Tailored crystalline microporous materials by post-synthesis modification
Valentin
Valtchev
*a,
Gerardo
Majano
b,
Svetlana
Mintova
a and
Javier
Pérez-Ramírez
*b
aLaboratoire Catalyse et Spectrochimie, ENSICAEN – Université de Caen – CNRS, 6 Bd du Maréchal Juin, 14050 Caen, France. E-mail: valentin.valtchev@ensicaen.fr; Tel: +33 231452733
bInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Wolfgang-Pauli-Strasse 10, CH 8093, Zurich, Switzerland. E-mail: jpr@chem.ethz.ch; Tel: +41 446337120
First published on 20th September 2012
Abstract
Crystalline microporous solids are an important class of inorganic materials with uses in different areas impacting our everyday lives, namely as catalysts, adsorbents, and ion exchangers. Advancements in synthesis have been invaluable in expanding the classical aluminosilicate zeolites to new unique framework types and compositions, motivating innovative developments. However, the inexhaustible post-synthetic options to tailor zeolite properties have been and will continue to be indispensable to realize emerging and to improve conventional applications. Starting from the routine drying and template removal processes that every zeolite must experience prior to use, a wide spectrum of treatments exists to alter individual or collective characteristics of these materials for optimal performance. This review documents the toolbox of post-synthetic strategies available to tune the properties of zeolitic materials for specific functions. The categorisation is based on the scale at which the alteration is aimed at, including the atomic structure (e.g. the introduction, dislodgment, or replacement of framework atoms), the micropore level (e.g. template removal and functionalisation by inorganic and organic species), and the crystal and particle levels (e.g. the introduction of auxiliary porosity). Through examples in the recent literature, it is shown that the combination of post-synthetic methods enables rational zeolite design, extending the characteristics of these materials way beyond those imposed by the synthesis conditions.
 Valentin Valtchev | Valentin Valtchev (Sofia, Bulgaria, 1959) studied geochemistry at the University of Sofia and completed his PhD thesis at the Bulgarian Academy of Sciences. He has worked as a post-doctoral and research fellow in the groups of Prof. A. Dyer (1992–1993) and Prof. J. Sterte (1995–1996). In 1996 he was promoted to senior researcher at the Bulgarian Academy of Sciences. Since 1997 he has been working in France. At present Dr Valtchev is Research Director at CNRS-France and leader of materials science group at the Laboratory of Catalysis and Spectroscopy in Caen. His research involves synthesis and modification of zeolites and other porous solids that can be used for molecular recognition, separation and catalysis. |
 Gerardo Majano | Gerardo Majano (San Salvador, El Salvador, 1980) studied chemistry at the Ludwig-Maximilians University (2005), Munich, Germany, and obtained his PhD at the Université de Haute-Alsace, Mulhouse, France (2008). After post-doctoral fellowships with Dr S. Mintova (2009) and Prof. D. de Vos (2010–2011) dealing with zeolite synthesis, modification and diverse applications, he is currently a post-doctoral fellow in the group of Prof. J. Pérez-Ramírez at the Institute for Chemical and Bioengineering of the ETH Zurich. His current interests are centred on the synthesis of composites by shaping and spray techniques comprising diverse microporous materials. |
 Svetlana Mintova | Svetlana Mintova (Radomir, Bulgaria, 1962) studied at the Technical University of Sofia, Bulgaria (1985) and received her PhD degree from the same university, Department of Physical Chemistry (1993). Her professional experience includes six years as C2 in University of Munich, Germany, two years as a Visiting Scholar in Purdue University, USA, and 18 months as a postdoc in Luleå University of Technology, Sweden. Since 2006 she has been working at CNRS, Laboratory of Catalysis and Spectroscopy, University of Caen, France, as a Research Director. Her research is devoted to nanosized porous materials (zeolites), films and assemblies directly related to new applications and processes. |
 Javier Pérez-Ramírez | Javier Pérez-Ramírez (Benidorm, Spain, 1974) studied chemical engineering at the University of Alicante, Spain (1997), and earned his PhD degree at TUDelft, the Netherlands (2002). He worked for Norsk Hydro and Yara International in Porsgrunn, Norway (2002–2005), and returned to academia as an ICREA research professor and a group leader at ICIQ in Tarragona, Spain (2005–2009). In 2010, he was appointed full professor and chair of Catalysis Engineering at the Institute for Chemical and Bioengineering of the ETH Zurich. His research focuses on the science and engineering of heterogeneous catalysis to design sustainable processes. |
1 Introduction
Porous solids have an important role in modern society since their applications range from chemical process industries to household and advanced uses in fields such as optics, electronics and medicine. In general, a solid skeleton comprising pores and/or voids is considered a porous material. In practice all solid materials can provide a porous medium, thus the chemical nature of porous solids is extremely rich covering all important groups of materials – inorganic and organic crystals, carbons, polymers, glasses, ceramics and metals. The International Union of Pure and Applied Chemistry (IUPAC) classifies porous materials according to their pore sizes as: (i) microporous, with pores less than 2 nm; (ii) mesoporous, with pores from 2 to 50 nm; and (iii) macroporous, with pores between 50 and 1000 nm.1 The pore size controls the accessibility to the pore volume, while the capacity is determined by the ratio between the skeleton and the empty space. A consequence of porous organisation is the high specific surface area of porous materials which can vary from several hundred to several thousand square meters per gram of solid. Another important characteristic that determines the properties of porous materials is their structural organisation. Based on this last criterion, porous solids are divided into two major groups, that is, crystalline and amorphous. It is important to note that the properties of porous materials depend on their chemical nature. Thus, the combination of pore characteristics, structural organisation and chemical composition determines the overall properties of a porous material and its possible areas of application.The present review deals with the properties of microporous crystalline zeolite-type materials. According to the classical definition, a zeolite is a crystalline, porous aluminosilicate mineral. The advances in zeolite synthesis have stretched the composition of zeolitic materials far beyond those of their natural counterparts and to the discovery of new families of microporous solids which do not exist in nature. Today this group includes several families of porous ordered materials with pore sizes below 2 nm. A periodic tectosilicate-type framework with pores of well-defined size distinguishes zeolite-like materials from microporous carbons, polymers and glasses. The regular atomic positioning in these materials permits tunable ordering of the active sites. Until recently, crystalline microporous materials were exclusively inorganic. However, during the last decade a new family of materials with structures built of organic linkers connecting metal cations, the so called metal–organic frameworks (MOFs), was established.2–4 In the present review the largest groups, i.e., aluminosilicates, substituted tetrahedral oxides and metal–organic frameworks, will be considered. Due to the complexity of the topic, octahedral5,6 and mixed octahedral–tetrahedral7 molecular sieves will not be included.
Zeolites are the most widely known and largely exploited member of the group of crystalline molecular sieves. A zeolite is a crystalline microporous oxide, whose framework is built of adjacent silicon and aluminium tetrahedron forming channels of microporous dimensions, where alkali or alkali-earth cations and water molecules are situated. The first zeolite mineral was discovered in 17568 and the first synthetic analogue of natural zeolite obtained in 1862.9 Although some developments took place in the mid-20th century, the flourishing period in zeolite science and practice started in the early sixties. Applications of these materials began after discovering that they can be obtained from very reactive initial systems under relatively mild hydrothermal conditions. Extensive work in industrial and academic laboratories resulted in the preparation of synthetic counterparts of zeolitic minerals and new framework types that did not exist naturally. Further, organic cations that allow raising the Si/Al ratio in the zeolite framework and synthesis of new framework topology were employed in zeolite crystallisation. This approach allowed the synthesis of the first high-silica zeolite, named beta.10 Sorption, catalytic and ion exchange properties of different types of zeolites were systematically studied. Most of these developments were prompted by R. M. Barrer and D. W. Breck, the founders of modern zeolite science.11–13 This period was also marked by the application of zeolites in fluid catalytic cracking. The replacement of amorphous aluminosilicate catalysts by zeolite Y, led to a revolution in catalytic cracking in terms of conversion and selectivity.14–16 In the seventies a very large number of organic molecules were tested as structure directing agents in zeolite crystallisation, which resulted in the synthesis of many new structure types and the preparation of high-silica and all-silica zeolites.17–19 The most important discovery in the eighties was the development of a new family of silica-free zeolite-like materials. Aluminophosphate molecular sieves, first synthesised by Union Carbide researchers, were further extended to silicon- and metal-containing counterparts.19–21
Ordered mesoporous materials were synthesised in the nineties, but this family is out of the scope of the present review since the framework walls are amorphous.22,23 In the dawn of the new millennium, the most important development was the synthesis of microporous materials with extra-large (>1 nm) pores. Classical zeolites usually comprise pores between 0.3 and 0.8 nm, thus a gap between zeolites and mesoporous materials (>2 nm) existed. The use of Ge as a co-structure directing agent that leads to the formation of smaller structural units and thus to more open framework types provided zeolites with pores in the range of 1–2 nm.24–26 This unique ability of germanium is due to the smaller Ge–O–Ge angle with respect to the one of Si–O–Si in the framework of zeolite-type materials. MOFs and related materials such as zeolite imidazolate frameworks (ZIFs) also possess very open frameworks with pore sizes substantially exceeding those of the classical zeolites.27,28
The number of microporous zeolite-type structures increases every year due to synthetic advancements. The zeolite family has already 201 framework types and a great part of them do not have a natural counterpart.29 The advances in synthesis have also allowed zeolite framework compositions to be stretched far beyond the limits observed in nature. At present the portfolio of microporous materials with well-defined periodic structures and various chemical compositions is large. Nevertheless, the industrial application of newly discovered microporous materials is very rare. Most industrial applications are based on materials known from the dawn of zeolite commercialisation. Up until now only about 5% of available zeolite structures has reached industrial use and amongst them six structures, namely FAU-, MOR-, MFI-, FER-, LTA- and BEA-type, cover more than 90% of all the applications. The limited number of zeolite structures employed in industrial processes is due to the stringent requirements that a particular zeolite should have in order to meet industrial scenarios. The intrinsic properties of a zeolite are imperative for a particular application, but the weight of economic, environmental and production issues is equally important in taking the final decision.30 Among the critical factors that determine the choice of microporous material for a particular application is the ability of a zeolite to undergo post-synthesis modifications.31,32 If we consider the most important area of zeolite application, viz. heterogeneous catalysis, there is not a single material that is employed without a preliminary treatment. The goal of post-synthesis modification is to maximize the performance of a zeolite catalyst, decreasing the impact of unfavourable characteristics and increasing those that offer a superior performance.33 Thus, the post-synthesis modifications are targeted at controlling the properties and distribution of the active sites, their accessibility, poisoning and regeneration. The latter aspects were revised by Kühl in the late nineties.31 Post-synthesis modifications often address the intrinsic features of the parent zeolite, for instance the thermal and hydrothermal stability, hydrophilic–hydrophobic properties, crystal size and level of agglomeration. Zeolite catalyst manufacturing also includes the shaping that is indispensable for practical applications. In other words, the preparation of bodies with size, pore structure and morphology that fulfil process requirements, including diffusion rates of reactants and products, as well as mechanical and attrition resistance. During these preparations binders of different nature and other additives are used that might further modify the properties of the zeolite. The last topic, however, is not included in the present review. Here we focus our analysis on the intrinsic properties of zeolites that can be modified in a controlled manner by post-synthesis methods. Silica-based microporous materials are revised first since their post-synthesis modification has been most widely studied. Zeolite-like (AlPOs, SAPOs and MeAPOs) and metal–organic framework (MOFs) materials are dealt with in a separate subsection. All key characteristics that determine the performance of crystalline microporous solids are addressed, including: (i) modifications at the micropore level; (ii) framework substitutions; and (iii) control of crystal features. To the best of our knowledge, a review collecting all these aspects has not been published to date.
2. Post-synthesis modification of zeolites: a door to versatility
The widening window of post-synthetic options enables the preparation of zeolites with application-specific properties. Given this versatility, one can easily understand why only a handful of framework types cover the whole spectra of current industrial applications.34 The toolbox of post-synthetic strategies has been categorised according to the level at which the alteration is aimed at, featuring (i) the micropore (e.g. template removal and functionalisation by inorganic and organic species), (ii) the framework (e.g. the introduction, extraction, or replacement of lattice atoms), and (iii) the crystal (e.g. change in size, morphology and introduction of meso- or macropores). The most common post-synthesis modifications and the resulting effects are summarised in Table 1. Throughout the sections, the reader will experience that a single post-synthesis treatment has often more than one important effect on the material. For example, the primary objective of high-temperature calcination of template-containing zeolites is the removal of the structure directing agent, but it may lead to secondary effects such as the extraction of framework aluminium and partial amorphisation (framework alteration) and crystal agglomeration. Besides, another up-front learning is that in most cases, a sequence of treatments is applied to make the zeolite ready for the specific use. Sometimes, each treatment incorporates distinct properties and occasionally, a certain post-treatment in the row ‘repairs’ some of the secondary changes induced by a previous treatment. For example, if a zeolite experiences dealumination during template removal, a further treatment can be applied to realuminate the extracted aluminium in the framework or to eliminate extraframework species. Thus almost every modification of a zeolite material is basically a combination of post-synthesis treatments. However, these conventional treatments have been trivialised to such a high level that they are no longer regarded separately. It should be mentioned that the transition from the particle (defined as an arrangement of crystals and/or crystals with additional phases such as binders) to the shaped body level by forming, strongly relates to their scale up, but it is beyond the scope of this review.| Level | Method | Goal | Treatment |
|---|---|---|---|
| Micropore | Thermal activation | Dehydration, template removal | ˙ Calcination in different atmospheres |
| Chemical activation | Template removal | ˙ Oxidative (H2O2, O3, UV) | |
| ˙ Dielectric-barrier discharge plasma | |||
| Functionalisation | Hybrid material production, pore and surface modification, passivation of unselective sites | ˙ Immobilisation of organics | |
| ˙ Covalent/electrostatic grafting of organics | |||
| ˙ Chemical liquid deposition | |||
| Metal deposition and incorporation | Creation of active sites (reduction, sintering), control of pore openings, passivation of unselective sites | ˙ Ion exchange | |
| ˙ Chemical vapour deposition | |||
| ˙ Impregnation | |||
| ˙ Ion beam implantation | |||
| Framework | Isomorphous substitution | Acidity modification | ˙ Hydrothermal, gas phase |
| Demetallation | Composition, acidity, and stability modification | ˙ Steaming | |
| ˙ Acid/base leaching | |||
| ˙ H2O2 and microwave irradiation | |||
| Secondary synthesis | Framework conversion, composite materials formation | ˙ Hydrothermal | |
| ˙ Steam assisted | |||
| Crystal/particle | Demetallation | Introduction of secondary porosity | ˙ Steaming |
| ˙ Acid/base leaching | |||
| ˙ H2O2 and microwave irradiation | |||
| Tribochemical treatment | Crystal/particle size modification | ˙ Milling | |
| Morphological constructions | Crystal/particle organisation | ˙ Aggregation | |
| ˙ Pillaring | |||
| ˙ Delamination | |||
| ˙ Secondary growth | |||
2.1 Micropore modifications
With increasing temperature, an SDA containing zeolite experiences a series of processes: (i) water/solvent removal, (ii) SDA degradation and removal, generally following a Hoffmann elimination mechanism, (iii) silanol condensation of surface or defect silanol groups and (iv) lattice degradation.
Water, the most common solvent in zeolite synthesis, presents different interactions in the zeolite that strongly depend on the experimental conditions, the framework type, the lattice chemistry and even the crystal size. The two main types are: physisorbed (stable up to 50–200 °C); and chemisorbed, coordinated to delocalised cationic sites (stable up to 350 °C). However, other neglected interactions can be rather complex as demonstrated by Siegel et al. who identified up to five desorption steps on Na–Mg zeolite A in the range of 80–370 °C by thermogravimetry and differential scanning calorimetry.35
While zeolites are widely regarded as structurally stable at high temperatures, solvent removal influences the crystal structure as shown by XRD studies of the Na-A zeolite where a reversible change from orthorhombic to cubic at 85 °C after dehydration at 400 °C has been observed.36 The change in unit cell dimension provoked by dehydration is inevitably accompanied by a reduction in pore size. Besides water, other common solvents such as n-hexane or benzene are also known to give rise to unit cell changes in MFI-type zeolites, from monoclinic to orthorhombic and pseudo tetragonal, respectively.37,38 Lattice framework changes during the dehydration of a Ni-Y zeolite have been correlated to the migration of the metal from the supercage to hexagonal prism sites by in situ EXAFS in the range of 100–400 °C.39 Water loss is also related to the migration of the metal, which is finally accommodated into the S1 hexagonal prism sites due to distortion of the lattice, making them inaccessible for reduction in H2 stream.40 Also changes in copper coordination were observed during sintering of Cu-ZSM-5 at 100–300 °C related to lattice expansion which directly affects the performance in catalytic NO decomposition.41 These changes are especially relevant for organised architectures such as thin films where thermal expansion behaviours have been investigated for mordenite, faujasite and zeolite A.42 They influence not only sorptive selectivity but also film integrity and adhesive properties.
Lattice chemistry can also be induced by water removal due to the formation of extraframework aluminium species and additional Lewis acid sites by local steam generation at high temperature. However, the extraframework aluminium may be reinserted into the structure after NH4+ or Na+ ion exchange.43 As a method for water removal, it has been found that in the case of zeolite A microwave treatment resulted in a partial conversion to carnegieite in contrast with the full amorphisation observed after conventional high temperature treatment.44
The removal of water overlaps with the removal of most organic SDAs in the mid-temperature range.45 Template removal by calcination is the most widely used thermal activation method for zeolites. The interaction between a SDA and the zeolite framework is rather strong as it occupies particular spatial positions in the intracrystalline pore volume. Due to the high temperatures required (>450 °C), there is a decrease in crystallinity and reactive site formation (EFAl) due to the hydrolytic breakage of T–O bonds. An accurate investigation of the influence of the calcination temperature and time on zeolite beta has been performed by Collignon et al. using X-ray photoelectron spectroscopy. The latter study supports acidity measurements by probe molecules and 27Al MAS NMR investigations of the transition of tetra- to octahedral aluminium through tri-coordinated species.46
An important side effect of calcination is the condensation of silanol groups which causes: (i) loss of intercrystal porosity and formation of particles/agglomerated crystalline domains, (ii) lower total surface area, (iii) difficult redispersion and (iv) higher hydrophobicity. Additionally, secondary treatments after calcination, can increase hydrophobicity as achieved by UV treatment of calcined films, resulting in higher contact angles for water wetting.47 Furthermore, the application of microwave irradiation as a secondary treatment for beta results in improved structure retention and preservation of strong acid sites following SDA removal in comparison to the traditional method.48
When used, the gas flow during calcination has a strong impact on the characteristics of zeolite material.49 While flow of air or nitrogen enhances the removal of debris from the pore network, the application of oxidative gases such as ozone, N2O and NO2 may reduce the calcination temperature required for SDA removal to the range of 200 °C.50 This has been successfully carried out with B-BEA, B-MFI and DDR zeolites.51,52 Low temperature calcination with ozone keeps the boron in the framework but the ozone itself cannot penetrate small-pore zeolites such as DDR making the treatment ineffective for such materials.
The most important goal of SDA removal is to generate accessible acid exchange sites. An accurate study on the development of acidity in zeolite beta and ZSM-5 was conducted by Toktarev et al.53 As-synthesised zeolite beta was calcined in vacuum at different temperatures and the acid site concentration was determined by IR measurements of both NH3 and CO sorption. A plot of the evolution of acid site formation for zeolite beta with an overlaid typical weight loss curve from thermogravimetry is presented in Fig. 1.
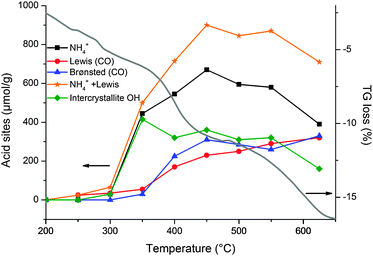 | ||
| Fig. 1 Evolution of acid site concentration in zeolite beta calcined under vacuum at different temperatures by IR measurements of NH3 and CO adsorption (plotted from data provided in ref. 53 and overlaid with a typical zeolite beta TG curve). Intercrystallite OH groups were determined as the difference between the Brønsted acid site concentrations determined by NH3 and by CO adsorption. | ||
This study provides valuable information on the different stages of the calcination process related to changes in lattice chemistry. First, chemisorbed water liberates surface silanol groups (<350 °C) while almost no acid sites are generated, and the SDA starts to degrade as reported earlier.45 Beyond 350 °C Brønsted and Lewis acid sites are generated due to SDA elimination, seen as a steep weight loss. The Brønsted acid site concentration reaches an optimum at 450 °C while the Lewis acid sites show an almost linear increase throughout the whole process. At the end of the process, condensation of surface silanol groups and the beginning of structural collapse are seen; the latter reflected in a slight increase of the Brønsted acid sites.
Thermal activation is essential to a great majority of post-synthetic treatment strategies. However, it has been trivialised to such an extent so that the effects presented are often not taken into account. Preservation of the chemical and structural stability of zeolites can be enhanced by optimising atmosphere, gas flow conditions, establishing zeolite-specific calcination profiles. In addition, other factors such as pelletisation and mode of calcination which influence greatly the outcome of the calcination process are rarely addressed.
Typically, a chemical treatment for SDA removal is carried out at mild temperatures (<100 °C) in the presence of a reactive compound that is able to extract, oxidize and/or otherwise decompose the occluded template. Differences, relevant to SDA removal, between mesoporous materials and zeolites are the nature of: (i) the SDA and (ii) the material itself. For instance, in mesoporous materials it is easier to dislodge the SDA (micelles of surfactants) with acid washing, which destroys the micelle structure while the large pore opening facilitates the removal. For zeolites, a simple protonation of the framework for facilitating removal is inefficient due to the shape, size and location of SDAs, smaller pore openings and resulting stronger SDA–framework interaction. Thus the employed methodologies generally require harsher conditions and higher concentrations of the reactive compound.
Solvent extraction with acids or salts has been carried out using diverse acids such as acetic acid, HCl, HNO3 or H2SO4, in aqueous or ethanolic media. In a common procedure 0.05 M H2SO4 is capable of removing up to 68% of the SDA from mesoporous MCM-41 in 1 h.55 This is more challenging for zeolites. The influence of the nature of the SDA and the zeolite in acid removal investigated by Jones et al.56 showed that harsher conditions and especially longer treatments are required (50% acetic acid at 80 °C for 24 h). The size relationship between the SDA and the pore opening also influenced the extraction. Complete removal of the tetraethylammonium cation from zeolite beta and the linear hexamethylenediamine from silicalite-1 was possible, while bis-piperidinium and tetraalkylammonium templates were impossible to remove completely from beta and silicalite-1, respectively. The framework chemistry and the resulting interaction with the SDA also play an important role, as seen in the increasing degree of SDA removal in the order of Al < B < Zn ≤ Si for zeolite beta.
An advantage of acid extraction is that charge balancing cations are exchanged at the same time, yielding the acid form. Still, the most important feature is that the material is left structurally intact. While mesoporous materials exhibit a small contraction of the walls, zeolites show almost no change in unit cell dimensions. Chemically, zeolites with heteroatoms such as aluminium and boron may experience a certain degree of demetallation depending on the conditions applied. At low temperatures the defects remain, leaving a hydrophilic material, while at high temperatures they may be healed resulting in a more hydrophobic zeolite, as demonstrated through 29Si Bloch decay NMR.56
Chemically harsher is the oxidative removal of SDA with radical generating species such as H2O2, H2O2 and Fe3+ (Fenton detemplation), ozone and UV radiation and combinations thereof.57,58 The difference between oxidants lies in their oxidation potentials: 1.77 eV for H2O2, 2.07 eV for O3 and 2.80 eV for OH˙ radicals. As known, UV radiation can deliver a mixture of them depending on the treatment conditions.
Peroxides, most commonly hydrogen peroxide but also peracids such as peracetic acid (1.81 eV), have been used for SDA removal from the layered zeolite MCM-56 and mesoporous MCM-41.59,60 For such materials, a 30 wt% solution of H2O2 with a liquid to solid ratio of 5 cm3 g−1 suffices after 20 h at 90 °C. The oxidizing properties of peroxides can be enhanced by the use of metals for catalysing the decomposition. Iron salts are preferred due to their low-cost and availability. Named Fenton detemplation, this method has been applied on zeolite beta with simultaneous Fe3+ ion exchange into the zeolite structure.61,62 The high oxidizing potential is responsible for the completeness of the process compared to the partial removal by treatment with only 30 wt% H2O2. Treatment with aqueous ammonia and H2O2 has also been successful in elimination of tetrapropylammonium from purified silicalite-1 colloids.63
Ozone can also be used for the generation of radicals in peroxide solutions.64 For SBA-15, the concentration of H2O2 can be lowered down to 0.5% and the detemplation can be carried out at 25 °C in 4 h using ozone. The pH in acidified media plays an important role in radical production where only a pH < 3.5 resulted in complete removal. Furthermore, ozone can also be used stand-alone, as it can be conveniently generated by UV irradiation during photochemical SDA removal from different materials such as silicalite-1, AlPO4-5 and Ge-ITQ-7.65,66 The effectiveness in retention of the structure, especially regarding heteroatoms, is highlighted by the fact that the resulting Ge-ITQ-7 remains structurally intact after extended exposure to air with moisture concentrations of up to 50%. The use of ozone also yields better results for structures such as thin films due to the lower thermal stress between particles themselves and the substrate interface.67 In addition to radical-generating environments, stable radicals themselves including species with free radicals such as 1-oxyl-2,2,6,6-tetramethylpiperidine, N2O or NO2 have been postulated but still require rather elevated temperatures (>200 °C).50,62
There are quite a number of other techniques for SDA removal. For instance, SDA decomposition can also be carried out by dielectric-barrier discharge plasma under room temperature conditions. This has been reported for zeolite beta, ZSM-5, MOF-5 and MCM-41.63,68–71 Supercritical CO2 and water have been investigated for SDA removal from mesoporous and zeolite-like materials.72,73 This is especially relevant for metal–organic frameworks, as it is sometimes the only method to access the porous framework while retaining structural stability.74 The process in the case of zeolites is limited to a maximum of 26% for Si-beta and 18% for Al-beta but reaches values above 95% for MCM-48. While not directly a zeolite modification, it is worthwhile to mention that it is possible to synthesize zeolites ZSM-5, ZSM-11, ZSM-12 and VPI-8 using a cyclic ketal as a degradable SDA which can be chemically cleaved after synthesis (Scheme 1), freeing the porous structure under mild conditions, while at the same time making possible the reuse of the template.75,76
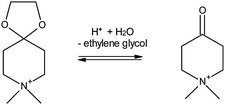 | ||
| Scheme 1 Template cleavage according to ref. 75, which allows re-use of the organic template. | ||
Chemical treatment of zeolites for SDA removal comprises effective and elegant methods which complement and represent an alternative to conventional thermal treatment. However, this group of methods is not currently applied on an industrial scale, probably due to cost issues. Nevertheless, the waste produced by both methodologies may have a similar impact on zeolite processing while chemical treatments yield generally more intact structures and can open new applications for unused zeolite frameworks.
As mentioned ion exchange, chemical vapour treatment and impregnation are preferred approaches for the deposition of metals in zeolites. Other methods that are widely used for oxides such as precipitation techniques are not used for zeolites due to clogging of the pores which leaves most of the structure unused.
2.1.3.1 Ion exchange. Ion exchange can be carried out in the liquid, gas and solid phases, and has been widely researched and exploited for water purification applications and generation of Brønsted and Lewis acid sites with enhanced metal dispersion.82,83 Ion interactions with the zeolite framework continue to yield catalytically interesting results, for instance, recently Ag-, Ni- and Co- ion exchanged titanosilicate ETS-10 has shown activity in the photocatalytic degradation of acetaldehyde, shifting the effective range into the visible light region.84 Additionally, improvement of the traditional aqueous ion exchange has generated new approaches for control of the resulting metal species, reduction of structural damage and increased efficiency for large or sensitive cations.
Non-aqueous methods have been explored for Li+ introduction (>90%) in sodalite using ethylene oxide-based oligomers under anhydrous conditions.85 Related to this, the use of polyethylene glycol has recently been expanded for Co2+, Mn2+ and Fe2+ on zeolite X with a high degree of exchange compared to solvents such as acetonitrile, dimethyl sulfoxide and formamide.86 This reduces structural damage and enhances activity for NO decomposition, although application of rigorously dried zeolites hinders exchange due to the preferential inclusion of the organic phase.
For more demanding cations, multivalent or too large, solid state ion exchange is applied. Complete exchange of In+ into zeolite beta was achieved by reducing the oxide together during the thermal decomposition of the SDA in an inert atmosphere.87 However, this process may eliminate Brønsted acid sites, resulting in lower activity toward acid-catalysed reactions. Additionally, lanthanide-exchanged zeolites can be used as luminescent materials; a brief review on this topic has been presented by Rocha and Carlos.88
As temperature variations do not influence greatly ion mobility, the majority of procedures are performed in the temperature range 25–90 °C. Nonetheless, microwave irradiation, which uses shorter times (<20 min), has been used to introduce copper in ZSM-5.89 Interestingly, the generated Cu-species show strong chemisorption of nitrogen even at room temperature. Microwave-induced solid-state ion exchange of MOR with Ni, Cu, and Co chlorides has also been reported.90 This led to the introduction of metals in defined positions as determined by Rietveld refinement of XRD data.
In spite of the fact that ion exchange requires several treatment steps, it can be applied to shaped bodies without compromising their stability. For instance, ion exchanged tubular Ru and Rh-MOR/Al2O3 membranes showed high conversion for partial oxidation of methane retaining structural stability.91 Likewise Pt–Co NaY-membranes for non-oxidative dehydrogenation of methane were obtained by successive treatment steps.92 Ion exchange may also be compatible for forms such as extrudates. This was carried out for obtaining the NH4+-form of ZSM-5/γ-Al2O3 extrudates after Al-reinsertion for regeneration during hexane cracking.93
2.1.3.2 Chemical vapour deposition. Chemical vapour deposition is a mild method for direct introduction of metal species in zeolites avoiding loss of intrinsic crystallinity and generation of extraframework sites, which is widely considered to be a special case of ion exchange. Nevertheless, the complexity of the method results in its limited use. Inclusion of diverse metals such as Al, V and Ti can be carried out by the generation of defects in the structure by complete or partial dealumination.94 The voids formed this way can then be occupied by the metal atom to be included by other methods such as impregnation, ion-exchange or CVD.95
Recently, inorganic zeolite-based electrides have been synthesised by introducing Cs, Rb, Na and K into all-silica ITQ-4, ITQ-7 and zeolite beta by chemical vapour deposition of the pure metals.96,97 The zeolites absorbed up to 40% alkali metal from the vapour phase and resulted in thermally stable electrides with strong reducing potential for water and aromatics.
2.1.3.3 Impregnation. In cases where ion-exchange, due to the size or charge of the metal ion, is not efficient, impregnation is the method of choice for metal deposition. The impregnation of metal species can be controlled by the pH of the employed solutions in the case of oxides and layered materials. For zeolites, the high number of acid sites combined with high specific surface area makes metal impregnation very efficient as the acid sites supersede the pH of the solution. Furthermore, metal impregnation can be used to introduce species in high-silica or all-silica molecular sieves. In this case, the predominant model of fixation is electrostatic adsorption on the silanol groups.98 Also, gold nanoparticles were stabilised in an ionic-liquid milieu (1-butyl-3-methylimidazolium tetrafluoroborate) for impregnation in the TS-1 zeolite which yields a catalyst with high activity for propylene epoxidation.99
The microporous nature of zeolites helps to stabilise complexes inside the network. However, due to the particular size of the pores mainly two approaches for deposition are currently in use: ion exchange and ship-in-a-bottle methodologies. Impregnation as used on oxide supports is not as effective in zeolites, while covalent bonding of complexes is complicated. Certainly, symmetric salen complexes have been tethered to porous polymers, but it results in distortion of the resulting complex, lowering its activity.100,101 Additionally, the use of asymmetric ligands for giving the system the desired freedom to form an optimal complex is limited as their synthesis is expensive. Also chiral molybdenum species have been tethered to mesoporous USY102 and TS-1103 has been grafted with phosphates which can coordinate to tungsten peroxides for limonene epoxidation. It is important to mention that due to the spatial needs for bulky complexes, there is a gap in investigations of zeolite topologies for immobilisation as the great majority has been carried on FAU- (Y and USY) and EMT-type zeolites.
Generally, the occluded complexes exhibit higher thermal stability and lower deactivation through limitation of intermolecular reactions. In addition, the confinement often helps to generate isolated metal centres as seen for Co(II) salen in USY where it remains stable at 25 °C, whereas the pure complex decomposes over −10 °C. In this case, the conversion in acetophenone transhydrogenation rises with temperature while retaining selectivity towards transhydrogenation.104
Confinement effects highly depend on numerous factors including the nature of the ligand, metal, alkali metal ions introduced and the topology of the zeolite. Though it is difficult to generalise, some trends can be observed in the confinement of salen complexes in zeolite Y.105–107 Being introduced by a ship-in-a-bottle encapsulation, all of the cases showed no leaching of the complexes and good performance after regeneration and reuse. The structure of the ligand strongly influences the accessibility and coordination sphere of the metal and thus the reactivity. However, due to the limited micropore space and shape, the trends are not as evident as for mesoporous materials. Other Schiff bases in zeolite Y have been applied to phenol hydroxylation with H2O2 providing conversion values above 95% and catechol selectivities up to 90%.108–110 A short linker chain between the chelating moieties favours a more comfortable fit inside the cages, as seen by IR spectroscopy and modelling. In spite of this, the occlusion strongly modifies the properties in a so far unpredictable manner. Mn(III) salen complexes have also been reported for more attractive uses in the epoxidation of olefins in EMT zeolites, ITQ-2 and zeolite Y.105,106,111 Here there is also a strong dependence on the spatial fit with the substrate used, which results in a limited conversion of 50%. Thus, epoxidation of (−)-α-pinene demonstrated on the mesoporous Y zeolite results in high stereoselectivity and full conversion.112
Complexes have a dynamic behaviour inside the zeolite network as proven by in situ EXAFS. Serna and Gates investigated Rh(C2H4) complexes in zeolite Y during the dimerisation and hydrogenation of ethylene.113 The complex has a dual nature depending on the changes of the C2H4/H2 feed, where it is reduced to clusters in H2-rich atmosphere and reverses to a complex in ethylene-rich feed even after many catalytic cycles. In addition, the coordination of Rh to transient species but more importantly to the oxygen groups of the zeolite structure was observed. This last observation also demonstrates the nature of the zeolite as a macro-ligand. Another example of dynamic changes inside zeolitic cavities has been seen in Rh(bpy)32+ complexes in zeolite Y, which have shown reorganisation under repeated cyclovoltammetric tests after 96 h of use.114 This has also been reported using Mössbauer spectroscopy on Fe(bpy)32+ occluded species by Umemura et al.115
In spite of all these examples, not only bulky complexes are of interest for immobilisation. Smaller complexes which do not suffer steric constraints can also profit from the confinement effect. For example, there is evidence that ferryl species (Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in Fe-ZSM-5 and Fe-beta are the active centres for Fenton-like reactions.116 The confinement, investigated through EPR, should ideally help to isolate the generated radical reactive species in the zeolite cavities conferring them remarkable longevity by limiting their recombination and deactivation, which readily happens in solution. This effect may help to control radical based reactions such as oligomerisation or polymerisation.
O) in Fe-ZSM-5 and Fe-beta are the active centres for Fenton-like reactions.116 The confinement, investigated through EPR, should ideally help to isolate the generated radical reactive species in the zeolite cavities conferring them remarkable longevity by limiting their recombination and deactivation, which readily happens in solution. This effect may help to control radical based reactions such as oligomerisation or polymerisation.
It is important to note the scarcity of reports on the effect of acid and cationic sites on the occluded complexes. While there are vague mentions of the “effect of the alkali cations” in most of the cases, there is no basic understanding of the observed effects. In addition, no study of the coordination and stabilisation effects of the framework Brønsted and Lewis sites on the complexes depending on quantity, framework type (besides FAU and EMT type zeolites) and treatment of the zeolite has been carried out.
Finally, the use of metal complexes does not only comprise their direct applications, but also their use as precursors for avoiding metal cluster agglomeration upon calcination, as demonstrated for mesoporous materials MCM-41, MCF, MSU and SBA-15 using Cr, Fe and Cu complexes.117 Superior reactivity of Pd for Heck coupling due to better dispersion at concentrations around 0.1 mol has also been proven for deposition on mordenite and zeolite Y among other typical substrates.118
Recently, the radical reduction of Ag+ in zeolite Y was achieved by sonochemical treatment in aqueous and alcoholic media under argon flow.127 The extent of reduction was controlled by regulation of the ultrasonic power while the particle size (1.2 m) was controlled by the concentration of the solution. An advantage of the method is that the reducing species, which are not gases or organic agents, are produced in situ.
2.1.6.1 Comparison of metal introduction methods of final zeolite modification – palladium in zeolites for C–C coupling reactions. Palladium has a special relevance as a metal for fine chemicals production in the pharmaceutical industry. And its activity depends on the method of introduction, for example metal introduced by complexes remains intact in the zeolitic pore network and has enhanced activity as seen in the Pd(0) and Pd(NH3)42+ in Y-zeolite for Heck C–C coupling reactions.128 Pd(NH3)42+ exchanged into the Na-Y zeolite is even active for copper-free Sonogashira C–C couplings.129
The confinement and nature of the active Pd species in zeolite micropore volume have also been studied and vary with its environment.130 For instance, the reactivity of Pd(NH3)42+ in three different zeolites for coupling of 4-bromoacetone with n-butyl acrylate decreased in the order Y > mordenite > ZSM-5. It is important to note that cluster formation has only been detected in zeolite Y following reaction. Additional advantages of Pd inclusion for the Heck coupling are the lower hydrodehalogenation of the starting product compared to traditional methods and the fact that aryl chlorides may be activated at high temperatures.118 This study revealed the significance of the appropriate reaction conditions, as the homogeneous nature and the switching of valences of Pd can lead to the generation of species which can be leached out. While amine coordinated Pd is retained better due to the ion exchange selectivity in zeolite Y, too higher loadings can lead to electrostatically loose complexes that experience leaching.
2.1.6.2 Fe-MFI for catalytic N2O abatement. The relevance of the metal introduction method to the final reactivity of a zeolite catalyst has been highlighted in a study of Fe-MFI catalysts for selective catalytic reduction of N2O. In this case, the catalyst was prepared by ion exchange, solid state ion exchange and isomorphous substitution (followed by steam activation).131 As expected, the Fe content of the zeolite increased following the trend in situ isomorphous substitution < ion exchange < solid state ion exchange (Table 2). In spite of extremely uniform Fe distribution in all cases, the resulting clustering after calcination generally follows the same trend and so does the N2O conversion which is more efficient on non-clustered, homogeneous Fe3+ sites in steamed samples and is mostly inactive for large Fe2O3 particles. The reactivity of the sample exchanged in the solid state is most likely due to the high iron content.
| Method | Si/Al | Si/Fe | Fe (wt%) | Fe distribution (%)a | Relative N2O conversion at 325 °C | ||
|---|---|---|---|---|---|---|---|
| Fe3+ | Fex3+Oy | Fe2O3 | |||||
| a From UV-Vis DRS analysis. | |||||||
| Isomorphous substitution + steaming | ∞ | 150 | 0.68 | 70 | 30 | — | 1.0 |
| Isomorphous substitution + steaming | 31 | 150 | 0.67 | 30 | 62 | 8 | 0.35 |
| Solid state ion exchange | 14 | 14 | 5.0 | 26 | 67 | 6 | 1.6 |
| Ion exchange | 37 | 65 | 1.4 | 15 | 52 | 33 | 0.26 |
While the proclivity of Fe to cluster and form diverse species can be controlled to a certain degree, it remains a challenging task and often results in the formation of large oxide nanoparticles outside the channels which are either inactive or unselective. More manageable approaches involve starting from isomorphously-substituted Fe-ZSM-5 followed by calcination132 and/or steaming for iron removal;133 methods that provide higher control over Fe speciation and have been used for clarification of single site properties.134,135 A complete review on the nature of Fe sites has been carried out by Zecchina et al. where not only the characterisation of the sites introduced by different methods is presented but also effects on Fe migration and other relevant reactions in addition to N2O reduction are discussed.136
2.1.6.3 Ag-MOR for radioactive iodine capture. Recently, the state of ion exchanged silver atoms in mordenite in the formation of AgI by selective gas sorption of radioactive iodine has been studied. In addition to radiological iodine capture the occlusion in the zeolite pores resulted in the lowest formation temperature reported for nanosized α-AgI, a super ion conducting phase, which cannot be achieved in bulk.137
2.1.6.4 Phosphorous modification. In order to avoid too much fragmentation, the phosphorous modification of zeolites will be considered in this section. It has been long known that phosphorus can be incorporated in zeolites by impregnation with phosphoric acid followed by calcination.138 The versatility of the method is based on the ability of phosphorous to act as an acidity modifier while increasing the steric constraints in the pores at high loadings.139 Weaker P–OH Brønsted acid sites are formed at the expense of strong sites by coordination of Al–OH–Si moieties or by extraction of Al (Scheme 2) or heteroatoms such as Fe and B.140–142 At high concentrations mild dealumination and formation of aluminium phosphates have been observed.143,144 Only minimal framework insertion occurs as Si–O–P bonds are relatively weak, but functionalisation of silanol groups has been verified by IR spectroscopy and P-species can be stabilised and incorporated during calcination, especially at high-temperature.145–148 In spite of the lower acidity of P-zeolites, it is interesting to note that in theoretical models the acidity of solid phosphoric acid (highly concentrated phosphoric acid on silicon phosphate) can be as high as or even higher than H-ZSM-5.149
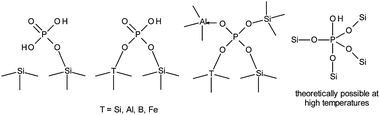 | ||
| Scheme 2 Possible P-coordination on zeolites not including non-covalently bonded forms. | ||
Generally the treatment is done using the H-form of the zeolite, as other cations/metals may disrupt acid site creation and the homogeneity of the pore size modification. Modification with phosphoric acid has attracted attention for the superior performance of P-modified ZSM-5 in FCC and MTO processes, as the modified acidity results in a lower amount of produced aromatics.150–152 Furthermore, the modification of zeolites Y, beta, mordenite and clinoptilolite has been reported.148,153 For zeolite extrudates the addition of phosphorous has to be carried out before the shaping stage as the binder hinders the incorporation.154
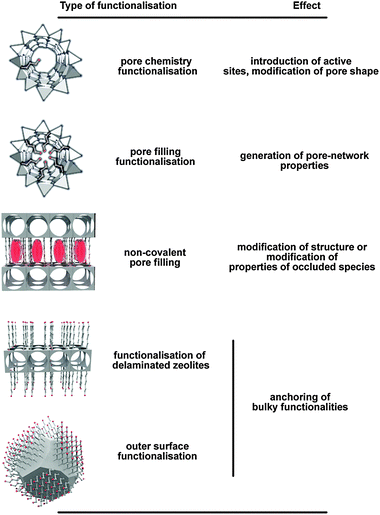 | ||
| Fig. 2 Schematic representation of different types of zeolite hybrids and desired effects. | ||
For large-pore zeolites, pore modification is known to enhance shape selectivity. In situ silylated zeolite beta with ethylcyclohexenyl, phenethyl and mercaptopropyl groups has been tested for shape selective catalysis after sulfonation of the anchored groups.155 Direct silylation may sometimes result in unwanted dealumination and damage of the structure as seen in the silylation of zeolite beta with large silanes.156 Direct silylation of zeolite beta with a mercaptopropylsilane can be carried out in supercritical CO2 due its lower viscosity and enhanced diffusivity. This material, after oxidation to the thiol groups, demonstrated strong acidity and high shape-selectivity for the ketalisation of cyclohexanone.157 For structures such as membranes, in situ deposition of Si-species during H2/CO2 separation enhanced the H2 selectivity without decrease in transport resistance.158
A step towards the enhancement of catalytic properties of zeolites has been achieved by the chiral induction method, where chirality is introduced by occlusion of a chiral inductor in the framework thus offering the system just a single degree of freedom to react. Two recent extensive reports from Sivaguru et al. have presented remarkable results using the cages of zeolite Y as chiral microreactors upon inclusion of ephedrine and methylene blue.159,160 Ephedrine-Na-Y yielded diastereomeric excess of 90% and 78% ee for photocyclisations which otherwise create racemic products in solution.159 Methylene blue has a similar effect for the enantioselective oxidation of carbamates, where 80% ee for the formation of methyl-desoxybenzoin has been achieved at room temperature.160 The conformational preference of the reactants was related to the interaction with occluded alkali-cations (Li, Na, K and Rb).
Functionalisation of the zeolite network is restricted by the dimensions of the organic compounds and the pores. Inspired by mercaptoamine-functionalised clays and silica gels, which display enhancement of their ion exchange properties, cysteamine and propylamine ion exchanged Na-clinoptilolite was used for Pb and Cd sequestration. However, in this case there is clear evidence of pore blockage hindering the ion exchange.161
Diffusion is certainly a limiting factor for pore functionalisation. However, it is not decisive for some applications. For example, the surface of zeolites has been routinely silylated with bulky silanes to increase hydrophobicity. A recent example is the surface silylation of HY with octadecylsilyl groups which remarkably increases its hydrothermal stability and results in enhanced performance in biomass related reactions in aqueous and biphasic media even at high temperature.162 Additionally, a careful silylation using tetraethylorthosilicate, so called chemical liquid deposition (CLD), has been used to deposit a thin silica layer over HZSM-5 crystals, thus passivating unselective sites on the surface and pore mouth, which results in enhanced sorptive properties.163,164 Recently, the in situ introduction of propylamine functionalities has led to sodalite nanocrystals with enhanced compatibility to solvents with drastically different polarities, whereas pure crystals agglomerate in dichloromethane and methyl-functionalised ones agglomerate in water.165 Also Na-Y functionalised by ionic liquid has been investigated for ion transport in fuel cells.166 In this case, the conductivity drastically increases and reaches high values when the pores of the zeolite are completely filled. This has been related to the enhancement of the ion mobility of sodium cations. This property has also been exploited in the creation of Nafion-sulfonated zeolite beta composites for direct methanol fuel cells.167,168
Basically the properties of any occluded molecule can also be exploited inside a zeolite matrix. Many other applications not related to catalysis can be carried out by filling the pore network of zeolite with oriented molecules, such as dye functionalised LTL-type crystals for imaging,169 host–guest antenna materials,170 and second harmonic generation for crystal detection and visualization of the inner crystal structure.171
2.2 Framework modifications
The framework topology of a microporous material determines the size and geometry of channels, the micropore volume and micropore area and thus its molecular sieving and sorption properties. This characteristic also determines to some extent the distribution of different cations (Si, Al,....) in tetrahedral positions and hence the catalytic activity of the zeolite. In general, the relatively rigid framework of zeolites does not allow substantial changes in channel size. Slight variations can be achieved by modification of framework composition and thus the channel size and unit cell volume. On the other hand, the changes in the framework composition (e.g. Si/Al ratio) have a much deeper impact on the physicochemical properties of the zeolites. The presence of aluminium in the zeolite framework introduces a negative charge which requires a charge balancing cation in micropore space. The lowest Si/Al ratio in zeolites is postulated by Lowenstein's rule,172 which forbids adjacent AlO4 tetrahedrons. The zeolites with composition close to Si/Al ∼ 1 are preferred as ion-exchangers. Finally, the ratio between framework cations determines hydrophilic–hydrophobic properties, as high silica zeolites are hydrophobic and low silica hydrophilic.173 This brief introduction reveals the crucial importance of zeolite framework composition for all important characteristics of zeolite materials and their potential applications.The in situ control of zeolite framework composition during synthesis is the most widely used approach to modulate to a certain extent the chemistry of the ultimate zeolite. However, a substantial change in the initial gel composition often leads to the formation of secondary phases or a mixture of solid materials which is highly undesirable. The use of SDAs usually leads to increase of the Si/Al ratio in the framework due to the larger size of organic with respect to alkali and alkali-earth cations.174 This approach, however, has also some limitations and thus it is not applicable to all zeolitic materials. Hence, methods that permit tuning of the zeolite framework composition, exceeding the limits imposed by synthesis conditions, are highly appreciated.
In the following subsections post-synthesis substitutions in zeolite frameworks are revised. Post-synthesis isomorphous substitutions can be divided into two large categories: (i) replacement of framework cations, and (ii) replacement of framework oxygen. Substitution of framework cations is the most largely used approach to modify zeolite properties. The process of isomorphous substitution can be performed consecutively, which means a two-step reaction where the original framework cation is firstly extracted from the framework and then in a second step another cation with appropriate size and charge incorporated. The reaction can also be performed in a single step, i.e., the single step extraction and incorporation of framework cations. Demetallation of the zeolite framework which results in lattice defects without destroying zeolite structure will also be briefly addressed.
Gas and liquid (hydrothermal) treatments are the two main approaches employed in post-synthesis substitution in zeolite frameworks. Gas-phase treatment includes the use of volatile chlorine compounds treatment at elevated temperatures (350–800 °C). Hydrothermal treatment is usually performed at moderate temperatures (60–170 °C) with very reactive, chlorine or fluorine salts of the replacing cation.
Barrer and Makki first reported that a substantial part of aluminium in clinoptilolite can be extracted with mineral acids without losing crystallinity.175 Another natural zeolite with STI-type structure was subjected to treatment with hot hydrochloric acid.176 The Si/Al ratio of the material was increased over 5.6 and impressive thermal stability (up to 1000 °C) was attained. It is worth noting that the process of aluminium extraction is more efficient when medium or high-silica zeolites are employed.177,178 For instance complete removal of framework aluminium in pentasil-type zeolites was achieved.179–185 An interesting study was performed by Omegna et al., who extracted aluminium from zeolite beta structure using hydrochloric acid.186 In a second step aluminium was reinserted in the framework employing aluminium isopropoxide. Careful analysis of the dealuminated and realuminated zeolite revealed that aluminium is preferentially inserted in the dealuminated crystallographic positions.
The extraction of a cation from a tetrahedral position in the zeolite framework results in the formation of a framework defect, generally denoted by the term “hydroxyl nest”. Contradictory statements can be found in the literature concerning the thermal stability of dealuminated zeolites. It is clear that upon heating dehydroxylation and local rearrangement of framework atoms leading to more stable Si–O–Si bonds take place. Thus, higher concentration of framework defects deteriorates thermal stability. For instance, after acid removal of about 80% of framework aluminium, the thermal stability of mordenite decreased.187 However, some authors claimed higher thermal stability of acid-leached materials. In general, the acid-leached material subjected to steaming has improved thermal stability. Hence, the improved thermal stability of some of dealuminated materials is most probably due to the presence of water steam formed during thermal treatment that may lead to “ultrastabilisation”. This issue is considered in Section 2.3 where steam dealuminaton and healing framework defects are revised.
Complexing agents have also been employed with success in zeolite dealumination. Kerr demonstrated that H4EDTA is a very efficient agent for removal of framework Al from Na-Y.188,189 According to this study about 50% of framework aluminium can be extracted without losing crystallinity. For the successful Al extraction, it is important to employ an acidic agent since no reaction occurs with the non-acidic counterpart (Na2H2EDTA). Obviously the hydrolysis of Si–O–Al bonds is the key factor determining the demetallation process. Collected data clearly pointed out that the dealumination with chelating agents is essentially an acid leaching process.190 Consequently other chelating agents were successfully tested in zeolite dealumination.191–195
2.2.2.1 Gas-phase substitution of framework cations. The first and probably the most spectacular example of gas-phase isomorphous substitution in the zeolite framework represents the work of Beyer and Belenykaja.197 Na-Y was treated with SiCl4 vapours at relatively high temperature (∼500 °C) resulting in full replacement of Al for Si in the FAU-type framework. The reaction includes several basic steps.198,199 First, SiCl4 reacts with Na+ cations leading to the formation of NaCl and coordinated −SiCl3. The latter further reacts with framework aluminium according to the equation [AlO4/2] + −SiCl3 → [SiO4/2] + AlCl3. A reaction by-product is NaAlCl4, which has to be washed out after the treatment. Controlling the process is a fairly demanding task since the reaction is strongly exothermic and the temperature rise may destroy the zeolite framework. Thus bed geometry, the amount of applied zeolite, the heating rate, SiCl4 flow and reaction temperature have to be optimised in order to accomplish the isomorphous substitution without zeolite amorphisation. Different aspects of the method, including the treatment conditions, the products of the reaction and their elimination, have been studied.200–203 These investigations provided a fairly complete picture of the advantages of the process.
Dealumination of zeolite Y was also successfully performed with SiHCl3 under the reaction conditions described above.197 Less successful was the use of SiF4, i.e., a rather moderate increase of the Si/Al ratio in zeolite Y and mordenite was observed.204 Aluminium in ZSM-20, which features an intimate intergrowth of FAU- and EMT-type zeolites, has also been successfully substituted by employing SiCl4 vapours.205
Aluminium substitution in zeolites with monodimensional channel systems was less efficient. Only 24% of aluminium in large pore mordenite was replaced.206 The replacement was much more modest for zeolite Ω.207 An interesting effect was observed during the treatment of zeolite L. Na-L was more successfully dealuminated with respect to the as-synthesised K-L, which was attributed to the smaller size of the sodium cation and thus improved penetration of SiCl4 in the channel system.208
Medium-pore zeolites are also less vulnerable to post-synthesis isomorphous substitution. The substitution of Al for Si by applying SiCl4 in ferrierite,209,210 ZSM-5,211,212 and MCM-22213 did not increase substantially the initial Si/Al ratio. Obviously, 10 membered ring systems impose diffusion limitations that block the reaction and thus only the peripheral part of the crystals is successfully treated.
Gas-phase treatment was employed not only for dealumination, but also for alumination of zeolites. Anderson et al. treated silicalite-1 in a flow of AlCl3 and N2.214 After zeolite dehydration at 400 °C the temperature was raised slowly to 500 °C and 600 °C and kept at this temperature for 10 h. 27Al MAS NMR investigation of the product unambiguously proved framework incorporation of aluminium. However, together with framework aluminium a substantial amount of extra-framework AlO6 species were detected. According to the authors, six coordinated aluminium neutralises the negative charge of framework aluminium. The authors did not discuss, however, the mechanism of aluminium incorporation. The two possible options are: (i) incorporation of aluminium in framework defects existing in parent materials; or (ii) replacement of silicon for aluminium during high temperature treatment. This question was addressed in the work of Yamagishi et al. who first estimated the number of defect sites by 18O exchange.215 The authors found a straight correlation between the number of Al atoms incorporated in the framework by post-synthesis treatment and the number of defect sites. This result unambiguously showed that the aluminium atoms transported by AlCl3 were inserted in hydroxyl nests in MFI-type structure.
Juttu and Lobo employed the same approach to incorporate zirconium in the structure of zeolite beta and SSZ-33.216 Boron analogues of these two large pore zeolites were treated with ZrCl4 vapours. The lower framework stability of boron was expected to facilitate the incorporation of Zr in zeolite structure. The results of the study, however, showed that Zr is partially grafted in the zeolite framework without occupying the tetrahedral position. A similar approach was employed by Niederer and Hoelderich, who also employed a boron analogue of zeolite beta in order to incorporate heteroatoms in the framework. Titanium (TiCl4), vanadium (VOCl3) and molybdenum (MoOCl3) (oxy)chlorides were used in the attempts for gas phase isomorphous substitution and the oxidation capabilities of resultant catalysts tested.217 Heteroatom-modified catalysts showed interesting catalytic properties. However, V and Mo were leached from the zeolite, which suggest that these two cations were not part of the zeolite framework. In contrast, [Ti]-beta remained stable and no leaching was detected.
2.2.2.2 Hydrothermal substitution of framework cations. The previous subsection described the post-synthesis isomorphous substitutions in the gas phase, where one-pot reaction of extraction and incorporation of framework cations takes place. The gas phase reaction requires relatively severe conditions, consequently alternative methods that could provide similar results under milder conditions are being sought.
Heteroatom substitutions under basic conditions. The hydrothermal isomorphous substitution of heteroatoms in the zeolite framework was achieved by employing strongly basic solutions. Xin-Sheng and Thomas substituted Si for Ga in all silica MEL-type materials.218 The same approach was applied to dealuminated zeolite Y by applying potassium aluminate solution.219 According to this study, the dealumination process is fully reversible, i.e., aluminium can be reinserted in the framework. This study was later revised by Bezman who suggested that the authors analysed namely the crystalline part of the product without taking into account the presence of an amorphous fraction.220 Kim et al. prepared V-containing mordenite by secondary synthesis and compared the physicochemical properties of the material with in situ synthesised V-mordenite.221 The physicochemical properties and catalytic activity of two materials are similar, suggesting vanadium incorporation in the framework. Sodium aluminate was used to improve the catalytic activity of zeolite beta in hydrogenation of benzene. After the alumination step, a higher concentration of Brønsted and Lewis sites was found.222 Boron was incorporated in the framework of ZSM-5 by Meier and Reschetilowski.223 The set of experimental results revealed a correlation between the level of isomorphous substitution and the alkalinity of the boron solution employed in secondary synthesis. This result raises the question of the mechanism of framework substitution upon secondary synthesis in alkaline media, which is not addressed in the quoted studies. Occupation of framework vacancies might be an option, but in this case the level of substitution would be very low. The other more plausible explanation is partial dissolution of the silica framework in basic media. Partial desilication of the framework leaving behind defect sites available for the substituting heterocations in the treating solution is the most likely replacing mechanism. The partial dissolution and transformation of the EMT-type zeolite into low silica EMT-FAU intergrowth support the last statement. Li and Armor treated EMT-type material with Si/Al = 3.3 with caustic solutions containing aluminium species.224 The Si/Al ratio in the resultant intergrown material was about 1, revealing substantial dissolution and recrystallisation of the parent zeolite.
Heteroatom substitutions under neutral and acidic conditions. Under neutral or acidic hydrothermal conditions the silica dissolution is minimised and thus can be better controlled than in basic media. Consequently, such conditions were largely used in the attempts of post-synthesis heteroatom incorporation in zeolite frameworks. Skeels and Breck developed a secondary synthesis method by employing ammonium hexafluoro salts.225 The method is based on the relative instability of the aluminium in the zeolite framework. The hydrothermal treatment involves compounds able to leach aluminium from the framework and at the same time serves as an external source providing the replacing cation. A soluble hexafluorosilicate was employed in order to replace aluminium in the framework of different types of zeolites.226 Later on, the method was employed for the incorporation of other heterocations in zeolite frameworks.227 For instance, (NH4)2TiF6 was used in the post-synthesis treatment of USY.228 The formation of extra strong Brønsted acid sites was associated with the incorporation of titanium in the zeolite framework.
Framework substitution according to the method above includes two competing reactions: (i) the extraction of aluminium from the framework; and (ii) the insertion of silicon or another cation in the lattice vacancy. The process is efficient when the reaction kinetics of the two processes are similar. Substantially higher rates of aluminium removal might lead to structure collapse, while too fast insertion of silicon could block the channel system. The process is also strongly dependent on the channel size and connectivity. Large pore zeolites with intersecting channels are more appropriate for secondary syntheses, while the framework substitution in medium and narrow pore materials is fairly limited. For instance, (NH4)2SiF6 treatment of ZSM-5 did not affect the total acidity of the zeolite. The obvious result was an increase in the para-selectivity of materials due to decreasing concentration of aluminium on the external surface.229 Ammonium hexafluorosilicate treatment on FER-type material increased only slightly the Si/Al ratio without improving the catalytic performance of the zeolite.230
Increase of Si/Al ratio in zeolites was also achieved by simple treatment with bifluoride salts.231 Relatively mild conditions, several hours of hydrothermal treatment at 75 °C, were employed in order to extract the Al from the zeolite framework. The authors suggested that during this reaction the silicon atoms derived from the zeolite structure heal the defects of the dealumination process. Healing framework defects by acid treatment has also been described by Jones et al.232 A series of almost defect-free zeolites (CIT-1, SSZ-33 and beta) were obtained by hydrothermal treatment of borosilicate analogues with acetic acid. Calcined molecular sieves were treated at temperatures above 100 °C for a week at pH ∼ 1.65. During hydrothermal treatment boron atoms were extracted from the framework and replaced with silicon dissolved from the parent material. Boron removal and healing of framework defects were most effective when the pH was slightly below the isoelectric point of silica (pH ∼ 2). Thus pure silica molecular sieves were produced by a secondary synthesis method without using an external silica source. Comparative studies performed with mineral acids (HCl, HNO3 and H2SO4) under the same conditions provided materials with lower microporosity. This effect was attributed to the higher solubility of silica in mineral acids and thus partial framework destruction.
Boron readily enters in the framework of tectosilicates. However, the relatively small size of this cation does not fit perfectly to the tetrahedral coordination thus leading to lower stability of the boron tetrahedron. The later fact has been used to facilitate in situ modification through the introduction of framework defects permitting the obtainment of framework compositions otherwise difficult to achieve.233 Acidic and even aqueous solutions are efficient hydrolysing agents for boron removal from zeolite frameworks. Silanol nests remaining after hydrothermal extraction of boron atoms were used by Chen and Zones to incorporate aluminium in structure that crystallizes as all-silica materials.234 The insertion of aluminium in the lattice of boron-containing zeolites was performed with aqueous solution of Al(NO3)3. The process obviously involves deboronation and incorporation of aluminium in silanol nests.235 These two steps can be performed in a sequential manner, where first boron is hydrolysed and extracted from crystals and in a second step the zeolite is subjected to alumination. Alternatively, the deboronation and alumination can be conducted in a single step. 11B MAS NMR analysis revealed that in both cases aluminated material is essentially boron-free.233 The set of complementary methods showed that both single- and two-step methods are equally efficient and lead to materials with similar characteristics. The successful isomorphous substitution of B3+ for Al3+ was confirmed by the catalytic performance of a series of SSZ-33 zeolites with different aluminium populations.236 This material with CON-type topology comprises intersecting 10- and 12-membered rings (MBR). Catalytic experiments revealed that the intracrystalline pore space of SSZ-33 acted as an ensemble of cages connected by 10- and 12-MBR. It is worth mentioning that the method showed high efficiency only in the case of large pore zeolites. Attempts to aluminate medium pore zeolites were not successful due to the bulkiness of the hydrated aluminium cation which hardly migrates through the channels.
Very recently, Tong and Koller demonstrated that the substitution of boron in the framework of zeolites can be controlled quantitatively.237 The extraction of boron strongly depends on the counterion compensating the framework charge. For instance, boron tetrahedra compensated by sodium are protected from hydrolysis while those compensated by a proton are vulnerable to extraction. Thus, regulating the ratio Na+/H+ in the parent material provides a close control of the level of deboronation and framework substitution that can be achieved.
Ge-silicate molecular sieves. Germanium provides small building units (3- and 4-membered rings) in the initial gel, which are indispensable for the synthesis of extra-large pore molecular sieves.238 Unfortunately, this element readily hydrolyses in a water vapour atmosphere and leaves the tetrahedral position in zeolite structures. Thus, once the organic structure directing agent is removed and Ge is exposed to ambient humidity most open frameworks, which are generally very rich in Ge, lose crystallinity. The fact that Ge is sensitive to the humidity in the air is particularly difficult to deal with, since immediately after template combustion the zeolite should remain in a water-free atmosphere. This roadblock makes it particularly difficult to apply post-synthesis methods that could be employed to replace germanium for a more stable framework cation. On the other hand, the stabilisation of these exciting new materials presents enormous interest from industrial and academic points of view.
The motivation to replace Ge in the framework of extra-large pore molecular sieves, first synthesized in ITQ-Valencia and later on in other laboratories, is related to three main issues: (i) stabilising the framework, (ii) incorporation of Brønsted acid sites, and (iii) recycling Ge.
A relatively simple post-synthesis method that could address the above issues was recently found. Valtchev and co-workers have developed a hydrothermal post-synthesis procedure where the preliminary removal of organic structure directing agents was not required.239,240 The procedure is based on a one-pot template extraction and germanium substitution. The following reaction depicts Ge4+ substitution for Al3+ in the zeolite framework, where R is the organic structure directing agent and M the cation that balances the negative charge of aluminium tetrahedron:

The model material employed for this study was a BEC-type zeolite with an intersecting three-dimensional 12-membered ring system. The method was first tested on micron-sized crystals and proved feasible.239 Germanium was partially substituted for aluminium and a part of the zeolite framework was stabilised. Careful analysis of experimental data revealed that due to the large crystal size only the peripheral part of zeolite crystals was stabilised. The process included partial extraction of the organic structure directing agent, which in this case was hexamethonium, thus perturbing close packing of the template molecules and opening enough space for diffusion of the species in and out from the structure. Noteworthy is that the partial extraction of the template is important for the successful isomorphous substitution, since the remaining molecules still stabilise the structure and slow down the kinetics of the process. The latter is particularly important since a very fast reaction would compromise the framework stabilisation. The process seems to be effective within the first several ten to hundred nanometres from the zeolite surface. Thus, using micron-sized crystals has the disadvantage that the core of the crystal remains unchanged. Therefore, the same approach was applied by employing nanosized crystals.240 Indeed, in this case full stabilisation of the zeolite framework was achieved. 27Al NMR showed that the major part of aluminium is in tetrahedral coordination. Gentle acid treatment allowed removal of extra framework Al species. The Si/Ge ratio in the material increased from 3 to 7 and the Si/Al ratio in the aluminated zeolite was about 20. The material was calcined at 600 °C and exposed to 67% humidity for a month without showing loss in crystallinity. The aluminated BEC-type zeolite was exchanged with K+, which revealed that after calcination 94% of aluminium atoms retained framework positions serving as ion exchange sites.
Physicochemical characterization of the intermediate products and stabilised BEC-type material provided some clues on the mechanism of the process. The effect of chemical treatment on the template occupying the zeolite channels and the mechanism of template extraction was studied by quantitative (1H)-13C cross polarization MAS NMR of the series of samples. The quantification of the related carbons showed a substantial decrease of template content (ca. 50%) in treated material. A closer look at the ratio between different species revealed a more significant decrease (ca. 65%) of –N–CH2 groups. The latter suggests breakage of the C–N bond between the N–(CH3)3 head group and the first carbon in the hydrocarbon chain. Thus, the decrease of template content in zeolite channels is a consequence of partial destruction and extraction of the hexamethonium cation.
Summarising all collected data, one can say that Ge substitution in zeolite frameworks resembles boron replacement as discussed above. The alumination of as-synthesised Ge-silicate molecular sieves includes the following parallel reactions: (i) partial decomposition and extraction of closely packed template molecules during the alumination–acid extraction step; (ii) hydrolysis of a part of framework Ge4+ cations leaving behind silanol nests with Si–O(H) groups positioned for the incorporation of other tetrahedral species; and (iii) incorporation of Al3+ in the framework position (Fig. 3). Amongst these reactions the extraction and reorganisation of template molecules are of primary importance for the access of hydrolysing species to the Ge positions in the framework. In other words, the key factor is the ability of the template to be decomposed in acidic media into species small enough to circulate through the channels and leave the micropore space. Simultaneous extraction of Ge and replacement for Al is also a reaction whose efficiency is controlled by the diffusion of the species in and out of the channel system. The results with nano-240 and micron-sized239 crystals revealed the importance of the pathway length for the successful framework substitution. Summarizing the above discussion one can say that the successful application of the present approach depends on two main factors, which are the successful degradation of the template in acidic media and the size of zeolite crystals.
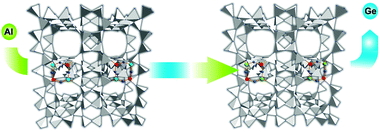 | ||
| Fig. 3 Schematic presentation of Ge substitution for Al in the BEC-type framework. | ||
Future studies will show the impact of the type of the channel system on the isomorphous substitution of Ge for Al in microporous zeolite-type frameworks. One could anticipate that the efficiency will not be very high in medium and narrow pore zeolites. The latter is not a big concern, since there are already a large number of stable aluminosilicate structures with small and medium size pores. Hence, the developed procedure is specifically targeted at large and extra-large pore Ge-silicate frameworks which through this post-synthesis method could be rendered useful for practical applications.
The possibility to prepare tectosilicate-like structures where oxygen is replaced by nitrogen was evoked by Barrer.12 The replacement of O for N would transform the formula of SiO2 into Si3N4 since each trivalent nitrogen is shared by three tetrahedra. β-Si3N4 (β-sialon) which also might include aluminium, magnesium, beryllium and other cations was obtained by solid state reactions at high temperatures. However, there is no report of the synthesis of microporous nitrogen equivalents of tectosilicates. At present, the substitution of oxygen for nitrogen in microporous type materials is achieved only by post-synthesis methods.
Substantial changes in catalytic activity are expected upon replacement of oxygen for nitrogen in a zeolite-type structure, in particular for base-catalysed reactions. The first example of nitrogen modified zeolite catalysts goes back to the early sixties when Young patented ammoniated zeolite catalysts.241 The parent zeolite was ammoniated at temperatures above 350 °C, which resulted in displacement of water from the zeolite structure. The ammonia is believed to be incorporated into the zeolite structure in the form of amide and/or imide groups bonded to framework atoms. Later on, in the early nineties porous nitrides and oxynitrides proved to be promising basic catalysts.242 The nitridation of crystalline microporous materials is now one of the approaches to introduce basic sites in zeolite catalysts,243 together with the exchange of large alkali cations244,245 and germanium substitution in the zeolite framework.246 Several groups have produced strongly basic materials by treating pre-formed zeolite crystals with amines.247,248
With the increase of the impact of biomass economy, interest in strongly basic zeolite catalysts has increased progressively. In the last few years a number of papers have addressed the subject providing valuable information on the exact position of ammonium compounds and the mechanism of catalytic reaction.249,250 Both theoretical and experimental studies have been performed to investigate the nitrogen substitution in zeolites. Agarwal et al. applied density functional theory to reveal the mechanism of nitrogen substitution in FAU- and MFI-type materials.251 The results of this study show that the mechanism of substitution strongly depends on the framework composition and the structure type. Thus, different configurations, such as a planar intermediate ring including pentavalent Si, can be observed in the course of the reaction. DFT calculations also reveal that overall nitridation barriers are relatively high, i.e., a high temperature is an indispensable condition for successful incorporation of nitrogen in the zeolite framework. However, the reverse process also requires relatively high energy, revealing the stability of nitridated catalysts. Results of theoretical calculations were complemented with experimental evidence. For instance, a detailed spectroscopic study performed by Srasra et al. revealed the effect of nitridation temperature on both the amount and chemical nature of nitrogen species incorporated in the framework.252 The use of different zeolite types and synthesis conditions complicate the straightforward evaluation of the efficiency of nitrogen substitution in zeolite frameworks. In order to find a reproducible and optimised synthesis protocol Hammond et al. performed a thorough study employing zeolite Y as a model material.253 The most important conclusions of this work were that: (i) higher temperatures produce higher levels of substitution and minimal changes in crystallinity; (ii) a high flow rate of ammonia is crucial to maintain the crystallinity and microporosity of the zeolite; and (iii) the absence of water prevents the dealumination of the framework.
2.2.3.1 F− anion interaction with the zeolite framework. A particular case of replacement in the zeolite framework represents the fluorine anion, which is used as a mineralising agent in the synthesis of high silica zeolites. The syntheses are performed under neutral or slightly basic conditions leading to almost defect-free crystals. Very often the fluoride anion is situated in the zeolite framework without building covalent interactions. F− anions reside in the small structural units, for instance in double four rings (D4R), having dual function as a stabiliser of the unit and an agent compensating the positive charge of the organic structure directing agent. An interesting case of fluorine substitution in the framework of germanium-silicate molecular sieves (ITQ-13 and ITQ-17) was recently published by Tuel et al.254,255 The authors demonstrated that F− situated in D4R can be fully exchanged with OH− by hydrothermal treatment. It is noteworthy that the reaction was performed in the presence of an organic structure directing agent. The substitution of the anions did not affect the crystallinity and framework composition of the zeolite.
2.3 Crystal features modification
Zeolite performance in catalytic and separation processes strongly depends on crystal features. Namely, the size of a crystal and the development of a crystal face giving access to a particular channel system determine the length of diffusion path and thus the kinetics of the reaction. At present, zeolite crystal morphology control remains a topic that is not properly addressed and future studies will show if zeolite crystal morphology could be subjected to rational design. Synthesis of smaller (nano) size crystals and preparation of zeolites comprising a secondary system of larger (meso- or macro-) pores remain the principle avenues to microporous materials with improved diffusion properties. Herein we highlight recent progress in the preparation of (i) mesoporous zeolites by demetallation, (ii) two-dimensional zeolites by pillaring and delamination approaches of layered zeolite precursors, (iii) nanosized zeolites by tribochemical treatments, and (iv) hierarchical morphological constructions built by nanocasting of preformed zeolite entities. In all cases, materials with higher accessibility and transport efficiency in catalytic applications are obtained.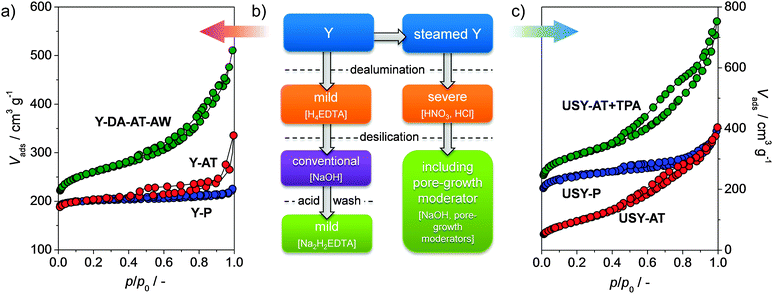 | ||
| Fig. 4 (a) and (c) N2 isotherms and derived textural parameters of Y and USY zeolites, respectively. (b) Strategies to design hierarchically structured FAU (Y and USY) zeolites by post-synthetic modifications. After desilication of Al-rich zeolites, the removal of remaining debris by a mild acid wash is crucial. On the other hand, upon alkaline treatment of Si-rich zeolites, the inclusion of pore-growth moderators such as TPA+ is highly beneficial. The N2 isotherms of the optimally treated samples, displaying a higher degree of intracrystalline mesoporosity and practically intact microporosity, contrast with the only alkaline-treated samples. In the latter case, there is either no mesoporosity development (for Y) or a complete amorphisation of the sample (for USY). Adapted from ref. 256. | ||
Over the last decade, desilication, which is the controlled leaching of the silicon framework by alkaline treatment, has been a major top-down approach for the creation of hierarchically structured zeolites.266,267 Six key focal points have been pursued to ensure amenability to industrial implementation of desilication, comprising the versatility, simplicity, cost-effectiveness, safety, efficiency, and scalability of the treatment. Here we concentrate on treatment versatility and scalability, two aspects that have been the subject of recent advancements. The versatility of desilication encompasses both framework structure and composition. Its effectiveness has now been demonstrated for over 12 distinct framework types and is achievable over the entire range of Si/Al ratios. This prompts the natural question: what gives rise to this remarkable versatility? The success of desilication can most likely be attributed to the number of parameters that may be tuned to optimize the resulting material. These include features inherent to the zeolite itself (e.g. framework type, Si/Al ratio, crystal size, morphology, presence of defects) and the treatment conditions (e.g. type and concentration of base used, the inclusion of pore growth moderators).267
However, it is rapidly becoming apparent that demetallation should not be regarded in terms of single treatments alone. Instead, the real strength lies in their strategic combination which presents an opportunity to precisely tailor not only porosity, but also composition and acidity. The ‘multitask’ challenge of demetallation is best illustrated by the approaches conceived to introduce intra-crystalline mesoporosity in zeolites Y and USY (Fig. 4).256 Standard alkaline treatment in aqueous sodium hydroxide proved to be highly sterile for this purpose. In the case of Y (Si/Al ∼ 2.6), the framework is very resistant to base leaching due to the high aluminium content. Accordingly, only minor increases in mesopore surface area were observed, even upon increasing the NaOH concentration to 5 M (sample Y-AT). On the other extreme, USY (Si/Al ∼ 30, prepared by steaming and acid leaching of the pristine Y) was readily leached in 0.2 M NaOH solution, resulting in significant mesoporosity development.
However, this occurred at the expense of widespread amorphisation and consequent reductions in micropore volume and crystallinity (sample USY-AT). Clearly, the preparation of hierarchical Y and USY zeolites requires a more elaborate exploitation of the post-synthetic modification toolbox. For zeolite Y, an optimal property modification was achieved by successive dealumination–desilication–dealumination. In the latter sequence, an initial mild dealumination step (DA) was executed to increase the bulk Si/Al ratio to ca. 4, thereby facilitating subsequent mesopore formation by desilication in alkaline media (AT). The last dealumination step comprised a mild acid wash (AW), aimed at the removal of species realuminated during alkaline treatment. The resulting hierarchical zeolite has a mesoporous surface area of over 250 m2 g−1 with a fully preserved micropore volume (sample Y-DA-AT-AW).256 A different strategy was required for the USY zeolite, relating to its lower stability in alkaline media. In this case, the hierarchical derivative was prepared by the inclusion of tetrapropylammonium cations in the alkaline solution. The latter species adsorb on the zeolite surface, protecting the framework during mesopore formation.256 The examples in Fig. 4 stress that in principle any zeolite can be made mesoporous while retaining an intact crystalline structure by appropriate selection of treatment methodology. Furthermore, it has been demonstrated that the acid and basic treatments can be easily scaled up.268
Besides the above-mentioned metal leaching treatments, a more exotic approach to ‘draw’ parallel macropores in zeolite crystals has been recently reported by Valtchev et al.269 by the use of a nuclear track imprinting technique. For this purpose, a high energy 238U ion beam was employed to form latent tracks in zeolite crystals, which were further subjected to attack with diluted HF solution and thus transformed into uniformly sized macropores traversing the entire crystal. The possibility of controlling the number of macropores per unit area of crystal surface and the improved catalytic activity of the resulting material in m-xylene conversion were demonstrated. This methodology provides a model material to understand the effect of a secondary pore system on the catalytic performance of hierarchical zeolites obtained by the top-down or bottom-up approach.
In pillared zeolites the original layers are permanently separated by the incorporation of thermally stable spacing components. Pillaring is typically achieved by the partial incorporation of a soluble precursor, such as a metal alkoxide, which is transformed into pillars by thermal treatment. The formation of amorphous silica pillars upon hydrolysis and calcination of TEOS (tetraethylorthosilicate) is one such example. An alternative kind of permanent separation involves the stabilisation of layered precursors in their expanded form by treatment with silylating agents. The ‘organic pillared’ zeolites formed represent novel layered inorganic–organic hybrid materials with integrated bifunctionality.
Although at least 10 framework types possess a known layered precursor, the MWW framework has, to date, been the major source of diversity and innovation in this area (Fig. 5). A significant achievement was the top-down conversion of a 3D zeolite framework into a lamellar product. This demonstrated that a lamellar precursor was not a prerequisite for the preparation of delaminated/pillared zeolites consequently widening the potential framework diversity.272 The 3D structure of the germanosilicate UTL, notable for its intersecting 14 MR and 12 MR wide pores, may be viewed as densely stacked 2D layers bridged, akin to pillars, by double four membered ring (D4R) units. Mild aqueous treatment of this zeolite induced elimination of the D4R units and conversion to the layered zeolite derivative (denoted IPC-1P). The lamellar architecture was further modifiable as other solids by the standard approaches detailed above.
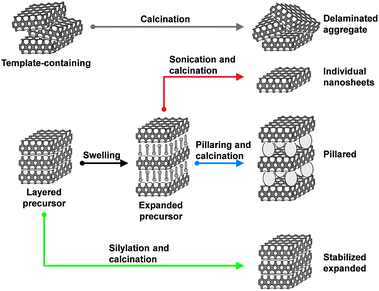
The preparation of self-supported zeolite structures was achieved namely by nanocasting of preformed zeolite nanocrystals. For instance zeolite fibres were produced by infiltration of zeolite nanocrystals into the ordered void spaces of macroscopic bacterial threads.279Bacillus subtilis and preformed silicalite-1 nanoparticles were, respectively, the organic and inorganic parts involved in this preparation. The swelling procedure gave a highly compacted network of silicalite-1 crystals after air-drying. Calcination at 600 °C removed both the structure directing agent (tetrapropylammonium) used in the zeolite synthesis and the supercellular template. The all silicalite-1 replica retained the fibre-like morphology of the bacterial template, where organised arrays of ca. 0.5 μm wide channels parallel to the fibre axis with channel walls of about 100 nm can be observed. Thus, a micro-macroporous material with fibre-like morphology was obtained. The above approach, i.e., to use preformed zeolite nanocrystals and a biological template in order to prepare complex materials was further extended by Mann and co-workers.280 Potato starch and silicalite-1 nanocrystals were used in the preparation of films, monoliths and foam structures with hierarchical porous organisation.
Synthetic macrotemplates have also been employed in the preparation of hierarchical polycrystalline zeolite structures. Tosheva et al. used the ability of anion exchange resins to attract negatively charged zeolite nanoentities and thus to form hierarchical zeolite structures with spherical shape.281
3. Post-synthesis modification of zeolite-related materials
Similarly to classical aluminosilicate zeolites, physicochemical properties of related families of crystalline microporous solids cannot be entirely controlled by synthesis parameters and post-synthesis modifications are often employed in order to tune their characteristics.3.1 Aluminophosphate molecular sieves
Physicochemical properties of aluminophosphate (AlPO) molecular sieves bare a close resemblance to their aluminosilicate counterparts. Therefore, basic post-synthesis treatments used in their modification are similar to those described in the section devoted to aluminosilicate zeolites.3.1.1.1 Thermal treatment. Open-framework aluminophosphates are synthesised hydrothermally and the inclusion of an organic structure-directing agent, usually an amine, is essential in producing the microporous framework. Some framework topologies are analogous to aluminosilicate zeolites while others are unique. The as-synthesised materials possess an open AlPO4 framework comprising template and water molecules residing within the micropore space. Calcination, usually performed in the temperature range of 500–600 °C, removes the template and all water molecules. Alternative methods, including ozone treatment, liquid extraction, acid treatment, or supercritical fluid extraction, are also applicable.
Negative thermal expansion, i.e., a contraction of the framework upon heating instead of the expected expansion, has been observed for a number of aluminophosphate structures. This phenomenon has important consequences, especially in the case of structures that are exposed to considerable temperature gradients. For instance, the control of the thermal expansion behaviour is of key importance for optical and electronic devices.282–285
Thermal and hydrothermal stability together with the force and distribution of active sites in functionalized aluminophosphates molecular sieves are the key factors that determine their application as acidic catalysts. In general, aluminophosphate frameworks are neutral and generation of active sites is achieved by substitution of Al3+ and P5+ for Si4+, Ti4+ and divalent transition metals. In order to generate a stable negative charge, the replacing cation has to withstand high temperature treatment without ejection from the framework or change of valence. For instance, transition of Fe2+ into Fe3+, yielding a charge neutral AlPO-5 framework, was reported.286 When the charge of a divalent cation is retained upon calcination the structure requires the presence of extra framework cations to preserve its charge neutrality. In the latter case, the organic cation is replaced during the calcination by a proton bonded to one of the framework oxygen atoms, resulting in the creation of Brønsted acid sites.
A very comprehensive multi-nuclear solid-state NMR investigation of H-SAPO-11, H-SAPO-18, H-SAPO-31 and H-SAPO-34 materials, thermally treated at high temperatures in synthetic air, was performed.287 It was shown that after calcination, the crystallinity of the samples was unchanged and the characteristic Bragg reflections were identical to the as-prepared samples. However, the removal of Si(4Al) species occurred. The corresponding silicon migration led to the formation of siliceous islands and aggregates. The concentration of bridging OH groups in silicoaluminophosphates decreased upon thermal treatment without changing the crystallinity of the framework and without causing dealumination. It is important to note that thermally induced dehydroxylation of bridging OH groups in silicoaluminophosphates was not accompanied by formation of defect OH groups as shown by 1H MAS NMR.288,289
The effects of calcination temperature and heating rate on the crystalline structure and morphology of nanosized AlPO-5 and Mn-AlPO-5 were studied.290 The purpose in this case was to minimize damage of textural mesoporosity during thermal treatment by controlling the regime of calcination. It was found that the slow heating rate during calcination of as-synthesised AlPO-5 was necessary in order to avoid the generation of Al2O3 and to preserve the mesoporosity.
A summary of frequently observed effects of high-temperature treatment on AlPO4-type molecular sieves is provided in Table 3. In general, aluminophosphate molecular sieves are much more sensitive to high temperature calcination with respect to their aluminosilicate counterparts. Coupled to the loss of crystallinity, undesired effects leading to lower catalytic activity of the material could occur. Therefore, precautions should be taken during thermal activation of aluminophosphate molecular sieves in order to retain the important physicochemical properties.
| Material | Result | Ref. |
|---|---|---|
| AlPO-34 | Contraction upon heating | 284 |
| AlPO-18 | ||
| SAPO-16 | Strong contraction of pores | 285 |
| AlPO-16 | ||
| Fe-AlPO-5 | Transition of Fe2+ to Fe3+ | 286 |
| H-SAPO-11 | Thermally induced dealumination | 287 |
| H-SAPO-18 | Formation of P(OAl)4 species in pores | |
| H-SAPO-31 | P migration and healing of framework vacancies | |
| H-SAPO-34 | Si migration resulting in formation of siliceous islands and aggregates | |
| Decrease of bridging OH groups | ||
| JDF-2 | Transformation in AlPO-53 | 288, 289 |
| AlPO-5 | Changes in local symmetry of P and Al atoms | 291–293 |
| SAPO-5 | Reversible framework changes upon dehydration | |
| EMM-8 | Removal of F− | |
| SAPO-40 | Irreversible reorganisation of Si atoms | 294–296 |
| AlPO-17 | Formation of octahedrally coordinated Al | |
| SAPO-17 | ||
| VPI-5 | Phase transformation into AlPO-8 | 297 |
3.1.1.2 Ion exchange. As already stated, the AlPO4 framework is neutral and no counter ions are present in the channels. Isomorphous substitution of P5+ for Si4+ or Al3+ for a bi-valent cation leads to introduction of a negative charge in the framework which is compensated by cations situated in the channel system. Such materials display ion exchange properties similar to aluminosilicate zeolites.298,299 The ion-exchange ability of AlPO-based molecular sieves is largely exploited in the modification of their physicochemical properties.
Ion-exchange properties of SAPO-34 were used to introduce Sr2+ and Ba2+ cations in the channel system, thereby significantly improving its CO2 sorptive capacity.300,301 The investigation showed that the introduction of Sr2+ and Ba2+ cations by a multi-step ion exchange procedure was more efficient and minimised undesired effects in the short-range structural order. Sr2+-SAPO-34 was also prepared via solid-state ion exchange.302 The obtained material demonstrated CO2 improved adsorptive properties, particularly at low partial pressure. A comprehensive study of liquid and solid-state exchanged forms of SAPO-34 revealed the higher efficiency of the latter approach. SAPO-34 membranes exchanged with alkaline earth cations were also used in the separation of CO2. The CO2/CH4 separation selectivity decreased in the following order Ca2+ > Mg2+ > Sr2+ > Ba2+.303,304 It is worth noting that gas permeability of SAPO-34 membranes is related to the size of the counter cation.305,306
A comparative study of low-pressure xenon adsorption on different cation exchanged forms of an AlPO, a SAPO and an aluminosilicate with CHA-type framework topology was performed.307 The Ca-form of aluminosilicate material showed superior performance with respect to AlPO4-based frameworks. SAPO-34 showed only modest increases in Xe uptake in comparison with its non-charged AlPO4 counterpart. More information on ion exchanged AlPO and SAPO molecular sieves is provided in Table 4.294–301
| Material | Counter cation | Modified properties | Ref. |
|---|---|---|---|
| a Solid-state ion exchange. b Non-aqueous solution ion-exchange. | |||
| AlPO-5 | Ti, Zn, Mg | Strong Brønsted acidity Increased reactivity in xylene isomerisation | 299, 314, 315 |
| SAPO-34 | Sr, Ba | Defects on the outer surface of the particles are observed | 300–307 |
| Ca, Li, Rb, Csa | Particles aggregation is observed | ||
| Ca, Mg, Sr, Bab | Adsorption capacity for CO2 is improved | ||
| CO2/CH4 separation selectivity is reduced | |||
| SAPO-34 | La, Y, K, Ce, Cr, Ni, Co, Mn, Ti, Fe, Cu, Mg, Ba | Higher selectivity to light olefins and lower methane formation in the MTO process and long catalytic lifetime | 316–319 |
| AlPO-11 | Pd | The 10-member rings are distorted | 320, 321 |
| SAPO-5 | Medium strong acidity is measured | ||
| SAPO-34 | High selectivity towards isobutene | ||
3.1.1.3 Metal introduction. Active sites in AlPOs based molecular sieves are often created or modified by metal ions employing the methods described in Section 2.1.3. A number of studies are devoted to the electronic properties associated with metal ions in AlPO frameworks and how the chemistry of the dopants is influenced by the crystalline environment.308,309 Metal-doped AlPO and SAPO materials show different and often improved performance with respect to their aluminosilicate counterparts. A highly selective catalyst Pt–Sn/SAPO-34 (CHA-type) demonstrated a new technological trend in light olefin production via the alkene dehydrogenation route.310,311 The dehydrogenation performance of Pt–Sn based catalysts was found to be largely dependent on the interactions between the active metal (Pt or Sn) and the support. Deactivation of Pt-based catalysts during the propane dehydrogenation process primarily resulted from the aggregation of Pt particles. The presence of Sn promotes Pt dispersion during catalyst manufacturing and significantly improves catalytic performance. Pt–Sn/SAPO-34 has also been used in a microreactor to optimize the operating parameters for maximizing propylene production.312,313
AlPO-5 samples were impregnated with different amounts of V2O5 (1–30 wt%) and subjected to calcination in the temperature range of 400–600 °C. A continuous decrease of specific surface area was observed with increasing vanadium. All of the samples showed an enhancement in dehydration reaction towards the ethyl acetate formation. Both, the level of V2O5 loading and the calcination temperature influenced the characteristics of the catalyst.322
Recently, metal deposition by plasma treatment has attracted a lot of attention. This approach offers important advantages such as reduced energy consumption and short preparation time. Most importantly, plasma deposition provides highly dispersed active species that exhibit enhanced catalytic activity. This approach allowed deposition of metals like Pt, Pd and Rh on AlPO-5.323 Palladium was employed to improve the isobutene selectivity of AEL-type (AlPO-11 and SAPO-11) molecular sieves. Both, Pd-AlPO-11 and Pd-SAPO-11 catalysts showed high selectivity towards isobutene. It is worth noting that the catalytic properties of Pd-SAPO-11 strongly depended on the Si content, as the maximum activity and isomerisation product selectivity were reached at Pd content around 0.10 wt%. Metal particles with a very narrow size distribution and an average size of 2.3 nm were prepared by plasma deposition and compared to the impregnated materials. Consequently, plasma treated AlPO-5 material demonstrated higher CO oxidation activity in comparison to the impregnated samples.324
The introduction of In3+, Ga3+, Cu2+, and Ni2+ into SAPO-34 by preparing a mechanical mixture of Me2O3 oxide and SAPO-type material was reported.325–328 The H-form of SAPO-34 was used, which after mixing with the metal was subjected to reduction in a H2 atmosphere. The solid state reaction led to the replacement of proton sites in SAPO-34 with Me+ cations and thus to modification of the catalytic properties.
3.1.1.4 Modification by organic compounds. Recent interest in mesoporous and microporous hybrid materials is based on the complementarity of the inorganic and organic parts of the system. The inorganic components can provide mechanical, thermal, or structural stability, while the organic functionalities are often more readily modified for specific applications in catalysis, separation, or sensing. For instance the separation selectivity of a membrane is based on the competitive adsorption between molecular species. In this respect, it is highly desirable to enhance the preferential adsorption of a particular molecule and thus to improve gas selectivity of the membrane. The latter was exemplified by the preparation of hybrid materials based on the CHA-type zeolite. SAPO-34 containing membrane modules were functionalised with ethylenediamine, hexylamine, and octylamine which modified their surface characteristics. Amine-modified membranes showed substantially improved CO2/CH4 and CO2/N2 selectivity.329
Other methods of post-synthesis modification include pore structure alteration of microporous materials by organometallic reagents. Both, solution and vapour deposition techniques were applied to SAPO-34 functionalized with dialkyl zinc compounds (ZnR2, R = Me, Et) (Fig. 6).330 A substantial change in the CHA-type framework was observed as a result of ZnMe2 modification. The formation of a new Zn–OH moiety was confirmed by NMR analysis. It was proposed that the Brønsted acid sites in H-SAPO-34 react with ZnMe2, thus forming methane and anchoring the Zn–Me species to the framework oxygen. Subsequent quenching with methanol converts Zn–Me into Zn–OMe, which was converted to Zn–OH upon heating. The presence of the Zn–OH species in the pores led to a reduction in the pore volume, as was measured by methanol sorption.
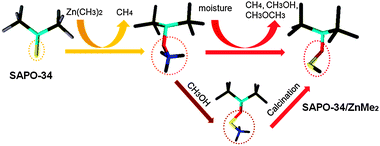 | ||
| Fig. 6 Post-synthesis modification of SAPO-34 by organometallic reagents. Adapted from ref. 330. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P = (2.6–0.5):l. These variations in composition suggested the presence of large amounts of extra framework species and thus the Si incorporation in the framework is difficult to be evaluated. A later study, again based on SiCl4 vapour treatment of AlPO-5, reported a more reasonable framework composition (1.0 A1 : 0.91 P : 0.026 Si), where namely P was substituted for Si.333 Ammonia temperature programmed desorption (TPD) proved the formation of strong acid sites. A substantial disadvantage of SiC14 vapour phase treatment is the deposition of silicon chloride and other products of chlorine interactions with the framework in the channel system of the molecular sieve.334 These species reside in the channels and upon washing can be hydrolysed to amorphous silica. Thus, materials subjected to SiCl4 treatment usually exhibit decreased adsorption capacity. On the other hand, the amount of silicon incorporated into the AlPO framework is very limited.
:P = (2.6–0.5):l. These variations in composition suggested the presence of large amounts of extra framework species and thus the Si incorporation in the framework is difficult to be evaluated. A later study, again based on SiCl4 vapour treatment of AlPO-5, reported a more reasonable framework composition (1.0 A1 : 0.91 P : 0.026 Si), where namely P was substituted for Si.333 Ammonia temperature programmed desorption (TPD) proved the formation of strong acid sites. A substantial disadvantage of SiC14 vapour phase treatment is the deposition of silicon chloride and other products of chlorine interactions with the framework in the channel system of the molecular sieve.334 These species reside in the channels and upon washing can be hydrolysed to amorphous silica. Thus, materials subjected to SiCl4 treatment usually exhibit decreased adsorption capacity. On the other hand, the amount of silicon incorporated into the AlPO framework is very limited.
Aqueous solutions of ammonium hexafluorosilicate have also been used for post-synthesis isomorphous substitution in AFI-type structure.335 The results clearly showed that the interaction of (NH4)2SiF6 with the AlPO-5 framework involves two consecutive processes: (1) creation of defects caused by the removal of Al and P, and (2) incorporation of Si in the framework, preferably at the Al sites, to produce SAPO-5. It remains unclear, however, how the framework is charge balanced if Al3+ is substituted for Si4+.
In conclusion, the post-synthesis isomorphous substitutions in AlPO4-type molecular sieves are still not very efficient and remain marginal amongst the methods employed for tuning physicochemical properties of this family of microporous solids.
3.2 Post-synthesis modification of MOFs
During the last decade, the so called metal–organic frameworks (MOFs) with zeolite-type structures have captured widespread attention due to their potential in gas storage, gas separation, and heterogeneous catalysis.336–338 These well-defined structures opened up the possibility to design hybrid materials combining more than one function within a well-defined micropore structure. In the quest of new functionalities post-synthesis methods have been widely used.339,340 Post-synthesis approaches allowed us to generate a series of MOFs from one parent material and to incorporate chemical functions that are either incompatible with in situ synthesis or that would have required ligands that cannot exist in solution phase.341–3473.2.1.1 Modification by organic species. Chemical modification of a MOF material was reported in 2000, shortly after this class of microporous solids was discovered.348 In this study the N-alkylation of pyridyl groups in the framework was achieved at room temperature by adding an excess amount of iodomethane to a suspension of the homochiral open-framework with the general formula [Zn3(μ3-O)(1-H)6]·2H3O·12H2O. The resultant material was referred to as D-POST-1. The N-alkylation resulted in modification of the pore size and shrinkage of the pore volume by 14%. In another study it was shown that more than one chemical reaction and more than one functional group can be introduced into a MOF structure by post-synthetic strategies.349 An important achievement was the functionalisation of MOFs with carboxylic acids since the resulting intermediate material can be further modified using a wide range of reactions to afford new MOFs with different capabilities.350,351 An example of a modified Zn based mixed-ligand MOF is presented in Fig. 7.352
 | ||
| Fig. 7 Top: a stick diagram showing a single network unit 1 subjected to post-synthesis functionalisation. Yellow polyhedra represent zinc ions. Carbon: grey; oxygen: red; nitrogen: blue. Bottom: a schematic representation of the reaction of unit 1 with succinic anhydride to give 1succ with free carboxylic acid groups tethered to the struts via ester linkages (copyright from ref. 352). | ||
Very recently, Lun et al. reported the thermal post-synthetic deprotection of a proline-functionalised cubic zinc(II) IRMOF.353 In this study the functionalised linker was first obtained via several synthetic steps in order to graft the protected proline. Then the framework was obtained by self-assembly and finally the protecting group was removed by heating the MOF at 165 °C in a solvent.
3.2.1.2 Modification by inorganic species. In addition to the modification of the framework itself, the host–guest chemistry of MOFs allows the implementation of desired properties by filling the cavities with guest species. In particular, doping MOFs with metal nanoparticles (NPs) is of interest for heterogeneous catalysis and hydrogen storage. An important aspect is whether the NPs are evenly distributed over the volume of the MOF crystallite with a size matching the dimensions of the channels and cavities, or they are accumulated preferentially at the outer surface regions with a pronounced tendency to substantially exceed the pore size.354,355
Different synthetic approaches, including hydro- and solvothermal treatment, and chemical vapour deposition, were employed in order to control the size of metal NPs in MOFs. Chemical vapour deposition allowed the encapsulation of very small gold nanoparticles in zeolitic imidazolate frameworks (ZIFs). The study showed how the network structure and especially the functional groups influenced the nanoparticle formation, controlling the size distribution and shape of the Au nanoparticles as well as their accessibility.356
A very particular procedure was employed to charge robust zeolite imidazolate frameworks (ZIF-8 and ZIF-90) with a volatile metal precursor [Au(CO)Cl].357–359 Gas phase infiltration was performed under static vacuum and after the loading procedure the tube was opened and the composite material was stored under a CO atmosphere at −30 °C. The reduction process of the precursor@ZIF samples was performed under hydrogen in a pressure jar at temperatures varying from 100 to 130 °C.360–362 Low-dose high-resolution transmission electron microscopy and electron tomography revealed a homogeneous distribution of Au nanoparticles throughout the ZIF matrix.
In addition to single clusters, Au–Ag core–shell nanoparticles were stabilised in ZIF-8.363,364 With the limitation effect of the pore structure of ZIF-8 Au@AgNPs were successfully restricted to 2–6 nm. The desolvated ZIF-8 was sequentially immersed in aqueous solutions of Au and Ag precursors, with respective reduction and drying thus yielding Au@Ag core–shell nanoparticles.365 Because of dielectric confinement, both Au and Ag NPs showed a pronounced surface plasmon resonance effect, giving optical properties remarkably different from those of their bulk counterparts. The features of Au–Ag core–shell nanoparticles were significantly modulated by different Au/Ag contents and preparation sequences. The bimetallic Au@Ag nanoparticles showed improved catalytic activity compared to monometallic gold and silver NPs.
Similarly to microporous aluminosilicate materials, the MOF-type crystalline solids offer a huge potential for a number of applications, such as chemical sensors, membranes and optical coatings.366,367 MOF films with well-defined thickness provided the basis for development of such applications. Shekhah et al. reported the post-synthesis modification of an extended film built of a layer-type MOF,368 consisting of [Cu2(NH2-bdc)2] sheets, where NH2-bdc is 2-amino-1,4-benzenedicarboxylate, connected by Dabco pillars. The modification of the dicarboxy-benzene ligand with NH2 groups did not change the crystallinity and the orientation of the microporous film. The MOF film was further modified by attaching synthons containing an isocyanate-group without modifying film characteristics.
Ferrocene-modified MOFs showed substantially different electrochemical properties.361,369 Besides, it was shown that the metallocene induced breathing effect was more pronounced in the MIL-53(Al) material in comparison to MIL-47(V), which was explained by the different bridging groups between the MO6 clusters. MOFs based on zinc(II) and aluminium(III) were post-synthetically functionalised with ferrocenyl through an amine or amide functional group providing materials with reversible electrochemical responses.370 It was demonstrated that ferrocenyl groups were tethered to the framework backbone. In organic media, well-defined and stable redox processes were observed to be associated with the oxidation and back-reduction of the ferrocenes occluded in the pores. Rapid hopping of charges across the MOF surface has been proposed to account for the magnitude and scan rate dependence of the signals. However, in aqueous media voltammetric responses for the ferrocene oxidation exhibit rapid decay due to dissolution of the functionalised MOF framework. The pH effect on the voltammetric responses has been interpreted in terms of a “framework effect” where hydroxide attack on the framework metal centre can “compensate” the positive charge from interstitial ferrocenium.
Metal–organic compounds have also been employed in post-synthesis functionalisation of MOFs with the quest to modify their catalytic properties. An example of a pure 2D cadmium MOF bearing binaphthol moieties by post-functionalisation with Ti(OiPr)4 was reported.371 It is worth noting that catalysts for asymmetric reactions can be produced within a non-chiral MOF by attaching an enantiomerically pure compound through post-synthesis reaction.357
A series of metallosalen-based MOFs have been prepared by the post-synthesis modification of Mn3+SO-MOF.372 Treatment of Mn-MOF with H2O2 resulted in the removal of the Mn3+ ions from the salen struts. Thus, the Mn3+SO-MOF was completely “demanganated” and the resulting dSO-MOF was further modified by remetallating the salen struts with a wide variety of metal ions, resulting in facile incorporation of different unsaturated metal centers as part of a chiral salen complex. These available MOFs have potential as salen-based catalysts and selective chemical sensors.
The above list of post-synthesis modifications of MOF-type microporous materials is not exhaustive, but it provides clear ideas about the unlimited opportunities for post-synthesis modification of metal–organic frameworks. In summary, the post-synthesis modification proved to be a useful strategy for preparing a variety of MOFs with long hydrophobic chains, free carboxylic acids, and non-structural metal centres.342,346,352,373,374
4. Summary and outlook
Flourishing developments in the field of crystalline microporous materials are strongly related to the advances in synthesis. Without any doubt many new exciting structures will be discovered. Nevertheless, the post-synthesis modification will always be an indispensable tool for tuning zeolite properties and pave the road for practical uses. Depending on the application (heterogeneous catalysts, ion exchangers or adsorbents), the post-synthesis modification may cover different structural properties, for instance the metal species in the pore system, the framework composition or the crystal morphology. Consequently, new approaches and combinations of synthesis methods have been developed during the last decade, which have extended the zeolite characteristics much beyond those imposed by the synthesis conditions.In this overview an unprecedented analysis of post-synthesis modifications used to modulate the properties of different families of crystalline microporous solids was provided. The limits of the in situ control were discussed and the possible post-synthesis interventions on key zeolite features addressed. Micropore-related modifications included dehydration/template removal and host–guest chemistry. Framework alteration was centred at post-synthesis substitutions in liquid and vapour phases. Crystal features tuning addressed the crystal size control by tribochemical methods, by a top-down approach and the formation of zeolite macrostructures employing pre-formed zeolite units.
The review also provides guidelines as to how and to what extent different families of microporous materials could be post-synthetically modified. For instance, the robust aluminosilicate zeolites are often subjected to severe treatments in an aggressive atmosphere at elevated temperatures. Furthermore, grinding and dissolution are used to modify the morphology of these materials. On the other hand, grafting of organic functionalities to zeolite frameworks and incorporation of large organic complexes in zeolite pore volume are difficult to achieve. The latter approach is much more applicable to metal–organic frameworks whose open structure and the presence of organic linkers offer ideal conditions for organic functionalisation. Thus, the post-synthesis modifications are pre-determined to some extent by the intrinsic characteristics of the microporous solids. This specificity should not be regarded as a limitation, but rather as a diversification that extends the portfolio of microporous solids and their possible area of application. Once scientists and engineers overcome the large-scale manufacture of a porous solid, the post-synthesis engine to tune selected properties in a very specific way will start functioning. The discovery of new materials through well-designed synthesis increases exponentially the post-synthesis possibilities to tailor specific properties. Therefore the future of post-synthesis modifications is bright.
Acknowledgements
VV acknowledges the support of French Research Agency (contract ANR-2010-BLAN-723) and Bulgarian National Science Fund (contract DTK 02-47). JPR acknowledges the Swiss National Science Foundation (Project Number 200021-134572). We thank Dr. S. Mitchell for comments on the manuscript.References
- IUPAC, Pure Appl. Chem., 1972, 31, 577–638 Search PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249–267 CrossRef CAS.
- Y. F. Shen, R. P. Zerger, R. N. DeGuzman, S. L. Suib, L. McCurdy, D. I. Potter and C. L. O'Young, Science, 1993, 260, 511–515 CAS.
- M. Nyman, A. Tripathi, J. B. Parise, R. S. Maxwell, W. T. A. Harrison and T. M. Nenoff, J. Am. Chem. Soc., 2001, 123, 1529–1530 CrossRef CAS.
- V. P. Valtchev, J. Chem. Soc., Chem. Commun., 1994, 261–262 RSC.
- A. F. Cronstedt, Proc. 9th Int. Zeolite Conf., 1993.
- H. de St. Claire Deville, C. R. Seances Acad. Sci., 1862, 54, 324 Search PubMed.
- R. L. Wadlinger, G. T. Kerr and E. J. Rosinski, US3308069, 1967 Search PubMed.
- R. M. Barrer, Zeolites and Clay Minerals as Sorbents and Molecular Sieves, Academic Press, London, 1978 Search PubMed.
- R. M. Barrer, Hydrothermal Chemistry of Zeolites, Academic Press, London, 1982 Search PubMed.
- D. W. Breck, Zeolite Molecular Sieves: Structure, Chemistry, and Use, John Wiley & Sons, New York, 1974 Search PubMed.
- C. J. Plank and E. J. Rosinski, US3140240, 1964 Search PubMed.
- D. W. Breck, US31300007, 1964 Search PubMed.
- J. Biswas and I. E. Maxwell, Appl. Catal., 1990, 63, 197–258 CrossRef CAS.
- R. J. Argauer and G. R. Landolt, US3702886, 1972 Search PubMed.
- E. M. Flanigen, J. M. Bennett, R. W. Grose, J. P. Cohen, R. L. Patton, R. M. Kirchner and J. V. Smith, Nature, 1978, 271, 512–516 CrossRef CAS.
- R. Szostak, Handbook of Molecular Sieves, Springer, New York, 1992 Search PubMed.
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146–1147 CrossRef CAS.
- E. M. Flanigen, R. L. Patton and S. T. Wilson, Stud. Surf. Sci. Catal., 1988, 37, 13–27 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson and E. W. Sheppard, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- J. Jiang, J. L. Jorda, J. Yu, L. A. Baumes, E. Mugnaioli, M. J. Diaz-Cabanas, U. Kolb and A. Corma, Science, 2011, 333, 1131–1134 CrossRef CAS.
- J. Sun, C. Bonneau, A. Cantin, A. Corma, M. J. Diaz-Cabanas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154–1157 CrossRef CAS.
- J. Jiang, J. L. Jorda, M. J. Diaz-Cabanas, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 122, 5106–5108 CrossRef.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- International Zeolite Association, Database of zeolite structures, http://www.iza-structure.org/databases/.
- B. Yilmaz and U. Müller, Top. Catal., 2009, 52, 888–895 CrossRef CAS.
- G. H. Kühl, in Catalysis and Zeolites – Fundamentals and Applications, ed. J. Weitkamp and L. Puppe, Springer, Berlin, 1999, pp. 81–197 Search PubMed.
- R. Szostak, in Stud. Surf. Sci. Catal, ed. H. van Bekkum, E. M. Flanigen, P. A. Jacobs and J. C. Jansen, Elsevier, 2001, vol. 137, pp. 261–297 Search PubMed.
- C. Martínez and A. Corma, Coord. Chem. Rev., 2011, 255, 1558–1580 CrossRef.
- W. Vermeiren and J. P. Gilson, Top. Catal., 2009, 52, 1131–1161 CrossRef CAS.
- H. Siegel, W. Schmitz, R. Schollner, A. Dyer and H. Enamy, Thermochim. Acta, 1983, 61, 329–340 CrossRef CAS.
- B. Belbéoch, M. Roulliay, R. Kahn and E. Cohen de Lara, Zeolites, 1983, 3, 99–101 CrossRef.
- H. Morell, K. Angermund, A. R. Lewis, D. H. Brouwer, C. A. Fyfe and H. Gies, Chem. Mater., 2002, 14, 2192–2198 CrossRef CAS.
- B. F. Mentzen and F. Lefebvre, Mater. Res. Bull., 1997, 32, 813–820 CrossRef CAS.
- J. W. Couves, J. M. Thomas, C. R. A. Catlow, G. N. Greaves, G. Baker and A. J. Dent, J. Phys. Chem., 1990, 94, 6517–6519 CrossRef CAS.
- E. Dooryhee, C. R. A. Catlow, J. W. Couves, P. J. Maddox, J. M. Thomas, G. N. Greaves, A. T. Steel and R. P. Townsend, J. Phys. Chem., 1991, 95, 4514–4521 CrossRef CAS.
- B. Ganemi, E. Björnbom, B. Demirel and J. Paul, Microporous Mesoporous Mater., 2000, 38, 287–300 CrossRef CAS.
- M. Noack, M. Schneider, A. Dittmar, G. Georgi and J. Caro, Microporous Mesoporous Mater., 2009, 117, 10–21 CrossRef CAS.
- J. A. van Bokhoven and N. Danilina, in Zeolites and Catalysis, ed. J. Cejka, A. Corma and S. I. Zones, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 283–300 Search PubMed.
- Z. Pilter, S. Szabó, M. Hasznos-Nezdei and E. Pallai-Varsányi, Microporous Mesoporous Mater., 2000, 40, 257–262 CrossRef CAS.
- E. Bourgeat-Lami, F. Di Renzo, F. Fajula, P. Hubert Mutin and T. Des Courieres, J. Phys. Chem., 1992, 96, 3807–3811 CrossRef CAS.
- F. Collignon, P. A. Jacobs, P. Grobet and G. Poncelet, J. Phys. Chem. B, 2001, 105, 6812–6816 CrossRef CAS.
- S. Eslava, F. Iacopi, M. R. Baklanov, C. E. A. Kirschhock, K. Maex and J. A. Martens, in Stud. Surf. Sci. Catal., ed. Z. G. J. C. Ruren Xu and Y. Wenfu, Elsevier, 2007, vol. 170, pp. 594–599 Search PubMed.
- J. He, X. Yang, D. G. Evans and X. Duan, Mater. Chem. Phys., 2003, 77, 270–275 CrossRef CAS.
- S. Palmery, F. Genoni, G. Spano, L. Dalloro, A. Cesana, R. Buzzoni, S. Palmari, F. Qinorni and G. Spanor, EP1101735-A, 2000 Search PubMed.
- E. Meretei, J. Halász, D. Méhn, Z. Kónya, T. I. Korányi, J. B. Nagy and I. Kiricsi, J. Mol. Struct., 2003, 651–653, 323–330 CrossRef CAS.
- J. Kuhn, M. Motegh, J. Gross and F. Kapteijn, Microporous Mesoporous Mater., 2009, 120, 35–38 CrossRef CAS.
- J. Kuhn, J. Gascon, J. Gross and F. Kapteijn, Microporous Mesoporous Mater., 2009, 120, 12–18 CrossRef CAS.
- A. Toktarev, L. Malysheva and E. Paukshtis, Kinet. Catal., 2010, 51, 318–324 CrossRef CAS.
- J. Patarin, Angew. Chem., Int. Ed., 2004, 43, 3878–3880 CrossRef CAS.
- S. Hitz and R. Prins, J. Catal., 1997, 168, 194–206 CrossRef CAS.
- C. W. Jones, K. Tsuji, T. Takewaki, L. W. Beck and M. E. Davis, Microporous Mesoporous Mater., 2001, 48, 57–64 CrossRef CAS.
- I. Melian-Cabrera, F. Kapteijn and J. A. Moulijn, Chem. Commun., 2005, 2744–2746 RSC.
- I. Melián-Cabrera, A. H. Osman, E. R. H. van Eck, A. P. M. Kentgens, E. Polushkin, F. Kapteijn and J. A. Moulijn, Stud. Surf. Sci. Catal., 2007, 170, 648–654 CrossRef.
- H. Xing, Y. Zhang, M. Jia, S. Wu, H. Wang, J. Guan, L. Xu, T. Wu and Q. Kan, Catal. Commun., 2008, 9, 234–238 CrossRef CAS.
- J. Kecht and T. Bein, Microporous Mesoporous Mater., 2008, 116, 123–130 CrossRef CAS.
- I. Melian-Cabrera, F. Kapteijn and J. A. Moulijn, Chem. Commun., 2005, 2178–2180 RSC.
- I. V. Melian Cabrera, F. Kapteijn and J. A. Moulijn, EP1690831-A1, 2005 Search PubMed.
- J. F. Gaynor and P. Vancleemput, US6660245-B1, 2002 Search PubMed.
- L. Xiao, J. Li, H. Jin and R. Xu, Microporous Mesoporous Mater., 2006, 96, 413–418 CrossRef CAS.
- A. N. Parikh, A. Navrotsky, Q. Li, C. K. Yee, M. L. Amweg and A. Corma, Microporous Mesoporous Mater., 2004, 76, 17–22 CrossRef CAS.
- A. Navrotsky and A. N. Parikh, US2004151651-A1, 2003 Search PubMed.
- Q. Li, M. L. Amweg, C. K. Yee, A. Navrotsky and A. N. Parikh, Microporous Mesoporous Mater., 2005, 87, 45–51 CrossRef CAS.
- T. L. M. Maesen, H. W. Kouwenhoven, H. van Bekkum, B. Sulikowski and J. Klinowski, J. Chem. Soc., Faraday Trans., 1990, 86, 3967–3970 RSC.
- Y. Liu, Y.-x. Pan, P. Kuai and C.-J. Liu, Catal. Lett., 2010, 135, 241–245 CrossRef CAS.
- Y. Liu, Y. Pan, Z.-J. Wang, P. Kuai and C.-J. Liu, Catal. Commun., 2010, 11, 551–554 CrossRef CAS.
- K. F. M. G. J. Scholle and W. S. Veeman, Zeolites, 1985, 5, 118–122 CrossRef CAS.
- S. Kawi, Chem. Commun., 1998, 1407–1408 RSC.
- Z. Huang, D. Y. Luan, S. C. Shen, K. Hidajat and S. Kawi, J. Supercrit. Fluids, 2005, 35, 40–48 CrossRef CAS.
- A. P. Nelson, O. K. Farha, K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2008, 131, 458–460 CrossRef.
- H. Lee, S. I. Zones and M. E. Davis, Nature, 2003, 425, 385–388 CrossRef CAS.
- H. Lee, S. I. Zones and M. E. Davis, J. Phys. Chem. B, 2004, 109, 2187–2191 CrossRef.
- P. Gallezot, in Molecular Sieves: Post-Synthesis Modification I, ed. H. G. Karge and J. Weitkamp, Springer Berlin/Heidelberg, 2002, vol. 3, pp. 257–305 Search PubMed.
- D. E. de Vos, B. F. Sels and P. A. Jacobs, in Advances in Catalysis, ed. B. C. Gates and H. Knözinger, Academic Press, 2001, vol. 46, pp. 1–87 Search PubMed.
- D. E. de Vos and P. A. Jacobs, Stud. Surf. Sci. Catal., 2001, 137, 957–985 CrossRef CAS.
- Synthesis of Solid Catalysts, ed. K. P. de Jong, Wiley-VCH, Weinheim, 2009 Search PubMed.
- Zeolites and Catalysis, Synthesis, Reactions and Applications, ed. J. Cejka, A. Corma and S. I. Zones, 2010 Search PubMed.
- R. P. Townsend and E. N. Coker, in Introduction to Zeolite Science and Practice, Stud. Surf. Sci. Catal., ed. H. Van Bekkum, E. M. Flanigen, P. A. Jacobs and J. C. Jansen, Elsevier, Amsterdam, 2001, vol. 137, pp. 467–524 Search PubMed.
- H. Karge and H. Beyer, in Molecular Sieves: Post-Synthesis Modification I, Springer Berlin Heidelberg, 2002, vol. 3, pp. 43–201 Search PubMed.
- S. Uma, S. Rodrigues, I. N. Martyanov and K. J. Klabunde, Microporous Mesoporous Mater., 2004, 67, 181–187 CrossRef CAS.
- G. M. Canfield, M. Bizimis and S. E. Latturner, J. Mater. Chem., 2007, 17, 4530–4534 RSC.
- G. M. Canfield, M. Bizimis and S. E. Latturner, Chem. Mater., 2009, 22, 330–337 CrossRef.
- Y. Neinska, R. M. Mihaly, V. Mavrodinova, C. Mincheva and H. K. Beyer, Phys. Chem. Chem. Phys., 1999, 1, 5761–5765 RSC.
- J. Rocha and L. D. Carlos, Curr. Opin. Solid. State. Mater. Sci., 2003, 7, 199–205 CrossRef CAS.
- Y. Kuroda, T. Okamoto, R. Kumashiro, Y. Yoshikawa and M. Nagao, Chem. Commun., 2002, 1758–1759 RSC.
- M. Zendehdel, G. Cruciani and M. Dondi, J. Porous Mater., 2011, 1–8 Search PubMed.
- A. Tavolaro and P. Tavolaro, Catal. Commun., 2009, 10, 586–591 CrossRef CAS.
- X. Gu, J. Zhang, J. Dong and T. M. Nenoff, Catal. Lett., 2005, 9–13 CrossRef CAS.
- J. N. Miale and C. D. Chang, United States Pat., US4596704, 1986 Search PubMed.
- D. Levin, C. D. Chang, S. Luo, J. G. Santiesteban and J. C. Vartuli, US6114551, 2000 Search PubMed.
- S. Dzwigaj and M. Che, Catal. Today, 2011, 169, 232–241 CrossRef CAS.
- A. S. Ichimura, J. L. Dye, M. A. Camblor and L. A. Villaescusa, J. Am. Chem. Soc., 2002, 124, 1170–1171 CrossRef CAS.
- D. P. Wernette, A. S. Ichimura, S. A. Urbin and J. L. Dye, Chem. Mater., 2003, 15, 1441–1448 CrossRef CAS.
- J. R. Regalbuto, in Synthesis of Solid Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 33–58 Search PubMed.
- M. Du, G. Zhan, X. Yang, H. Wang, W. Lin, Y. Zhou, J. Zhu, L. Lin, J. Huang, D. Sun, L. Jia and Q. Li, J. Catal., 2011, 283, 192–201 CrossRef CAS.
- B. B. De, B. B. Lohray, S. Sivaram and P. K. Dhal, Tetrahedron: Asymmetry, 1995, 6, 2105–2108 CrossRef CAS.
- L. Canali, E. Cowan, C. L. Gibson, D. C. Sherrington and H. Deleuze, Chem. Commun., 1998, 2561–2562 RSC.
- A. Corma, A. Fuerte, M. Iglesias and F. Sánchez, J. Mol. Catal. A: Chem., 1996, 107, 225–234 CrossRef CAS.
- E. Duprey, J. Maquet, P. P. Man, J. M. Manoli, M. Delamar and J. M. Brégeault, Appl. Catal., A, 1995, 128, 89–96 CrossRef CAS.
- E. Möllmann, P. Tomlinson and W. F. Hölderich, J. Mol. Catal. A: Chem., 2003, 206, 253–259 CrossRef.
- M. J. Sabater, A. Corma, A. Domenech, V. Fornes and H. Garcia, Chem. Commun., 1997, 1285–1286 RSC.
- S. B. Ogunwumi and T. Bein, Chem. Commun., 1997, 901–902 RSC.
- P.-P. Knops-Gerrits, D. E. De Vos and P. A. Jacobs, J. Mol. Catal. A: Chem., 1997, 117, 57–70 CrossRef CAS.
- M. R. Maurya, S. J. J. Titinchi and S. Chand, J. Mol. Catal. A: Chem., 2004, 214, 257–264 CrossRef CAS.
- M. R. Maurya, S. J. J. Titinchi, S. Chand and I. M. Mishra, J. Mol. Catal. A: Chem., 2002, 180, 201–209 CrossRef CAS.
- M. R. Maurya, S. J. J. Titinchi and S. Chand, J. Mol. Catal. A: Chem., 2003, 201, 119–130 CrossRef CAS.
- V. Ayala, A. Corma, M. Iglesias, J. A. Rincón and F. Sánchez, J. Catal., 2004, 224, 170–177 CrossRef CAS.
- C. Schuster, E. Möllmann, A. Tompos and W. F. Hölderich, Catal. Lett., 2001, 74, 69–75 CrossRef CAS.
- P. Serna and B. C. Gates, J. Am. Chem. Soc., 2011, 133, 4714–4717 CrossRef CAS.
- E. Briot, F. Bedioui and K. J. Balkus Jr., J. Electroanal. Chem., 1998, 454, 83–89 CrossRef CAS.
- Y. Umemura, Y. Minai and T. Tominaga, Chem. Commun., 1993, 1822–1823 RSC.
- R. Gonzalez-Olmos, F. Holzer, F. D. Kopinke and A. Georgi, Appl. Catal., A, 2011, 398, 44–53 CrossRef CAS.
- P. Kuśtrowski, L. Chmielarz, R. Dziembaj, P. Cool and E. F. Vansant, J. Phys. Chem. B, 2005, 109, 11552–11558 CrossRef.
- K. Köhler, M. Wagner and L. Djakovitch, Catal. Today, 2001, 66, 105–114 CrossRef.
- H. Yamashita, M. Honda, M. Harada, Y. Ichihashi, M. Anpo, T. Hirao, N. Itoh and N. Iwamoto, J. Phys. Chem. B, 1998, 102, 10707–10711 CrossRef CAS.
- H. Yamashita and M. Anpo, Catal. Surv. Asia, 2004, 8, 35–45 CrossRef CAS.
- M. Anpo, M. Takeuchi, K. Ikeue and S. Dohshi, Curr. Opin. Solid State. Mater. Sci., 2002, 6, 381–388 CrossRef CAS.
- K. Shimizu, K. Sugino, K. Kato, S. Yokota, K. Okumura and A. Satsuma, J. Phys. Chem. C, 2007, 111, 1683–1688 CAS.
- J. de Graaf, A. J. van Dillen, K. P. de Jong and D. C. Koningsberger, J. Catal., 2001, 203, 307–321 CrossRef CAS.
- Y. Wang, H. Wu, Q. Zhang and Q. Tang, Microporous Mesoporous Mater., 2005, 86, 38–49 CrossRef CAS.
- J.-J. Zou, Y.-p. Zhang and C.-J. Liu, Langmuir, 2006, 22, 11388–11394 CrossRef CAS.
- M. Rivallan, I. Yordanov, S. Thomas, C. Lancelot, S. Mintova and F. Thibault-Starzyk, ChemCatChem, 2010, 2, 1074–1078 CrossRef CAS.
- J. Talebi, R. Halladj and S. Askari, J. Mater. Sci., 2010, 45, 3318–3324 CrossRef CAS.
- L. Djakovitch and K. Koehler, J. Am. Chem. Soc., 2001, 123, 5990–5999 CrossRef CAS.
- L. Djakovitch and P. Rollet, Tetrahedron Lett., 2004, 45, 1367–1370 CrossRef CAS.
- M. Dams, L. Drijkoningen, B. Pauwels, G. Van Tendeloo, D. E. De Vos and P. A. Jacobs, J. Catal., 2002, 209, 225–236 CrossRef CAS.
- J. Pérez-Ramírez, M. Santhosh Kumar and A. Brückner, J. Catal., 2004, 223, 13–27 CrossRef.
- L. V. Pirutko, V. S. Chernyavsky, A. K. Uriarte and G. I. Panov, Appl. Catal., A, 2002, 227, 143–157 CrossRef CAS.
- J. Pérez-Ramírez, F. Kapteijn, J. C. Groen, A. Doménech, G. Mul and J. A. Moulijn, J. Catal., 2003, 214, 33–45 CrossRef.
- J. Pérez-Ramírez, J. Catal., 2004, 227, 512–522 CrossRef.
- J. Pérez-Ramírez, F. Kapteijn and A. Brückner, J. Catal., 2003, 218, 234–238 CrossRef.
- A. Zecchina, M. Rivallan, G. Berlier, C. Lamberti and G. Ricchiardi, Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 RSC.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS.
- S. A. Butter and W. W. Kaeding, US3972832-A, 1976 Search PubMed.
- J. A. Lercher and G. Rumplmayr, Appl. Catal., 1986, 25, 215–222 CrossRef CAS.
- C. de las Pozas, W. Kolodziejski and R. Roque-Malherbe, Microporous Mater., 1996, 5, 325–331 CrossRef CAS.
- G. Rodriguez-Fuentes, L. C. de Ménorval, E. Reguera and F. Chávez Rivas, Microporous Mesoporous Mater., 2008, 111, 577–590 CrossRef CAS.
- J.-F. Gu, X.-J. Zhang, J.-Z. Wang, J. Xu, F. Deng and Z.-Y. Yuan, Stud. Surf. Sci. Catal., 2008, 174A, 209–212 CrossRef CAS.
- J. Caro, M. Bülow, M. Derewinski, J. Haber, M. Hunger, J. Kärger, H. Pfeifer, W. Storek and B. Zibrowius, J. Catal., 1990, 124, 367–375 CrossRef CAS.
- A. Corma, V. Fornes, W. Kolodziejski and L. J. Martineztriguero, J. Catal., 1994, 145, 27–36 CrossRef CAS.
- G. Sastre, D. W. Lewis and C. R. A. Catlow, J. Phys. Chem., 1996, 100, 6722–6730 CrossRef CAS.
- Z. Liu, Z.-X. Chen, W. Ding, G.-J. Kang and Z. Li, J. Mol. Struct. (THEOCHEM), 2010, 948, 99–101 CrossRef CAS.
- M. Göhlich, W. Reschetilowski and S. Paasch, Microporous Mesoporous Mater., 2011, 142, 178–183 CrossRef.
- A. F. Costa, H. S. Cerqueira, J. M. M. Ferreira, N. M. S. Ruiz and S. M. C. Menezes, Appl. Catal., A, 2007, 319, 137–143 CrossRef CAS.
- T. R. Krawietz, P. Lin, K. E. Lotterhos, P. D. Torres, D. H. Barich, A. Clearfield and J. F. Haw, J. Am. Chem. Soc., 1998, 120, 8502–8511 CrossRef CAS.
- N. Nesterenko, S. Van Donk, D. Minoux and J. Dath, EP2348004-A1; WO2011089262-A1; WO2011089263-A1, 2011 Search PubMed.
- D. Minoux, N. Nesterenko, S. Van Donk, W. Vermeiren, B. E. Nesterenko N, B. E. Vermeiren W, B. E. Minoux D and B. E. Van D S, WO2009016156-A1; EP2039427-A1; EP2175991-A1; CN101795766-A; US2010256431-A1; IN201000511-P1, 2009 Search PubMed.
- G. Zhao, J. Teng, Z. Xie, W. Jin, W. Yang, Q. Chen and Y. Tang, J. Catal., 2007, 248, 29–37 CrossRef CAS.
- W. Kolodziejski, V. Fornés and A. Corma, Solid State Nucl. Magn. Reson., 1993, 2, 121–129 CrossRef CAS.
- N. Xue, R. Olindo and J. A. Lercher, J. Phys. Chem. C, 2010, 114, 15763–15770 CAS.
- C. W. Jones, M. Tsapatsis, T. Okubo and M. E. Davis, Microporous Mesoporous Mater., 2001, 42, 21–35 CrossRef CAS.
- W. O. Parker, A. de Angelis, C. Flego, R. Millini, C. Perego and S. Zanardi, J. Phys. Chem. C, 2010, 114, 8459–8468 CAS.
- Y. Shin, T. S. Zemanian, G. E. Fryxell, L.-Q. Wang and J. Liu, Microporous Mesoporous Mater., 2000, 37, 49–56 CrossRef CAS.
- Z. Tang, J. Dong and T. M. Nenoff, Langmuir, 2009, 25, 4848–4852 CrossRef CAS.
- J. Sivaguru, A. Natarajan, L. S. Kaanumalle, J. Shailaja, S. Uppili, A. Joy and V. Ramamurthy, Acc. Chem. Res., 2003, 36, 509–521 CrossRef CAS.
- J. Sivaguru, T. Poon, R. Franz, S. Jockusch, W. Adam and N. J. Turro, J. Am. Chem. Soc., 2004, 126, 10816–10817 CrossRef CAS.
- U. Wingenfelder, B. Nowack, G. Furrer and R. Schulin, Water Res., 2005, 39, 3287–3297 CrossRef CAS.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570–8578 CrossRef CAS.
- S. Zheng, H. R. Heydenrych, A. Jentys and J. A. Lercher, J. Phys. Chem. B, 2002, 106, 9552–9558 CrossRef CAS.
- S. J. Reitmeier, O. C. Gobin, A. Jentys and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 533–538 CrossRef CAS.
- D. Li, J. Yao, H. Wang, N. Hao, D. Zhao, K. R. Ratinac and S. P. Ringer, Microporous Mesoporous Mater., 2007, 106, 262–267 CrossRef CAS.
- S. Ntais, A. M. Moschovi, F. Paloukis, S. Neophytides, V. N. Burganos, V. Dracopoulos and V. Nikolakis, J. Power Sources, 2011, 196, 2202–2210 CrossRef CAS.
- Z. Chen, B. Holmberg, W. Li, X. Wang, W. Deng, R. Munoz and Y. Yan, Chem. Mater., 2006, 18, 5669–5675 CrossRef CAS.
- J. C. McKeen, Y. S. Yan and M. E. Davis, Chem. Mater., 2008, 20, 5122–5124 CrossRef CAS.
- M. Busby, H. Kerschbaumer, G. Calzaferri and L. De Cola, Adv. Mater., 2008, 20, 1614–1618 CrossRef CAS.
- G. Calzaferri, S. Huber, H. Maas and C. Minkowski, Angew. Chem., Int. Ed., 2003, 42, 3732–3758 CrossRef CAS.
- M. B. J. Roeffaers, R. Ameloot, M. Baruah, H. Uji-i, M. Bulut, G. De Cremer, U. Müller, P. A. Jacobs, J. Hofkens, B. F. Sels and D. E. De Vos, J. Am. Chem. Soc., 2008, 130, 5763–5772 CrossRef CAS.
- W. Löwenstein, Am. Mineral., 1964, 33, 1008–1013 Search PubMed.
- V. Valtchev and S. Mintova, in Inorganic Encyclopedia, ed. C. M. Lukehart and R. A. Scott, Wiley & Sons, Chichester, 2008, pp. 63–80 Search PubMed.
- R. Szostak, Molecular Sieves: Principles of Synthesis and Identification, Blackie Academic and Professional, London, 2nd edn, 1998 Search PubMed.
- R. M. Barrer and M. B. Makki, Can. J. Chem., 1964, 42, 1481–1487 CrossRef CAS.
- X.-W. Cheng, Y. Zhong, J. Wang, J. Guo, Q. Huang and Y.-C. Long, Microporous Mesoporous Mater., 2005, 83, 233–243 CrossRef CAS.
- S. P. Zhdanov and B. G. Novikov, Dokl. Akad. Nauk. SSSR, Ser. Khim., 1966, 16, 1107–1110 Search PubMed.
- F. Hernandez, R. Ibarra and F. Figueras, Acta Phys. Chem., 1985, 31, 81–88 CAS.
- F. Wolf and H. John, Chem. Technol., 1973, 25, 736 CAS.
- J. Kornatowski, M. Rozwadowski, A. Gutsze and K. E. Wisniewski, Stud. Surf. Sci. Catal., 1989, 46, 567–574 CrossRef.
- H. G. Karge and V. Dondur, J. Phys. Chem., 1990, 94, 765–772 CrossRef CAS.
- M. J. Van Niekerk, J. C. Q. Fletcher and C. T. O'Connor, J. Catal., 1992, 138, 150–163 CrossRef CAS.
- A. W. O'Donovan, C. T. O'Connor and K. R. Koch, Microporous Mater., 1995, 5, 185–202 CrossRef CAS.
- M. Maache, A. Janin, J. C. Lavalley, J. F. Joly and E. Benazzi, Zeolites, 1993, 13, 419–426 CrossRef CAS.
- E. B. Lami, F. Fajula, D. Anglerot and T. Des Courieres, Microporous Mater., 1993, 1, 237–245 CrossRef CAS.
- A. Omegna, M. Vasic, J. Anton van Bokhoven, G. Pirngruber and R. Prins, Phys. Chem. Chem. Phys., 2004, 6, 447–452 RSC.
- H. K. Beyer, I. M. Belenykaja, I. W. Mishin and G. Borbely, Stud. Surf. Sci. Catal., 1984, 18, 133–140 CrossRef CAS.
- G. T. Kerr, J. Phys. Chem., 1968, 72, 2594–2596 CrossRef CAS.
- G. T. Kerr, J. Phys. Chem., 1969, 73, 2780–2782 CrossRef CAS.
- B. Sulikowski, J. Phys. Chem., 1993, 97, 1420–1425 CrossRef CAS.
- R. Beaumont and D. Barthomeuf, J. Catal., 1972, 26, 218–225 CrossRef CAS.
- P. E. Pickert, US3640681, 1972 Search PubMed.
- A. Wiecznikowski and B. Rzepa, Rocz. Chem., 1977, 51, 1955 Search PubMed.
- G. Harvey, R. Prins, R. Crockett and E. Roduner, J. Chem. Soc., Faraday Trans., 1996, 92, 2027–2033 RSC.
- M. R. Apelian, A. S. Fung, G. J. Kennedy and T. F. Degnan, J. Phys. Chem., 1996, 100, 16577–16583 CrossRef CAS.
- T. Tatsumi, in Zeolites and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 713–743 Search PubMed.
- H. K. Beyer and I. Belenykaja, Stud. Surf. Sci. Catal., 1980, 5, 203–210 CrossRef CAS.
- H. K. Beyer, I. M. Belenykaja, F. Hange, M. Tielen, P. J. Grobet and P. A. Jacobs, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2889–2901 RSC.
- J. A. Martens, H. Geerts, P. J. Grobet and P. A. Jacobs, J. Chem. Soc., Chem. Commun., 1990, 1418–1419 RSC.
- J. Klinowski, J. M. Thomas, C. A. Fyfe, G. C. Gobbi and J. S. Hartman, Inorg. Chem., 1983, 22, 63–66 CrossRef CAS.
- J. R. Sohn, S. J. DeCanio, P. O. Fritz and J. H. Lunsford, J. Phys. Chem., 1986, 90, 4847–4851 CrossRef CAS.
- B. Sulikowski, G. Borbely, H. Beyer, H. G. Karge and I. W. Mishin, J. Phys. Chem., 1989, 93, 3240–3243 CrossRef CAS.
- H. Miessner, H. Kosslick, U. Lohse, B. Parlitz and V. A. Tuan, J. Phys. Chem., 1993, 97, 9741–9748 CrossRef CAS.
- F. P. Gortsema and B. M. T. Lok, Eur. Pat. Appl. 100544, 1983 Search PubMed.
- V. Fulop, G. Borbely, H. K. Beyer, S. Ernst and J. Weitkamp, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2127–2139 RSC.
- J. Klinowski, J. M. Thomas, M. W. Anderson, C. A. Fyfe and G. C. Gobbi, Zeolites, 1983, 3, 5–7 CrossRef CAS.
- J. Klinowski, M. W. Anderson and J. M. Thomas, J. Chem. Soc., Chem. Commun., 1983, 525–526 RSC.
- P. Bartl, B. Zibrowius and W. Hoelderich, Chem. Commun., 1996, 1611–1612 RSC.
- M. W. Anderson and J. Klinowski, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 1449–1469 RSC.
- P. Cañizares, A. Carrero and P. Sánchez, Appl. Catal., A, 2000, 190, 93–105 CrossRef.
- S. Namba, A. Inaka and T. Yashima, Chem. Lett., 1984, 817–820 CrossRef CAS.
- S. Namba, A. Inaka and T. Yashima, Zeolites, 1986, 6, 107–110 CrossRef CAS.
- S. Unverricht, M. Hunger, S. Ernst, H. G. Karge and J. Weitkamp, Stud. Surf. Sci. Catal., 1994, 84, 37–44 CrossRef CAS.
- M. W. Anderson, J. Klinowski and L. Xinsheng, J. Chem. Soc., Chem. Commun., 1984, 1596–1597 RSC.
- K. Yamagishi, S. Namba and T. Yashima, J. Phys. Chem., 1991, 95, 872–877 CrossRef CAS.
- G. G. Juttu and R. Lobo, Catal. Lett., 1999, 62, 99–106 CrossRef CAS.
- J. P. M. Niederer and W. F. Hölderich, Appl. Catal., A, 2002, 229, 51–64 CrossRef CAS.
- L. Xin-Sheng and J. M. Thomas, J. Chem. Soc., Chem. Commun., 1985, 1544–1545 RSC.
- X. Liu, J. Klinowski and J. M. Thomas, J. Chem. Soc., Chem. Commun., 1986, 582–584 RSC.
- R. D. Bezman, J. Chem. Soc., Chem. Commun., 1987, 1562–1563 RSC.
- G.-J. Kim, D.-S. Cho, K.-H. Kim, W.-S. Ko, J.-H. Kim and H. Shoji, Catal. Lett., 1995, 31, 91–102 CrossRef CAS.
- J. Ma, N. Ma, Z. Xue, Y. Wang and R. Li, Stud. Surf. Sci. Catal., 2008, 174A, 257–260 CrossRef CAS.
- B. Meier and W. Reschetilowski, J. Anal. Chem., 1994, 349, 145–147 CAS.
- H.-X. Li and J. N. Armor, Microporous Mater., 1997, 9, 51–57 CrossRef CAS.
- G. W. Skeels and D. W. Breck, Proc. 6th Int. Zeolite Conf., Reno, USA, 1984 Search PubMed.
- D. W. Breck, H. Blass and G. W. Skeels, US4503023, 1985 Search PubMed.
- S. Valencia and A. Corma, US5968473, 1999 Search PubMed.
- Y. Wang, A. Zhang, Q. Xu and R. Chen, Appl. Catal., A, 2001, 214, 167–177 CrossRef CAS.
- J. M. Silva, M. F. Ribeiro, F. R. Ribeiro, N. S. Gnep, M. Guisnet and E. Benazzi, React. Kinet. Catal. Lett., 1995, 54, 209–215 CrossRef CAS.
- P. Cañizares and A. Carrero, Appl. Catal., A, 2003, 248, 227–237 CrossRef.
- G. W. Skeels, D. M. Chapman-Snyder and E. M. Flanigen, US5100644, 1992 Search PubMed.
- C. W. Jones, S.-J. Hwang, T. Okubo and M. E. Davis, Chem. Mater., 2001, 13, 1041–1050 CrossRef CAS.
- C.-Y. Chen and S. I. Zones, in Zeolites and Catalysis, Wiley-VCH, 2010, pp. 155–170 Search PubMed.
- C. Y. Chen and S. I. Zones, US64689501, 2002 Search PubMed.
- C. Y. Chen and S. I. Zones, Stud. Surf. Sci. Catal., 2001, 135, 211–218 CrossRef.
- C. W. Jones, S. I. Zones and M. E. Davis, Microporous Mesoporous Mater., 1999, 28, 471–481 CrossRef CAS.
- H. T. T. Tong and H. Koller, Microporous Mesoporous Mater., 2012, 148, 80–87 CrossRef CAS.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef CAS.
- L. Tosheva, N. Mahé and V. Valtchev, Stud. Surf. Sci. Catal., 2007, 170, 616–621 CrossRef.
- F. Gao, M. Jaber, K. Bozhilov, A. Vicente, C. Fernandez and V. Valtchev, J. Am. Chem. Soc., 2009, 131, 16580–16586 CrossRef CAS.
- D. A. Young, US3644200, 1972 Search PubMed.
- R. W. Chorley and P. W. Lednor, Adv. Mater., 1991, 3, 474–485 CrossRef CAS.
- S. Ernst, M. Hartmann, S. Sauerbeck and T. Bongers, Appl. Catal., A, 2000, 200, 117–123 CrossRef CAS.
- T. Yashima, K. Sato, T. Hayasaka and N. Hara, J. Catal., 1972, 26, 303–312 CrossRef CAS.
- A. Corma, V. Fornés, R. M. Martín-Aranda, H. García and J. Primo, Appl. Catal., 1990, 59, 237–248 CrossRef CAS.
- A. Corma, R. M. Martín-Aranda and F. Sánchez, J. Catal., 1990, 126, 192–198 CrossRef CAS.
- K. Yamamoto, Y. Sakata, Y. Nohara, Y. Takahashi and T. Tatsumi, Science, 2003, 300, 470–472 CrossRef CAS.
- A.-J. Han, H.-Y. He, J. Guo, H. Yu, Y.-F. Huang and Y.-C. Long, Microporous Mesoporous Mater., 2005, 79, 177–184 CrossRef CAS.
- K. D. Hammond, F. Dogan, G. A. Tompsett, V. Agarwal, W. C. Conner, C. P. Grey and S. M. Auerbach, J. Am. Chem. Soc., 2008, 130, 14912–14913 CrossRef CAS.
- M. Srasra, S. Delsarte and E. Gaigneaux, Top. Catal., 2009, 52, 1541–1548 CrossRef CAS.
- V. Agarwal, G. W. Huber, W. C. Conner Jr. and S. M. Auerbach, J. Catal., 2010, 269, 53–63 CrossRef CAS.
- M. Srasra, S. p. Delsarte and E. M. Gaigneaux, J. Phys. Chem. C, 2010, 114, 4527–4535 CAS.
- K. D. Hammond, M. Gharibeh, G. A. Tompsett, F. Dogan, A. V. Brown, C. P. Grey, S. M. Auerbach and W. C. Conner, Chem. Mater., 2009, 22, 130–142 CrossRef.
- X. Liu, U. Ravon and A. Tuel, Chem. Mater., 2011, 23, 5052–5057 CrossRef CAS.
- X. Liu, U. Ravon and A. Tuel, Angew. Chem., Int. Ed., 2011, 50, 5900–5903 CrossRef CAS.
- D. Verboekend, G. Vilé and J. Pérez-Ramírez, Adv. Funct. Mater., 2012, 22, 916–928 CrossRef CAS.
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev. Sci. Eng., 2003, 45, 297–319 CAS.
- G. T. Kerr, J. Phys. Chem., 1967, 71, 4155–4156 CrossRef CAS.
- G. T. Kerr, J. Catal., 1969, 15, 200–204 CrossRef CAS.
- U. Lohse, H. Stach, H. Thamm, W. Schirmer, A. A. Isirikjan, N. I. Regent and M. M. Dubinin, Z. Anorg. Allg. Chem., 1980, 460, 179–190 CrossRef CAS.
- J. Lynch, F. Raatz and P. Dufresne, Zeolites, 1987, 7, 333–340 CrossRef CAS.
- A. H. Janssen, A. J. Koster and K. P. de Jong, J. Phys. Chem. B, 2002, 106, 11905–11909 CrossRef CAS.
- W. Lutz, D. Enke, W.-D. Einicke, D. Täschner and R. Kurzhals, Z. Anorg. Allg. Chem., 2010, 636, 2532–2534 CrossRef CAS.
- P. Kortunov, S. Vasenkov, J. Kärger, R. Valiullin, P. Gottschalk, M. Fé Elía, M. Perez, M. Stöcker, B. Drescher, G. McElhiney, C. Berger, R. Gläser and J. Weitkamp, J. Am. Chem. Soc., 2005, 127, 13055–13059 CrossRef CAS.
- D. A. Cooper, T. W. Hastings and E. P. Hertzenberg, US5601798, 1997 Search PubMed.
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- D. Verboekend and J. Perez-Ramirez, Catal. Sci. Technol., 2011, 1, 879–890 CAS.
- J. Pérez-Ramírez, S. Mitchell, D. Verboekend, M. Milina, N.-L. Michels, F. Krumeich, N. Marti and M. Erdmann, ChemCatChem, 2011, 3, 1731–1734 CrossRef.
- V. Valtchev, E. Balanzat, V. Mavrodinova, I. Diaz, J. El Fallah and J.-M. Goupil, J. Am. Chem. Soc., 2011, 133, 18950–18956 CrossRef CAS.
- W. J. Roth and J. Čejka, Catal. Sci. Technol., 2011, 1, 43–53 CAS.
- K. Varoon, X. Zhang, B. Elyassi, D. D. Brewer, M. Gettel, S. Kumar, J. A. Lee, S. Maheshwari, A. Mittal, C.-Y. Sung, M. Cococcioni, L. F. Francis, A. V. McCormick, K. A. Mkhoyan and M. Tsapatsis, Science, 2011, 334, 72–75 CrossRef CAS.
- W. J. Roth, O. V. Shvets, M. Shamzhy, P. Chlubná, M. Kubü, P. Nachtigall and J. Čejka, J. Am. Chem. Soc., 2011, 133, 6130–6133 CrossRef CAS.
- T. Wakihara, K. Sato, S. Inagaki, J. Tatami, K. Komeya, T. Meguro and Y. Kubota, ACS Appl. Mater. Interfaces, 2010, 2, 2715–2718 CAS.
- T. Wakihara, R. Ichikawa, J. Tatami, A. Endo, K. Yoshida, Y. Sasaki, K. Komeya and T. Meguro, Cryst. Growth Des., 2011, 11, 955–958 CAS.
- V. Valtchev, S. Mintova and L. Konstantinov, Zeolites, 1995, 15, 679–683 CrossRef CAS.
- S. Mintova, V. Valtchev and L. Konstantinov, Zeolites, 1996, 17, 462–465 CrossRef CAS.
- L. T. Tosheva, J. P. Sterte and V. P. Valtchev, US6569400, 2003 Search PubMed.
- V. Valtchev, J. Mater. Chem., 2002, 12, 1914–1918 RSC.
- B. Zhang, S. A. Davis, N. H. Mendelson and S. Mann, Chem. Commun., 2000, 781–782 RSC.
- B. Zhang, S. A. Davis and S. Mann, Chem. Mater., 2002, 14, 1369–1375 CrossRef CAS.
- L. Tosheva, V. Valtchev and J. Sterte, Microporous Mesoporous Mater., 2000, 35–36, 621–629 CrossRef CAS.
- D. A. Woodcock, P. Lightfoot, L. A. Villaescusa, M.-J. Díaz-Cabañas, M. A. Camblor and D. Engberg, Chem. Mater., 1999, 11, 2508–2514 CrossRef CAS.
- M. M. Martínez-Iñesta and R. F. Lobo, J. Phys. Chem. B, 2005, 109, 9389–9396 CrossRef.
- M. Amri and R. I. Walton, Chem. Mater., 2009, 21, 3380–3390 CrossRef CAS.
- M. Amri, G. J. Clarkson and R. I. Walton, J. Phys. Chem. C, 2010, 114, 6726–6733 CAS.
- C. Zenonos, G. Sankar, F. Cora, D. W. Lewis, Q. A. Pankhurst, C. R. A. Catlow and J. M. Thomas, Phys. Chem. Chem. Phys., 2002, 4, 5421–5429 RSC.
- A. Buchholz, W. Wang, M. Xu, A. Arnold and M. Hunger, Microporous Mesoporous Mater., 2002, 56, 267–278 CrossRef CAS.
- B. H. Wouters, T. Chen and P. J. Grobet, J. Phys. Chem. B, 2001, 105, 1135–1139 CrossRef CAS.
- E. Brunner, H. Ernst, D. Freude, M. Hunger, C. B. Krause, D. Prager, W. Reschetilowski, W. Schwieger and K. H. Bergk, Zeolites, 1989, 9, 282–286 CrossRef CAS.
- H. Ren and F. Xin, Catal. Commun., 2006, 7, 848–854 CrossRef CAS.
- R. W. Broach, S. T. Wilson and R. M. Kirchner, Microporous Mesoporous Mater., 2003, 57, 211–214 CrossRef CAS.
- M. Afeworki, G. Cao, D. L. Dorset, K. G. Strohmaier and G. J. Kennedy, Microporous Mesoporous Mater., 2007, 103, 216–224 CrossRef CAS.
- S. Antonijevic, S. E. Ashbrook, S. Biedasek, R. I. Walton, S. Wimperis and H. Yang, J. Am. Chem. Soc., 2006, 128, 8054–8062 CrossRef CAS.
- A. Meden, C. Baerlocher and L. B. McCusker, Microporous. Mater., 1997, 11, 247–251 CrossRef.
- J. P. Lourenço, M. F. Ribeiro, F. R. Ribeiro, J. Rocha, Z. Gabelica and E. G. Derouane, Microporous. Mater., 1995, 4, 445–453 CrossRef.
- B. Zibrowius and U. Lohse, Solid State Nucl. Magn. Reson., 1992, 1, 137–148 CrossRef CAS.
- M. B. Kenny, K. S. W. Sing and C. R. Theocharis, J. Chem. Soc., Chem. Commun., 1991, 974–975 RSC.
- F. Corà and C. R. A. Catlow, J. Phys. Chem. B, 2003, 107, 11861–11865 CrossRef.
- D. R. Pyke, P. Whitney and H. Houghton, Appl. Catal., 1985, 18, 173–190 CrossRef CAS.
- A. G. Arévalo-Hidalgo, J. A. Santana, R. Fu, Y. Ishikawa and A. J. Hernández-Maldonado, Microporous Mesoporous Mater., 2010, 130, 142–153 CrossRef.
- M. E. Rivera-Ramos, G. J. Ruiz-Mercado and A. J. Hernández-Maldonado, Ind. Eng. Chem. Res., 2008, 47, 5602–5610 CrossRef CAS.
- A. G. Arévalo-Hidalgo, N. E. Almodóvar-Arbelo and A. J. Hernández-Maldonado, Ind. Eng. Chem. Res., 2011, 50, 10259–10269 CrossRef.
- S. Li, J. L. Falconer and R. D. Noble, J. Membr. Sci., 2004, 241, 121–135 CrossRef CAS.
- T. L. Chew, A. L. Ahmad and S. Bhatia, Chem. Eng. J., 2011, 171, 1053–1059 CrossRef CAS.
- S. Li, M. A. Carreon, Y. Zhang, H. H. Funke, R. D. Noble and J. L. Falconer, J. Membr. Sci., 2010, 352, 7–13 CrossRef CAS.
- M. Hong, S. Li, H. F. Funke, J. L. Falconer and R. D. Noble, Microporous Mesoporous Mater., 2007, 106, 140–146 CrossRef CAS.
- C. G. Saxton, A. Kruth, M. Castro, P. A. Wright and R. F. Howe, Microporous Mesoporous Mater., 2010, 129, 68–73 CrossRef CAS.
- J. Sanz, J. M. Campelo and J. M. Marinas, J. Catal., 1991, 130, 642–652 CrossRef CAS.
- J. M. Campelo, A. Garcia, D. Luna and J. M. Marinas, J. Catal., 1986, 102, 299–308 CrossRef CAS.
- Z. Nawaz and F. Wei, J. Ind. Eng. chem., 2010, 16, 774–784 CrossRef CAS.
- Z. Nawaz, X. Tang, Q. Zhang, D. Wang and W. Fei, Catal. Commun., 2009, 10, 1925–1930 CrossRef CAS.
- Z. Nawaz, X. Tang, Y. Wang and F. Wei, Ind. Eng. Chem. Res., 2009, 49, 1274–1280 CrossRef.
- Z. Nawaz and W. Fei, Ind. Eng. Chem. Res., 2009, 48, 7442–7447 CrossRef CAS.
- N. Z. Logar, N. N. Tušar, G. Mali, M. Mazaj, I. Arčon, D. Arčon, A. Rečnik, A. Ristić and V. Kaučič, Microporous Mesoporous Mater., 2006, 96, 386–395 CrossRef CAS.
- N. Z. Logar, N. N. Tušar, A. Ristić, G. Mali, M. Mazaj and V. Kaučič, in Ordered Porous Solids, ed. V. Valtchev, S. Mintova and M. Tsapatsis, Elsevier, Amsterdam, 2009, pp. 101–126 Search PubMed.
- Z. Zhu, M. Hartmann and L. Kevan, Chem. Mater., 2000, 12, 2781–2787 CrossRef CAS.
- D. R. Dubois, D. L. Obrzut, J. Liu, J. Thundimadathil, P. M. Adekkanattu, J. A. Guin, A. Punnoose and M. S. Seehra, Fuel Process. Technol., 2003, 83, 203–218 CrossRef CAS.
- L. Xu, Z. Liu, A. Du, Y. Wei and Z. Sun, Stud. Surf. Sci. Catal., 2004, 147, 445–450 CrossRef CAS.
- D. Obrzut, P. Adekkanattu, J. Thundimadathil, J. Liu, D. Dubois and J. Guin, React. Kinet. Catal. Lett., 2003, 80, 113–121 CrossRef CAS.
- Y. Wei, G. Wang, Z. Liu, C. Sun and L. Xu, Stud. Surf. Sci. Catal., 2001, 135, 3822–3829 CAS.
- Y. Chen, Y. Wu, L. Tao, B. Dai, M. Yang, Z. Chen and X. Zhu, J. Ind. Eng. Chem., 2010, 16, 717–722 CrossRef CAS.
- M. M. M. Abd El-Wahab, A. E.-A. A. Said and S. S. Al-Shihry, Monatsh. Chem., 2004, 135, 357–370 CrossRef CAS.
- S. Hinokuma, M. Okamoto, E. Ando, K. Ikeue and M. Machida, Catal. Today, 2011, 175, 593–597 CrossRef CAS.
- S. Hinokuma, K. Murakami, K. Uemura, M. Matsuda, K. Ikeue, N. Tsukahara and M. Machida, Top. Catal., 2009, 52, 2108–2111 CrossRef CAS.
- Y. Neinska, C. Minchev, L. Kosova and V. Kanazirev, Stud. Surf. Sci. Catal., 1995, 94, 262–269 CrossRef CAS.
- Y. Neinska, C. Minchev, R. Dimitrova, N. Micheva, V. Minkov and V. Kanazirev, Stud. Surf. Sci. Catal., 1994, 84, 989–996 CrossRef CAS.
- R. Dimitrova, Y. Neinska, M. Mihályi, G. Pal-Borbély and M. Spassova, Appl. Catal., A, 2004, 266, 123–127 CrossRef CAS.
- M.-A. Djieugoue, A. M. Prakash and L. Kevan, J. Phys. Chem. B, 1999, 103, 804–811 CrossRef CAS.
- S. R. Venna and M. A. Carreon, Langmuir, 2011, 27, 2888–2894 CrossRef CAS.
- K. Wang, G. Cao, G. J. Kennedy, M. Afeworki, R. E. Bare and R. B. Hall, J. Phys. Chem. C, 2011, 115, 18611–18617 CAS.
- J. L. Brinen and M. G. White, J. Catal., 1990, 124, 133–147 CrossRef CAS.
- V. R. Choudhary, M. Y. Pandit and S. D. Sansare, J. Chem. Soc., Chem. Commun., 1989, 1343–1344 RSC.
- C. R. Theocharis, M. R. Gelsthorpe and D. Yeates, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2641–2646 RSC.
- H. X. Li and M. E. Davis, J. Phys. Chem., 1992, 96, 331–334 CrossRef CAS.
- B. Herreros and J. Klinowski, J. Phys. Chem., 1995, 99, 9514–9518 CrossRef CAS.
- A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS.
- Y.-J. Zhang, T. Liu, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2009, 132, 912–913 CrossRef.
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368–12369 CrossRef CAS.
- Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4635–4637 CrossRef CAS.
- J. S. Costa, P. Gamez, C. A. Black, O. Roubeau, S. J. Teat and J. Reedijk, Eur. J. Inorg. Chem., 2008, 2008, 1551–1554 CrossRef.
- K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef CAS.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354–14355 CrossRef CAS.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS.
- O. K. Farha, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2008, 47, 10223–10225 CrossRef CAS.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2007, 130, 806–807 CrossRef.
- T. Gadzikwa, G. Lu, C. L. Stern, S. R. Wilson, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2008, 5493–5495 RSC.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS.
- Z. Wang and S. M. Cohen, Angew. Chem., Int. Ed., 2008, 47, 4699–4702 CrossRef CAS.
- S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H.-C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS.
- L. Ma and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 3637–3640 CrossRef CAS.
- T. Gadzikwa, O. K. Farha, K. L. Mulfort, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2009, 3720–3722 RSC.
- D. J. Lun, G. I. N. Waterhouse and S. G. Telfer, J. Am. Chem. Soc., 2011, 133, 5806–5809 CrossRef CAS.
- S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456–8460 CrossRef CAS.
- M. J. Ingleson, J. Perez Barrio, J.-B. Guilbaud, Y. Z. Khimyak and M. J. Rosseinsky, Chem. Commun., 2008, 2680–2682 RSC.
- S. Uchida, R. Kawamoto, H. Tagami, Y. Nakagawa and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 12370–12376 CrossRef CAS.
- J.-P. Zhang and S. Kitagawa, J. Am. Chem. Soc., 2008, 130, 907–917 CrossRef CAS.
- D. Esken, S. Turner, O. I. Lebedev, G. Van Tendeloo and R. A. Fischer, Chem. Mater., 2010, 22, 6393–6401 CrossRef CAS.
- S. Hermes, F. Schroder, S. Amirjalayer, R. Schmid and R. A. Fischer, J. Mater. Chem., 2006, 16, 2464–2472 RSC.
- X.-C. Huang, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS.
- J. Zhang, Y. Tang, L. Weng and M. Ouyang, Nano Lett., 2009, 9, 4061–4065 CrossRef CAS.
- X. Liu, A. Wang, X. Yang, T. Zhang, C.-Y. Mou, D.-S. Su and J. Li, Chem. Mater., 2008, 21, 410–418 CrossRef.
- H.-L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 CrossRef CAS.
- O. Shekhah, J. Liu, R. A. Fischer and C. Woll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- D. Zacher, O. Shekhah, C. Woll and R. A. Fischer, Chem. Soc. Rev., 2009, 38, 1418–1429 RSC.
- O. Shekhah, H. K. Arslan, K. Chen, M. Schmittel, R. Maul, W. Wenzel and C. Woll, Chem. Commun., 2011, 47, 11210–11212 RSC.
- M. Meilikhov, K. Yusenko and R. A. Fischer, Dalton Trans., 2010, 39, 10990–10999 RSC.
- J. E. Halls, A. Hernan-Gomez, A. D. Burrows and F. Marken, Dalton Trans., 2012, 41, 1475–1480 RSC.
- C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS.
- A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2011, 133, 13252–13255 CrossRef CAS.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS.
- C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 9492–9493 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |