22 (Scheme 7). The ratio 21:22 depends on the solvent and in particular on the tether: 20a forms selectively the syn product 21a, whereas the more flexible CH2–O–CH2 tether gives both stereoisomers: 20c → 21c, 22c.37
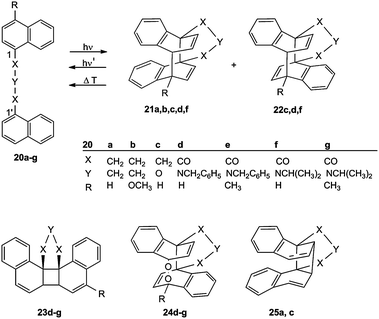 | ||
| Scheme 7 Generation and cleavage of naphthalene–naphthalene biplanemers (21, 22) and obtained side products 23–25. | ||
In the case of the tethers 20d–g, the [2 + 2] cycloadducts 23 are the sole or at least the major products and the [4 + 4] cycloadducts the minor products.33 In the presence of oxygen, the epidioxides 24 were obtained.33 A consecutive Cope rearrangement can generate cyclobutane derivatives (21a,c → 25a,c).36,37 The bichromophores 20 generate upon irradiation an intramolecular excimer/exciplex. The following σ bond formation between C-1 and C-1′ leads to a diradical whose syn form is the precursor of 21 and 23, whereas its anti form can generate 22 and 24.33
3.4 Anthracene–benzene biplanemers
As soon as an anthracene chromophore is part of the bichromophoric system, intra- and intermolecular cycloadditions can occur. Let us consider here anthracene–benzene systems A–B connected by a CH2–O–CH2 tether. Scheme 8 summarizes the possible reaction routes.38,39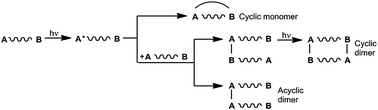 | ||
| Scheme 8 Intra- and intermolecular photocycloadditions of bichromophoric anthracene (A)–benzene (B) systems. | ||
The dimerization to open-chained systems represents moreover the first step in the formation of polymers. Dimers and polymers are preferentially formed when highly concentrated solutions of B–A–B compounds are used.38,40 In diluted solutions (c < 3.3 × 10−3 mol L−1) in benzene, cyclic monomers are formed – often in quantitative yields. The shorter tether A–CH2–O–B does not permit the photocyclization.41Scheme 9 summarizes our experiments with compounds 26a–r.2,8,14,38,39,42,43
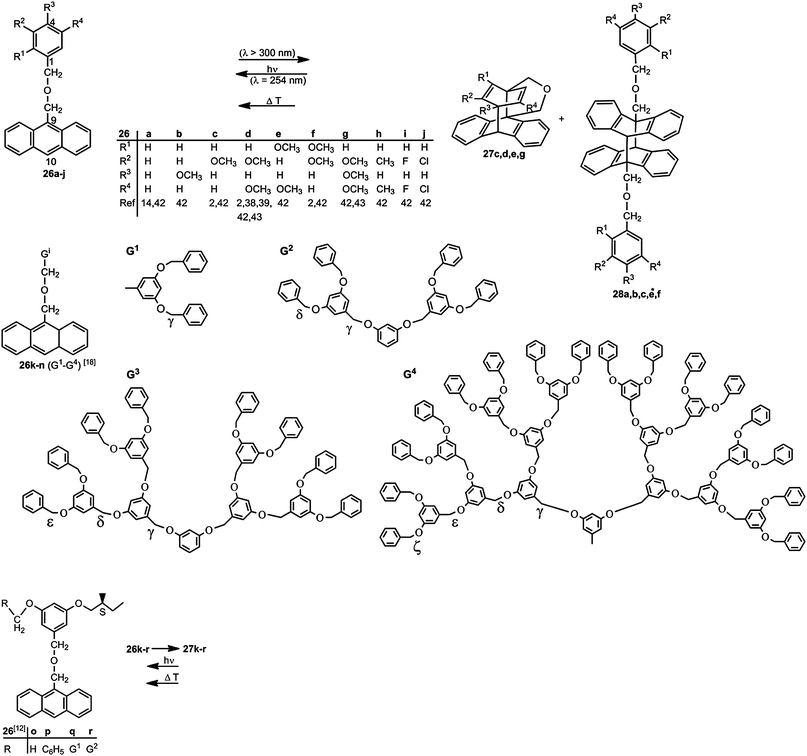 | ||
| Scheme 9 Formation and cleavage of anthracene–benzene biplanemers. | ||
The cycloisomerization 26 → 27 requires electron rich benzene rings, in particular at the para-position C-4 which is connected to C-10 of the anthracene moiety for the formation of a new σ bond in 27. Therefore 26a, 26b and 26f, 26h, 26i and 26j do not show photocyclization.42 Irradiation leads in these cases to anthracene dimers 28 of the type B–A–A–B (Scheme 8). The dimers 28a, b, e and f were studied in detail. Their side chains have anti-position (head-to-tail).42 Compound 26c with one methoxy group gives approximately a 1:1-mixture of cyclization product 27c and anthracene dimer 28c.2,42
The series 26k–n represents a dendrimer series having two dendrons of the Fréchet type (Scheme 9).14 Irradiation at λ > 300 nm affords quantitatively the biplanemers 27k–n. The energy transfer from the dendrons to the core (antenna effect) makes these systems highly attractive as optical switches.14
Fig. 3 visualizes for the dendrimer of 4th generation the peripheral energy migration (EM) from chain to chain and the focal energy transfer (ET) along a chain. In principle, there are in 26 three fairly independent regions of chromophores: anthracene (Chr-1), 1,3,5-trisubstituted benzene rings (Chr-2), and terminal benzene rings (Chr-3).
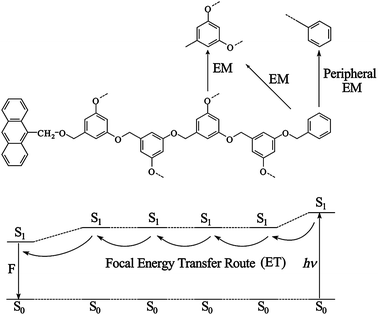 | ||
| Fig. 3 Downhill focal energy-transfer (ET) route and peripheral energy migration (EM) in the dendrimer 26n (fourth generation). | ||
The ratio of the three chromophores depends on the generation i:Chr-1:Chr-2:Chr-3 = 1:(2i-1):2i (i = 0–4). Compound 26a can be regarded as generation zero of the dendrimer series. The focal energy transfer is a (multistep) “downhill” process (1 to 5 steps). According to the fluorescence enhancement, the quantum yield of the energy transfer (λexc = 260 nm, 3 × 10−7 M solution in CH2Cl2) increases from the 1st to the 4th generation from 9 ± 1 to 14 ± 1%. The fluorescence quantum yield itself decreases with increasing generation.14 Unsubstituted anthracene shows under these conditions a quantum yield of 27%. Increasing generation leads to an increasing number of vibrational modes and consequently to an enhanced radiationless decay.
The focal energy transfer enhances the fluorescence, but it does not change the photostationary state because the reverse process 27k–n → 26k–n is obviously more efficient upon irradiation at λ = 260 nm. We attribute this to a high quantum yield of the cycloreversion and to an additional energy transfer from the benzene rings to the cyclohexadiene chromophore (Chr-4) in 27k–n. The photostationary state for the irradiation at λ = 260 or 254 nm is completely on the side of 26k–n as it is at λ ≥ 320 nm completely on the side of 27k–n (Scheme 10).14
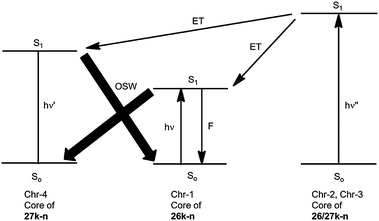 | ||
| Scheme 10 Optical switching (OSW) and its amplification by energy transfer (ET) in the systems 26k–n/27k–n with photoreactive cores and light-harvesting dendrons (ν < ν′ ≤ ν′′). | ||
Fig. 4 exhibits the decline and the recovery of the anthracene absorption (Chr-1) of the optical switch 26l ⇄ 27l. The parameters given for the maximum at 394 nm concern the irradiation time in seconds. A fatigue at λ ≥ 320 nm and for the thermal recovery was not observed. The irradiation at 254 nm has a slow fatigue, which became visible after more than 20 cycles.
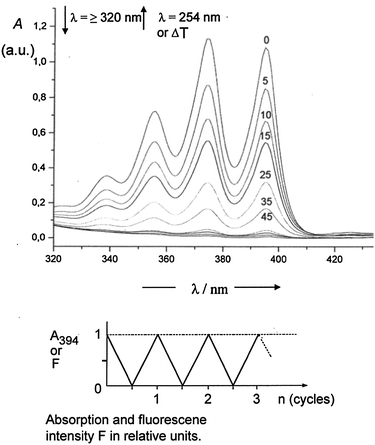 | ||
| Fig. 4 Upper part: reduction of the typical anthracene absorption (Chr-1) by irradiation (λ ≥ 320 nm) of a 4 × 10−3 M solution of 26l in benzene within 45 s. Subsequent irradiation at λ = 254 nm or heating leads to the corresponding recovery of the anthracene absorption. Lower part: change of absorption at 394 nm or fluorescence intensity (relative units) within three cycles of optical switching. | ||
A chiroptical switch could be obtained on the basis of compounds 26p–r ⇄ 27p–r (Schemes 9 and 11).8
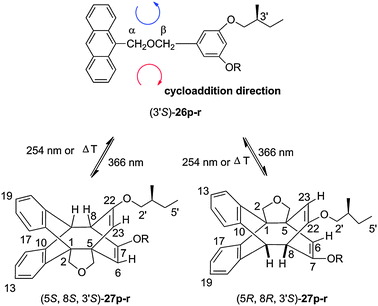 | ||
| Scheme 11 Photocycloisomerization, photocleavage and thermal cycloreversion 26p–r ⇆ 27p–r (the thermal cycloreversion was performed at 60 °C). | ||
The intramolecular cycloaddition leads to the diastereomeric 1:1-mixture (5S,8S,3′S)-27p–r/(5R,8R,3′S)-27p–r. The conversion of model compound 26o was less complete.8 The two equivalent cycloaddition directions are shown in Scheme 11. According to the optical switch (Scheme 10), the optical rotation values of 4.0 × 10−3 M solutions of (3′S)-26p–r in benzene can be reversibly modulated by light. Fig. 5 demonstrates that the optical rotation value [α]D20 of 26p increases upon irradiation at λ = 366 nm and returns to its initial magnitude upon irradiation at λ = 254 nm. A fatigue cannot be seen within six cycles.
![Optical rotation values [α]D20 in the switching cycles 26p ⇄ 27p. Irradiation at λ = 366 nm (—), irradiation at λ = 254 nm (....). The irradiation period for the complete conversion 26p → 27p was 1 min and for the complete reversion 27p → 26p was about 2 min.8](/image/article/2013/CS/c2cs35271k/c2cs35271k-f5.gif) | ||
| Fig. 5 Optical rotation values [α]D20 in the switching cycles 26p ⇄ 27p. Irradiation at λ = 366 nm (—), irradiation at λ = 254 nm (....). The irradiation period for the complete conversion 26p → 27p was 1 min and for the complete reversion 27p → 26p was about 2 min.8 | ||
The cyclization products 27, which have an enol ether structure, can be hydrolysed to the corresponding mono- and diketones, but 27g shows additionally to the formation of 30g and 32g a rearrangement of the protonated species 28g → 29g, so that finally the originally saturated ether function of 27g is cleaved (Scheme 12). The structure determination of ketone 31g was based on an X-ray measurement.43
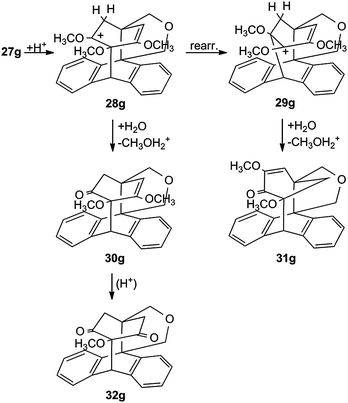 | ||
| Scheme 12 Acidic enol ether cleavage of the trimethoxy biplanemer 27g. | ||
3.5 Anthracene–naphthalene biplanemers
The generation of anthracene–naphthalene biplanemers 34a–r is shown in Scheme 13. It is often accompanied by the formation of anthracene head-to-tail dimers – analogous to 26 → 28.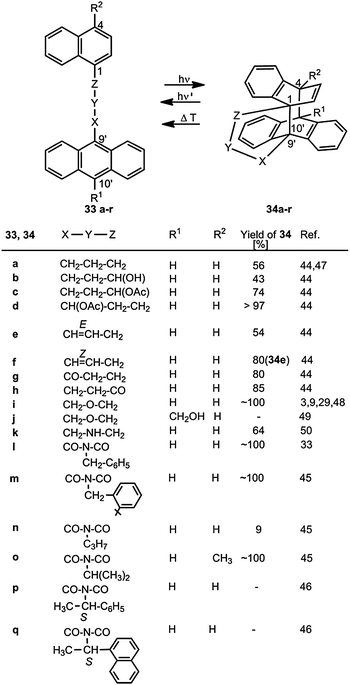 | ||
| Scheme 13 Generation and cycloreversion of anthracene–naphthalene biplanemers (the numbering in 34 does not correspond to the nomenclature). | ||
Substituted linkers as in the examples 33b,c,d lead to diastereomeric products 34b–d.44 The substituents OH or OAc can have the syn or the anti position to the remaining benzene ring of the naphthalene moiety. The cis configuration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in 33f is preserved in product 34f but not the trans configuration of 33e. Conjugated enone tethers CO–CHCH do not permit the cyclization process.
C double bond in 33f is preserved in product 34f but not the trans configuration of 33e. Conjugated enone tethers CO–CHCH do not permit the cyclization process.
Kohmoto et al.45,46 studied in detail the reactions of 33l–q, which have iminodicarbonyl tethers. The reactivity in the solid state depends on the crystal structure – in particular on the distances C-1–C-9′ and C-4–C-10′ and the angles between the planes of the two arene ring systems. Whereas the benzyl system 33l and the 2-tert-butylbenzyl system 33m show a topochemically controlled cycloisomerization, the corresponding 2-methylbenzyl, 4-methylbenzyl and 4-fluorobenzyl compounds do not.45 Moreover, 33l crystallizes in the chiral space group P212121, which is a precondition for an absolute asymmetric synthesis. The product (chiral space group P21) was obtained in the absolute configuration (1R,4S)-34l with an enantiomeric excess ee of 82%. The ee value for the reaction 33m → 34m is close to 100%.45
A different situation is present in 33p and 33q. These starting compounds contain (S)-configured chiral centers in the R substituents of the CO–NR–CO linker. Therefore, a diastereoselectivity can be expected. Table 1 summarizes the results for the photoreaction in the solid state and in solution.46 The reversal of the de, measured for 34p in the crystalline state and in acetone, is remarkable.
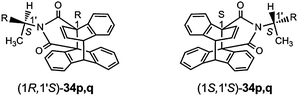

A novel type of chiral T-photochromic system was found by Kohmoto et al.17 by utilizing the thermally reversible photocyclization of anthracenes which bear at the 9- and the 10-position iminodicarbonyl linkers to naphthalene units. The compound with N-benzyl groups for example forms in the solid state and also at room temperature for a long period in solution a foldamer in which the naphthalene and anthracene moieties are twisted against each other. Racemization of the helical structures occurs very slowly. The N-(1S)-1-(1-naphthyl)ethyl system 35 was used for the switch of the induced circular dichroism of the CD active anthracene derivate and its CD inactive (Δε ≈ 0) photoisomer 36. A fatigue within 9 cycles could not be found17 (Scheme 14).
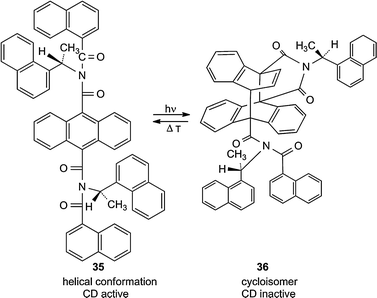 | ||
| Scheme 14 Chiral photochromic system 35/36. The irradiation in solution was performed at λ > 290 nm (25 s) and the heating at 60 °C (20 min) in order to switch Δε (271 nm) between its maximum and zero.17 | ||
3.6 Anthracene–anthracene biplanemers
The photodimerization of anthracenes and its reversion were studied intensively and reported in several excellent reviews,27–31 so that Section 3.6 can be kept short and focused on new applications.A vast number of bichromophoric systems 37a–d with three-atom tethers have been studied (Scheme 15). The intramolecular cycloaddition is the only photoreaction in diluted solution and often also in the crystalline state. The yields of 38 are normally high to excellent and in some cases quantitative when the irradiation wavelength is restricted to the typical anthracene absorption (λ ≥ 300 nm). The fatigue of the photochemical or thermal cycloreversion in optical switches is in most cases low.
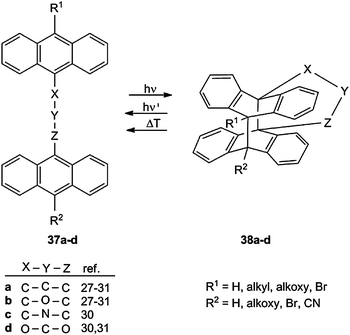 | ||
| Scheme 15 Generation and cleavage of anthracene–anthracene biplanemers with 3-atom tethers. | ||
Some special cases shall be mentioned here. Becker and Amin51 found that diastereomers (X–Y–Z: CH(OH)–CH2–CH(CH3) or CH(CH3)–CH(CH3)–CO) give the same yields and quantum yields for the cycloisomerization 37 → 38 – irrespective of preferred conformations. A carbonyl group in tether type a causes an intersystem crossing and consequently an intramolecular [4π + 4π] cycloaddition in the triplet state T1, which can also be populated by biacetyl sensitization. The quantum yield for the triplet reaction can be extraordinarily high (up to 72%).51 When the tether contains an (E)-configured double bond, first photoisomerization to the (Z)-isomer has to take place.
Compound 37b with R1 = R2 = C6H5 is an example which exhibits an excimer fluorescence but fails to cycloisomerize.31 Cyano groups (R2 = CN) permit an efficient cycloisomerization but the reverse thermal process works already at room temperature.52
In contrast to COC- and OCO-tethers, CNC-tethers are less flexible. Consequently, the corresponding cyclization reactions are less effective and have low quantum yields. Although compounds 38a–d with three-atom tethers represent the majority of anthracene–anthracene biplanemers, shorter and longer tethers were successfully used as well. Scheme 16 gives a survey over “butterfly” compounds 39–41 with one- and two-atom tethers and Scheme 17 over systems 42 with four-atom and longer tethers.
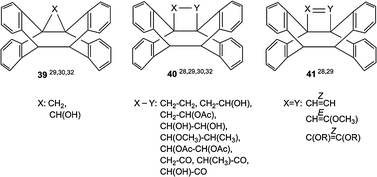 | ||
| Scheme 16 Anthracene–anthracene biplanemers with one- or two-atom tethers. | ||
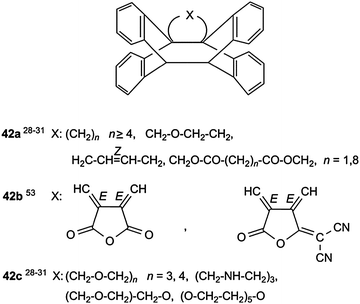 | ||
| Scheme 17 Anthracene–anthracene biplanemers with four-atom or longer tethers. | ||
The olefinic double bonds in 41 and 42 must have a suitable configuration Z or E, as shown in Schemes 16 and 17, respectively.
Compound 40 (X–Y: CH2–CH2) can be regarded as the head-to-head dimer (H–H) of the 9-anthrylmethyl radical. The corresponding head-to-tail dimer (H–T) is called lepidopterene (40′). The dimerization leads in both cases to the formation of three σ bonds (Scheme 18).31,54–56
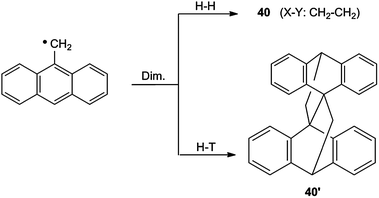 | ||
| Scheme 18 Dimerization of 9-anthrylmethyl radicals. | ||
Apart from the structures 42, which represent head-to-head dimers, head-to-tail dimers can be formed, provided that the tethers are long enough.57
Sterically congested bis(9-anthryl)methane derivatives, such as 2,2-bis(9-anthryl)lactic acid esters, do not show a [4π + 4π] cycloaddition reaction. They enter into an intramolecular [2π+4π] photocycloaddition.58 The quantum yields for the photocycloisomerization of the dianthracene series with (CH2)n tethers are high for n = 1–3, for longer tethers they become very low and approach zero (Table 2).
Mattay et al.11,12,15,16,21–23 synthesized resorcin[4]arenes such as 43 and related compounds, which bear anthracene chromophores on opposite benzene rings. Irradiation at the anthracene absorption at 350 nm transforms 43a–e to the corresponding cycloisomers by intramolecular [4 + 4] cycloadditions; 43f shows a decomposition upon irradiation. A head-to-head regiochemistry could be established for the short linker X = CH2–O–CH2. DFT calculations predict head-to-tail adducts for the other systems 43b–e. The reverse thermal reaction occurs for these longer linkers already at room temperature; however, for 43a 60 °C is needed (Scheme 19).
![Photochromic resorcin[4]arenes 43 which form a “pocket” by intramolecular [4π + 4π] cycloaddition of the two anthracene units A.](/image/article/2013/CS/c2cs35271k/c2cs35271k-s19.gif) | ||
| Scheme 19 Photochromic resorcin[4]arenes 43 which form a “pocket” by intramolecular [4π + 4π] cycloaddition of the two anthracene units A. | ||
The photoswitchable cavitand led to an interesting study by AFM single-molecule force spectroscopy.23 The AFM technique can differentiate between the two states of the host–guest system. Moreover, it was shown, with ammonium ions as guests, how dynamic force spectroscopy can be used for the determination of complex decomposition rates and molecular parameters such as the depth of the pocket of these molecular tweezers.
The idea to put a “lid” on calix[4]arenes by attaching two photoswitchable anthracene units was first realized by Shinkai et al.61–63 The ionophoric cavities complex alkali cations which are squeezed out by the anthracene cycloisomerization. The selectivity of alkali ion complexation depends significantly on the length of the linkers, which are differently attached in 43 and 44 (Scheme 20).
![Light-switched ionophoric calix[4]arenes.](/image/article/2013/CS/c2cs35271k/c2cs35271k-s20.gif) | ||
| Scheme 20 Light-switched ionophoric calix[4]arenes. | ||
Optical recording connected with luminescence readout can also be realized with optical switches which contain metal complexes. Near-field scanning optical microscopy proved to be a powerful tool for this purpose. Ruthenium complex 45, embedded in an inert polymer film of PMMA, was irradiated with a 400 nm “write” laser. The process of intramolecular cycloaddition 45 → 46 corresponds to a two-dimensional data storage (Scheme 21). The absorption spectrum of the unexposed film shows a broad band at 460 nm which can be correlated with a RuII based metal-to-ligand charge transfer transition. The corresponding emission band at 610 nm has a low intensity, which is strongly increased after the irradiation. This can be used for the readout, triggered by 488 nm light. The experiment proved an areal data density of greater than 0.1 Gbit inch−2 (16 Mbit cm−2) in this write-once read many (WORM) optical storage medium.24
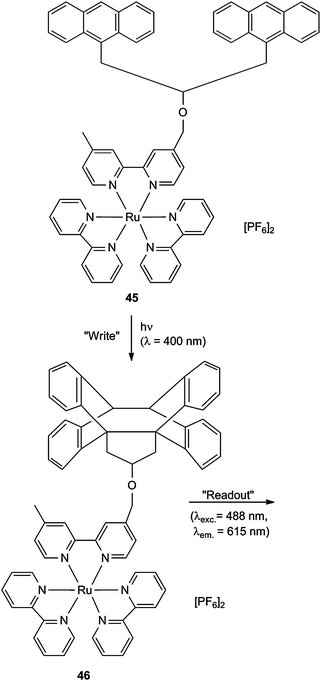 | ||
| Scheme 21 Optical recording for the WORM technology (instead of the CH2O segment in the tether, a COO function can be used). | ||
A similar system, which is based on a ReI complex 47, is shown in Scheme 22. The cyclomerization 47 → 48 stops the quenching of the excited rhenium moiety by energy transfer to the anthracene units and increases therefore the ReI based emission.19,20
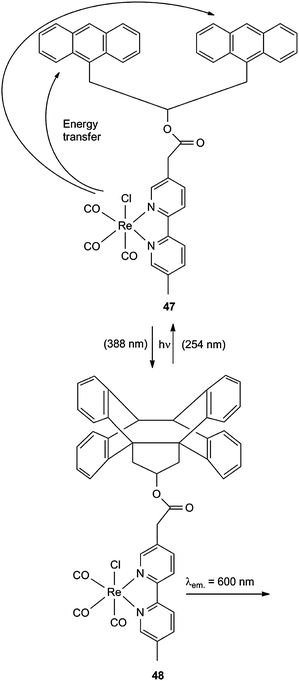 | ||
| Scheme 22 Optical switch for luminescence readout. | ||
A double complexation is possible with the photochromic system 49 ⇄ 50. The quantum yield for the cycloisomerization increases significantly, when Na+ is complexed by the oxygen atoms in the tether, or when HgCl2 is complexed by the bipyridyl unit or when both complexations are performed (Scheme 23).25 Low energy conformations of the bipyridyl have non-interacting chains on different sides. Both complexations force the compound to adapt the conformation shown in Scheme 23, which permits the intramolecular cycloaddition of the anthracene moieties (49 → 50). The thermal cleavage 49 → 50, on the other hand, is only influenced by the Na+ complexation in the crown ether unit. The rate constant of the cycloreversion at room temperature is much higher in the absence of Na+.
 | ||
| Scheme 23 Influence of the complexations on the formation and the cleavage of the intramolecular anthracene–anthracene cycloadducts. | ||
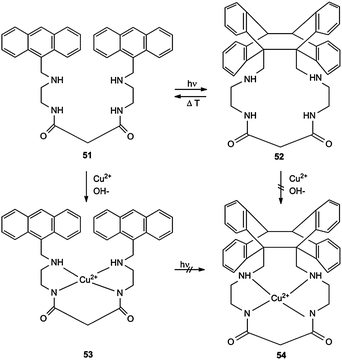
Another type of photoswitchable system was realized for the dioxotetramin ligand 51 and its Cu2+ complex 53 (Scheme 24).7 Compound 51 shows the normal cycloisomerization to 52 which can be thermally reverted. 51 acts as a selective receptor for Cu2+ ions at pH 6.5–7.0. Other dications, such as Ni2+, Co2+, Fe2+, Zn2+ and Mn2+, are not complexed. The Cu2+ complex 53 exhibits a typical absorption centered at 510 nm, but it does not permit photocycloisomerization to 54. Obviously, the square planar complex structure keeps the anthracene ring systems too far apart from each other. Cyclic structure 52 does not complex Cu2+, either. The new ligand 51 can also be used for a photoinduced pKa modulation.7
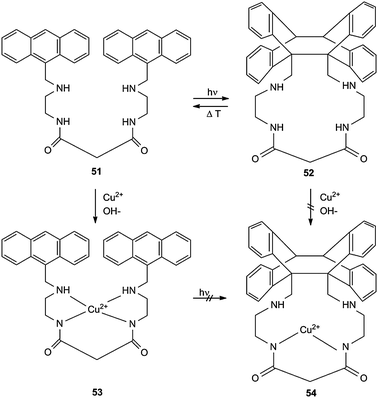 | ||
| Scheme 24 Photoswitchable selective receptor for CuII. | ||
The aza-crown ether 55 binds group 1 metal cations in a 1:1 stoichiometry (Scheme 25).26 The 2-(9-anthryloxy)ethyl pendants on the pyramidal N atoms can be oriented to the same side or to different sides of the coronand. The Na+ complex of 55 prefers the trans geometry, whereas the Rb+ complex has cis oriented pendants. Upon irradiation the coronand 55 forms a cryptand 56 whose metal binding constants are much weaker than those of 55. Table 3 shows the effect of complexation on the photocycloisomerzation and its thermal reversion.
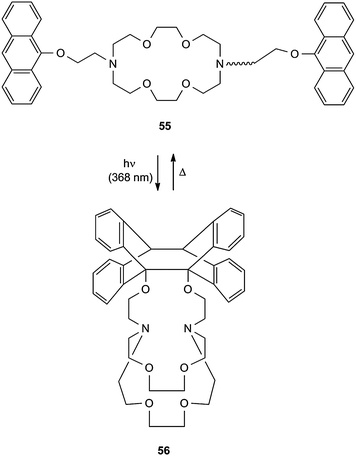 | ||
| Scheme 25 Switch between a coronand and a cryptand structure. | ||
|
|
|
|||
|---|---|---|---|---|
| ϕ [%] | k [10−6 s−1] | k′ [10−6 s−1] | ||
| 55 | 11 | 9 | 56 | 3.4 |
| Na+ ⊂ 55 | 5 | 4.0 | Na+ ⊂ 56 | 0.34–0.7 |
| K+ ⊂ 55 | 8 | 6.5 | K+ ⊂ 56 | 2.3 |
| Rb+ ⊂ 55 | 7 | 3.8 | Rb+ ⊂ 56 | 170–255 |
| Cs+ ⊂ 55 | 2–3 | 2.4 | Cs+ ⊂ 56 | 6.7–8.5 |


3.7 Unusual anthracene–anthracene biplanemers
The tethers of all these bichromophoric systems are normally fixed at the 1-position of naphthalene and the 9-position of anthracene. However, there is formation of biplanemers with tethers at other positions. De Schryver et al.64,65 studied the intramolecular photocycloadditions of 57 to 58 and 59. The proportion of regioisomer 59 increases with increasing numbers n (Scheme 26).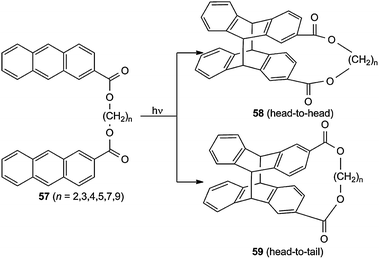 | ||
| Scheme 26 Cycloisomerization of bisanthracenes tethered at the 2-position. | ||
Even bichromophoric anthracenes, which are linked by a chain, which is fixed at both 9-positions, do not always give the normal [4π + 4π] cycloadduct. Steric hindrance causes in 60 the reaction of a central with an outer benzene ring (Scheme 27) via the corresponding unsymmetrical excimer.66 The same type of [4π + 4π] cycloaddition occurs with one Si(CH3)2.67
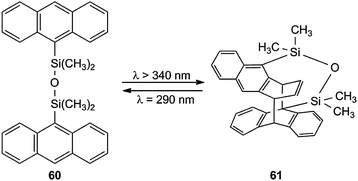 | ||
| Scheme 27 Unsymmetrical biplanemers 61 obtained from bisanthracene 60. | ||
This type of unsymmetrical biplanemers seems to be more general, but only very few examples have been studied.
4. Conclusions
In the middle of the 19th century Fritzsche68 detected the light sensitivity of anthracene, a compound which he called “photene”. The present article shows that the inherent [4π + 4π] photocycloaddition could be extended to tethered bichromophores in which benzene, naphthalene or anthracene ring systems can be involved. Hence, in recent years the scope of this reaction type could be significantly extended. Tethered higher benzenoid aromatics, such as the naphthacene (tetracene) and pentacene derivatives 62, 63, have never been studied in this context, but will presumably react in the same way (Scheme 28).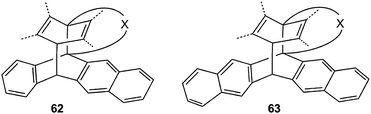 | ||
| Scheme 28 Unknown biplanemers. | ||
The tethered bichromophores, discussed in Section 3, form biplanemers upon irradiation at λ ≥ 270 nm. The reverse photoreaction, a [2π + 2π + 2σ + 2σ] cycloreversion occurs at λ = 254 nm. The photochromic behaviour is complemented by a thermal back reaction. The T-photochromic system has normally a slower fatigue than the genuine P-photochromic system. Both systems can be used in optical and chiroptical switches. Additionally a light harvesting effect can be applied in dendritic compounds, whose dendrons provide an antenna effect.
Apart from different UV/Vis absorption ranges of the bichromophores before and after irradiation, light emission can be used for the “reading” after the “writing”. Normally, fluorescence can be measured for the open form of the bichromophore and not for its cycloisomer. However, an opposite fluorescence behaviour is possible for biplanemers which contain a metal complexation.
Other applications of optical switches of biplanemers make use of systems which work as molecular tweezers or pockets.
In previous years, photonic devices, lithographic patterning, imaging processes, data processing, data storage and sensor techniques were in the focus of this “old photoreaction” with “new compounds and new applications”.
Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, the National Natural Science Foundation of China (20872038, 21072064) and the Natural Science Foundation of Guangdong Province (06025664, 1035 1064101000000) for financial support.References
- K. K. Baldridge, T. R. Battersby, R. VernonClark and J. S. Siegel, J. Am. Chem. Soc., 1997, 119, 7048–7054 CrossRef CAS.
- S. Dobis, D. Schollmeyer, C. Gao, D. Cao and H. Meier, Eur. J. Org. Chem., 2007, 2964–2969 CrossRef CAS.
- D. Cao and H. Meier, Tetrahedron Lett., 2005, 46, 4975–4977 CrossRef CAS.
- R. Srinivasan and K. H. Carlough, J. Am. Chem. Soc., 1967, 93, 4932–4936 CrossRef.
- R. S. H. Liu and G. S. Hammond, J. Am. Chem. Soc., 1967, 93, 4936–4944 CrossRef.
- F. Hirayama, J. Chem. Phys., 1965, 42, 3163–3171 CrossRef CAS.
- G. Dacarro, P. Ricci, D. Sacchi and A. Taglietti, Eur. J. Inorg. Chem., 2011, 1212–1218 CrossRef CAS.
- W. Liu, D. Cao, J. Peng, H. Zhang and H. Meier, Chem.–Asian J., 2010, 5, 1896–1901 CrossRef CAS.
- D. Shykind, R. Bristol, J. Roberts, J. Blackwell and Y. Borodovsky, Proc. SPIE-Int. Soc. Opt. Eng., 2010, 7639 Search PubMed (Pt. 2, Adv. in Resist Mat. and Process. Technol. XXVII), 76391Y/1–76391Y/10.
- R. Bristol, D. Shykind, S. Kim, Y. Borodovsky, E. Schwartz, C. Turner, G. Masson, K. Min, K. Esswein and J. M. Blackwell, et al. , Proc. SPIE-Int. Soc. Opt. Eng., 2009, 7273 Search PubMed (Pt. 1, Adv. in Resist Mat. And Process. Technol. XXVI), 727307/1–727307/12.
- C. Schaefer, A. B. Rozhenko and J. Mattay, Photochem. Photobiol. Sci., 2009, 8, 1187–1194 CAS.
- M. Letzel, C. Schaefer, F. R. Novara, M. Speranza, A. B. Rozhenko, W. W. Schoeller and J. Mattay, J. Mass Spectrom., 2008, 43, 1553–1564 CrossRef CAS.
- D. Anselmetti, F. W. Bartels, A. Becker, B. Decker, R. Eckel, M. McIntosh, J. Mattay, P. Plattner, R. Ros, C. Schaefer and N. Sewald, Langmuir, 2008, 24, 1365–1370 CrossRef CAS.
- D. Cao, S. Dobis, C. Gao, S. Hillmann and H. Meier, Chem.–Eur. J., 2007, 13, 9317–9323 CrossRef CAS.
- C. Schaefer, B. Decker, M. Letzel, F. Novara, R. Eckel, R. Ros, D. Anselmetti and J. Mattay, Pure Appl. Chem., 2006, 78, 2247–2259 CrossRef CAS.
- C. Schaefer and J. Mattay, Photochem. Photobiol. Sci., 2004, 3, 331–333 CAS.
- H. Masu, I. Mizutani, T. Kato, I. Azumaya, K. Yamaguchi, K. Kishikawo and S. Kohmoto, J. Org. Chem., 2006, 71, 8037–8044 CrossRef CAS.
- V. Ferri, M. Scoponi, C. A. Bignozzi, D. S. Tyson, F. N. Castellano, H. Doyle and G. Redmond, Nano Lett., 2004, 4, 835–839 CrossRef CAS.
- P. Belser, S. Bernhard, C. Blum, A. Beyeler, L. De Cola and V. Balzani, Coord. Chem. Rev., 1999, 190, 155–169 CrossRef.
- A. Beyeler, P. Belser and L. De Cola, Angew. Chem., 1997, 109, 2878–2881 ( Angew. Chem., Int. Ed. Engl. , 1997 , 36 , 2779–2781 ) CrossRef.
- M. Michelswirth, M. Raekers, C. Schaefer, J. Mattay, M. Neumann and U. Heinzmann, J. Phys. Chem. B, 2010, 114, 3482–3487 CrossRef CAS.
- C. Schaefer, F. Struebe, S. Bringmann and J. Mattay, Photochem. Photobiol. Sci., 2008, 7, 1457–1462 CAS.
- C. Schaefer, R. Eckel, R. Ros, J. Mattay and D. Anselmetti, J. Am. Chem. Soc., 2007, 129, 1488–1489 CrossRef CAS.
- D. S. Tyson, C. A. Bignozzi and F. N. Castellano, J. Am. Chem. Soc., 2002, 124, 4562–4563 CrossRef CAS.
- G. McSkimming, J. H. R. Tucker, H. Bouas-Laurant and J. P. Desvergne, Angew. Chem., 2000, 2251–2253 ( Angew. Chem., Int. Ed. , 2000 , 39 , 2167–2169 ) CrossRef.
- G. McSkimming, J. H. R. Tucker, H. Bouas-Laurant, J. P. Desvergne, S. J. Coles, M. B. Hursthouse and M. E. Light, Chem.–Eur. J., 2002, 8, 3331–3342 CrossRef CAS.
- H. D. Becker, Pure Appl. Chem., 1982, 54, 1589–1604 CrossRef CAS.
- H. D. Becker, Chem. Rev., 1993, 93, 145–172 CrossRef CAS.
- H. Bouas-Laurant, A. Castellan and J. P. Desvergne, Pure Appl. Chem., 1980, 52, 2633–2648 CrossRef.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC and references therein.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC and references therein.
- A. K. Sundaresan, S. Jockusch, Y. Li, J. R. Lancaster, S. Banik, P. Zimmerman, J. M. Blackwell, R. Bristol and N. J. Turro, Photochem. Photobiol. Sci., 2010, 9, 1082–1084 CAS.
- S. Kohmoto, T. Kobayashi, J. Minami, X. Ying, K. Yamaguchi, T. Karatsu, A. Kitamura, K. Kishikawa and M. Yamamoto, J. Org. Chem., 2001, 66, 66–73 CrossRef CAS.
- G. Vermeersch, N. Febvay-Garot, S. Caplain, A. Couture and A. Lablache-Combier, Tetrahedron Lett., 1979, 20, 609–612 CrossRef.
- D. Cao, S. Xu, C. Gao and H. Meier, Tetrahedron Lett., 2006, 47, 2759–2763 CrossRef CAS.
- E. A. Chandross and C. J. Dempster, J. Am. Chem. Soc., 1970, 92, 703–704 CrossRef CAS.
- R. Todesco, J. Gelan, H. Martens, J. Put, N. Boens and F. C. De Schryver, Tetrahedron Lett., 1978, 31, 2815–2818 CrossRef.
- D. Cao, S. Dobis and H. Meier, Tetrahedron Lett., 2002, 43, 6853–6855 CrossRef CAS.
- D. Cao, C. Gao, S. Dobis and H. Meier, Ganguang Kexue Yu Guang Huaxue, 2002, 20, 401–404 CAS.
- D. Cao and H. Meier, Angew. Chem., 2001, 113, 193–195 ( Angew. Chem., Int. Ed. , 2001 , 40 , 186–188 ) CrossRef.
- C. Gao, D. Cao and L. Zhu, Ganguang Kexue Yu Guang Huaxue, 2004, 22, 103–107 CAS.
- D. Cao, C. Gao, S. Dobis and H. Meier, Synthesis, 2007, 1995–2001 CAS.
- D. Cao, S. Dobis, D. Schollmeyer and H. Meier, Tetrahedron, 2003, 59, 5323–5327 CrossRef CAS.
- Y. Mori and K. Maeda, J. Chem. Soc., Perkin Trans. 2, 1996, 113–119 RSC.
- S. Kohmoto, Y. Ono, H. Masu, K. Yamaguchi, K. Kishikawa and M. Yamamoto, Org. Lett., 2001, 3, 4153–4155 CrossRef CAS.
- S. Kohmoto, H. Masu, C. Tasuno, K. Kishikawa, M. Yamamoto and K. Yamaguchi, J. Chem. Soc., Perkin Trans. 1, 2000, 4464–4468 RSC.
- E. A. Chandross and A. H. Schiebel, J. Am. Chem. Soc., 1973, 95, 611–612 CrossRef CAS.
- J. P. Desvergne, N. Bitit, A. Castellan and H. Bouas-Laurent, J. Chem. Soc., Perkin Trans. 2, 1983, 109–114 RSC.
- T. Okamoto, Y. Imamura, O. Urakawa and Q. Tran-Cong, J. Photochem. Photobiol., A, 1999, 129, 147–156 CrossRef CAS.
- Y. Mori and K. Maeda, Bull. Chem. Soc. Jpn., 1997, 70, 869–875 CrossRef CAS.
- H. D. Becker and K. A. Amin, J. Org. Chem., 1989, 54, 3182–3188 CrossRef CAS.
- A. Castellan, J.-M. Lacoste and H. Bouas-Laurent, J. Chem. Soc., Perkin Trans. 2, 1979, 411–419 RSC.
- H. G. Heller and M. J. Ottaway, J. Chem. Soc., Chem. Commun., 1995, 479–480 RSC.
- M. Horiguchi and Y. Ito, Tetrahedron, 2007, 63, 12286–12293 CrossRef CAS.
- M. Horiguchi and Y. Ito, J. Org. Chem., 2006, 71, 3608–3611 CrossRef CAS.
- W. Adam, K. Schneider, M. Stapper and S. Steenken, J. Am. Chem. Soc., 1997, 119, 3280–3287 CrossRef CAS.
- C. H. Tung, L.-Z. Wu, Z.-Y. Yuan and N. Su, J. Am. Chem. Soc., 1998, 120, 11594–11602 CrossRef CAS.
- H. D. Becker, E. Hammarberg, B. W. Skelton and A. H. White, Tetrahedron Lett., 1989, 30, 2137–2140 CrossRef CAS.
- W. R. Bergmark, G. Jones II, R. E. Reinhardt and A. M. Halpern, J. Am. Chem. Soc., 1978, 100, 6665–6673 CrossRef CAS.
- A. Castellan, J.-P. Desvergne and H. Bouas-Laurent, Chem. Phys. Lett., 1980, 76, 390–397 CrossRef CAS.
- G. Deng, T. Sakaki, Y. Kawahara and S. Shinkai, Tetrahedron Lett., 1992, 33, 2163–2166 CrossRef CAS.
- G. Deng, T. Sasaki and S. Shinkai, J. Polym. Sci., Part A, 1983, 31, 1915–1919 CrossRef.
- G. Deng, T. Sasaki, K. Nakashima and S. Shinkai, Chem. Lett., 1992, 1287–1290 CrossRef CAS.
- N. Boens, M. DeBrackeleire, J. Huybrechts and F. C. De Schryver, Z. Phys. Chem. (München, Ger.), 1976, 101, 417–428 CrossRef CAS.
- F. C. De Schryver, M. DeBrackeleire, S. Toppet and M. Van Schoor, Tetrahedron Lett., 1973, 14, 1253–1256 CrossRef.
- J. Ferguson, A. Castellan, J.-P. Desvergne and H. Bouas-Laurent, Chem. Phys. Lett., 1981, 78, 446–450 CrossRef CAS.
- K. Takahashi, Y. Takenoshi, S. Yagai, A. Kitamura and T. Karatsu, J. Photopolym. Sci. Technol., 2010, 23, 789–794 CrossRef CAS.
- J. Fritzsche, Bull. Acad. Imp. Sci. St.-Petersbourg, 1866, 9, 406–419 Search PubMed; Z. Chem., 1866, 9, 139–144 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |