DOI:
10.1039/C2CS35069F
(Critical Review)
Chem. Soc. Rev., 2013,
42, 387-410
Hybrid materials based on lanthanide organic complexes: a review
Received 9th March 2012
First published on 13th September 2012
Abstract
A great deal of research has been carried out on lanthanide organic complex-based organic–inorganic hybrid materials in the last decade. This critical review begins with a formulation of the fundamentals of lanthanide organic complex luminescence, and presents various current designed ideas, synthetic routes, luminescence behaviors and potentials of the latest advanced (a) sol–gel materials, (b) mesoporous materials, (c) titania materials, (d) ionic liquids and ionogels, (e) polymers, and (f) bifunctional magnetic–optical composites based on lanthanide organic complexes. Finally, challenges and opportunities for further improvement of organic–inorganic hybrid luminescent materials based on lanthanide organic complexes will be discussed.
 Jing Feng | Dr Jing Feng received her BSc in Chemistry from Inner Mongolia University in 2004 and then joined the group of Professor Hongjie Zhang at Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (CAS), where she earned her PhD in inorganic chemistry in 2009. She is working as an assistant professor under the direction of Prof. Zhang at Changchun Institute of Applied Chemistry, CAS. Her current research interests include lanthanide organic–inorganic hybrid materials and upconversion luminescence nanomaterials for applications in drug release and fluorescence imaging. |
 Hongjie Zhang | Prof. Hongjie Zhang received his MSc from Changchun Institute of Applied Chemistry, CAS in 1985. Then, he worked as an assistant professor at Changchun Institute of Applied Chemistry, CAS from 1985–1989. He then studied at Université de Bordeaux I, Laboratoire de Chimie du Solide du CNRS (France), where he received his PhD in Solid State Chemistry and Material Sciences in 1993. He joined Changchun Institute of Applied Chemistry, CAS, as a professor in 1994. His current research interests involve lanthanide organic–inorganic hybrid materials, electroluminescent devices, and functional nanomaterials. He has published over 350 papers in peer-reviewed international journals. |
1. Introduction
Since Weissman discovered that excitation may be accomplished in the lanthanide (Ln) complex under suitable conditions, through light absorption by other constituents of the lanthanide complex, with subsequent transfer of energy to the lanthanide ion,1 numerous lanthanide complexes have been studied in detail.2–4 Formation of a complex between a lanthanide ion and certain organic ligands has a double beneficial effect, both protecting metal ions from vibrational coupling and increasing the light absorption cross section by “antenna effects”.5–8 Typically, the coordination number requirement of a lanthanide ion is 8 or 9, resulting from the relatively large atomic radius of lanthanide. The lanthanide complex can emit a characteristic luminescence based on a specific lanthanide ion in the visible (400–800 nm) or near-infrared (NIR) (800–1700 nm) spectral regions. Eu3+ (red light) and Tb3+ (green light) ions are typical ions that emit visible light. In addition, Sm3+ emits orange light, Tm3+ blue light and Dy3+ white or near white light in the visible region. Lanthanide ions, such as Pr3+, Nd3+, Sm3+, Dy3+, Ho3+, Er3+, Tm3+, and Yb3+ show NIR emissions. That is, some lanthanide ions are pure visible or NIR emitting ions, and some could emit both visible and NIR light. In recent years, research toward NIR emitting lanthanide systems has become increasingly popular. Interest in the photophysical properties of these NIR lanthanide ions mainly stems from the following aspects. For instance, the Yb3+ ion emission at about 1000 nm has potential in various analytical and chemosensor applications, since biological tissues and fluids (e.g. blood) are relatively transparent; the Nd3+ ion has two important emissions occurring in the NIR region, one at 1.06 μm can be used in laser systems, and the other at 1.3 μm matches one of the telecommunication windows for amplification commonly used for long-distance communication. Furthermore, the corresponding Er, Ho and Tm emissions at around 1.5 μm match the other telecommunication windows for amplification. Besides the above lanthanides, La3+, Gd3+, Lu3+, and Y3+ complexes only show the luminescence of the ligands. For the complexation of lanthanide ions, the commonly used ligands are β-diketones (1,3-diketones), aromatic carboxylic acids, macrocyclic ligands and non-charged ligands (such as 1,10-phenanthroline, bathophenanthroline, 2,2′-bipyridine and triphenylphosphine oxide). However, as we know, lanthanide complexes have been excluded from practical applications, mainly due to their poor thermal stability and low mechanical strength.9,10 One feasible solution is to introduce the complexes in a stable rigid matrix, for example, a silica- or titania-based material. Incorporation of lanthanide complexes into these matrices has not only improved the photo and thermal stabilities of the complexes, but also avoids self-quenching, due to the concentration effect. Lanthanide complex-based organic–inorganic hybrid materials have attracted much attention because of the genuine expectation that their properties will be altered by changing the synthetic approach, the host materials and the lanthanide ions. Up to now, lanthanide complexes have been successfully incorporated into sol–gel, mesoporous silica, titania, ionic liquids, polymers, mixed (such as silica-polymer mixed matrix) matrices, LB films, liquid crystals, zeolites, and functional hosts (such as magnetic silica nanospheres). Previously, related reviews have been reported by Bünzli,11,12 Binnemans,13–16 Carlos,17 Sanchez,18–20 and Okamoto.21 More recently, much interesting and significant research has appeared in this field. The present review summarizes various current designed ideas, synthetic routes, luminescence behaviors and potential of the latest advances in selective types of such hybrids, including lanthanide complex-based sol–gel materials, mesoporous materials, titania materials, ionic liquids (or ionogels), polymers, and bifunctional magnetic–optical composites, especially the latest developments in this field. The main point differentiating this review from previous reviews is: the lanthanide organic complex is the basic unit in the hybrid materials mentioned. The present review gives an overview of such kinds of hybrid materials and presents representative examples for the different kinds of hybrid materials, based on lanthanide organic complexes.2. Energy transfer mechanism in the lanthanide organic complex
The commonly accepted mechanism of lanthanide organic complex luminescence is the energy transfer from the organic ligands to the lanthanide ion, the so-called “indirect excitation” or “antenna effects”. The energy transfer process (Fig. 1) is as follows: (1) the lanthanide complex is excited from the singlet S0 ground state to the singlet S1 excited state by absorbing the energy; (2) the energy of the S1 excited state is transferred to the triplet excited state of the ligands through intersystem crossing; (3) the intramolecular energy transfer from the triplet state of the ligands to one of the excited 4f states of the Ln3+ ions; (4) lanthanide luminescence is produced by intra-4f transitions within the ion.22 Dexter suggested that the suitability of the energy difference (ΔE) between the resonance level of the Ln3+ ion and the triplet state of the organic ligand is a critical factor for efficient energy transfer.23 The energy transfer rate constant decreases due to the diminution in the overlap between the donor and the acceptor when the energy difference is too big. And energy back-transfer will occur from the Ln3+ ion to the resonance level of the triplet state of the ligand when the reverse condition happens. Generally, the energy gap must be at least 10 kBT (2000 cm−1) to effectively preclude energy back-transfer.24 In order to investigate the energy transfer from the organic ligands to the Ln3+ ion, the information about the triplet energy level of the organic ligands and the accepting energy levels of the Ln3+ ion should be cleared up in advance. The triplet energy level of the ligand could be obtained through the phosphorescence spectrum of the corresponding gadolinium (Gd) complex, since Gd has no energy levels below 312 nm (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1) and it cannot accept energy from the ligand. After excitation, the Gd complex emits the singlet fluorescence of the ligand, or the triplet phosphorescence, or both. The phosphorescence spectrum of the Gd complex could be obtained at room temperature after the solutions had been degassed (removal of oxygen which can efficiently quench the phosphorescence) with dry nitrogen or at 77 K without being degassed.25–27 Thus, the triplet-state energy level of the ligand could be measured and calculated from the shortest-wavelength phosphorescence band of the phosphorescence spectrum for the corresponding Gd complex. Then, the energy transfer from the suitable organic ligand to the lanthanide ions would provide an effective way to excite the high quantum efficiency, long-lived, sharply spiked emission. These characteristics make the lanthanide complexes particularly valuable in sensors, displays and optical fibers, and enable them to be employed for applications such as fluoroimmunoassay and fluorescence microscopy. Some reviews and books have expounded lanthanide photophysics and the luminescence of lanthanide complexes.11,12,28,29
000 cm−1) and it cannot accept energy from the ligand. After excitation, the Gd complex emits the singlet fluorescence of the ligand, or the triplet phosphorescence, or both. The phosphorescence spectrum of the Gd complex could be obtained at room temperature after the solutions had been degassed (removal of oxygen which can efficiently quench the phosphorescence) with dry nitrogen or at 77 K without being degassed.25–27 Thus, the triplet-state energy level of the ligand could be measured and calculated from the shortest-wavelength phosphorescence band of the phosphorescence spectrum for the corresponding Gd complex. Then, the energy transfer from the suitable organic ligand to the lanthanide ions would provide an effective way to excite the high quantum efficiency, long-lived, sharply spiked emission. These characteristics make the lanthanide complexes particularly valuable in sensors, displays and optical fibers, and enable them to be employed for applications such as fluoroimmunoassay and fluorescence microscopy. Some reviews and books have expounded lanthanide photophysics and the luminescence of lanthanide complexes.11,12,28,29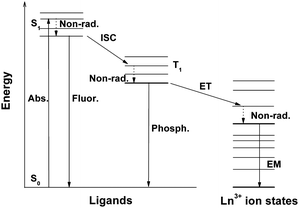 |
| | Fig. 1 Simplified energy diagram of the lanthanide organic complex system. Abs. = absorption, Fluor. = fluorescence, Phosph. = phosphorescence, EM = lanthanide (Ln3+) ion emission, ISC = intersystem crossing, ET = energy transfer, S = singlet, T = triplet, Non-rad. = nonradiative transitions. | |
3. Sol–gel hybrid materials based on the lanthanide organic complex
Sol–gel derived hybrid materials exhibits great potential in photonics, as they can combine the optical qualities of silica, its thermal stability and mechanical strength, together with the optical characteristics of active organic molecules. The starting reagent of silicate sol–gel materials is Si(OR)4, where R represents different kinds of organic groups, such as ethyl and methyl. Hydrolysis and condensation of Si(OR)4 create the Si–O–Si framework. The hydrolysis could be acid- or base-catalyzed. The typical scheme for hydrolysis and condensation of Si(OR)4 is as follows:Hydrolysis:
| Si(OR)4 + H2O → (HO)Si(OR)3 + ROH |
| (HO)Si(OR)3 + H2O → (HO)2Si(OR)2 + ROH |
| (HO)2Si(OR)2 + H2O → (HO)3Si(OR) + ROH |
| (HO)3Si(OR) + H2O → (HO)4Si + ROH |
Condensation:
A: ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OR + HO–Si → Si–O–Si+ ROH Si–OR + HO–Si → Si–O–Si+ ROH |
| B: Si–OH + HO–Si → Si–O–Si+ HOH |
In general, the sol–gel process involves the formation of the homogeneous precursor solution, sol, gel, aging, desiccation, and densification. The sol–gel method is superior in mild reaction conditions, and is feasible for controlling the composition of various shapes and convenient preparation routes. Meanwhile, the method offers the possibility of tailoring the final properties of the materials, for instance the refractive index, phonon energy, and transparency through suitable choice of organic and inorganic components.30–34 Sol–gel hybrid materials composed of both inorganic and organic components enable both inorganic and organic dopants to be incorporated with relatively high stability. Therefore, sol–gel materials are potential hosts for luminescent lanthanide complexes. At present, methods for making sol–gel hybrid materials based on lanthanide organic complexes involve impregnation, doping and chemical immobilization. The main shortcomings of the impregnation approach are the clustering and instability of lanthanide complexes in the silica host, since the lanthanide complexes just penetrate the interspaces of the silica framework. The doping approach includes the conventional sol–gel and in situ technique. The so-called conventional sol–gel technique is used to dissolve the preformed lanthanide complexes in a solvent that is compatible with silicon alkoxide sol–gel preparative techniques. The weak point is that the chelates cannot be fully homogeneously dispersed into a sol–gel glass to obtain a transparent monolithic composite, due to their low solubility and/or stability in the sol–gel precursor solutions. For the in situ technique, the organic ligands and lanthanide ion are mixed together into sol–gel precursor solutions. Thus, all the compositions are mixed at a molecular level, and the lanthanide complex is formed during the sol–gel process.35–38 By using the above mentioned methods, the lanthanide complexes and the sol–gel matrix were connected through weak interactions, such as van der Waals contacts, hydrogen bonding, or electrostatic forces.39,40 Using the chemical immobilization method, the lanthanide ion, the functionalized ligands (as the linkage), and (or not) other organic ligands were mixed in the precursor solution. Together with the in situ technique, the lanthanide organic complex was synthesized in situ through the sol–gel process and covalently bonded to the silica matrix.41–47 Therefore, the problems of phase separation, inhomogeneous dispersion, and optical quenching of the dopants could be avoided effectively. In general, lanthanide complex-based sol–gel hybrid materials were achieved under weak acidic or neutral hydrolysis and condensation. That is because lanthanide complexes are unstable and easily form lanthanide hydroxide under basic conditions. In recent years, the impregnation method and doping of preformed lanthanide complexes into the silica host were less used because of their inherent defects.48 Great efforts have been devoted to preparing the lanthanide complex-based sol–gel hybrid materials by the in situ technique and the chemical immobilization method. Different shapes (such as films, fibers and monoliths) of the sol–gel hybrid materials could be fabricated easily to satisfy the demands of practical applications.
3.1 In situ technique
As mentioned above, by using the in situ technique, all the compositions are mixed at a molecular level. The concentration quenching of lanthanide complexes could be reduced. In reality, many lanthanide complexes have been successfully synthesized into the hybrid materials in situ and a great number of desirable results such as good thermal stability, good mechanical resistance and excellent colorimetric purity have been obtained. For instance, the synthesis of the NIR luminescent lanthanide (Er3+, Nd3+, and Yb3+) complexes—with dibenzoylmethane (DBM) as the primary ligand and 1,10-phenanthroline (phen) as the synergic agent—and the synthesis of a homogeneous, transparent hybrid gel containing the corresponding complex (LnDP gel) via an in situ approach were reported.40,49 The radiative properties (such as radiative transition rates, emission cross sections, fluorescence branching ratios, and radiative lifetime) of trivalent lanthanide ions in the host matrix were predicted from optical absorption measurements and by using the Judd–Ofelt theory.50,51 The large value of the parameter Ω2 for both ErDP and NdDP indicates the presence of covalent bonding between the central lanthanide ion and the surrounding ligands, as is reasonable. On the basis of the experimental data and Judd–Ofelt analysis, it could be concluded that the ErDP gel has the possibility to be developed as a tunable laser and a broad-band optical amplifier in the 1.54 μm wavelength region and the Nd3+ ion in the NdDP gel can be considered as a possible candidate for a solid state laser transition. Recently, some less studied lanthanide complex (such as dysprosium, holmium, and thulium complexes) doped sol–gel materials have been reported. Our group developed a series of holmium (Ho) and thulium (Tm) complex doped sol–gel hybrid materials.52 The materials all show characteristic near-infrared luminescence of the corresponding Ln3+ ions. The radiative properties of the Ho3+ ion in the Ho(tta)3bipy (tta = thenoyltrifluoroacetonate, bipy = 2,2′-bipyridine) doped gel are described by Judd–Ofelt theory. Three Judd–Ofelt intensity parameters are Ω2 = 3.70 × 10−20 cm2, Ω4 = 0.65 × 10−20 cm2, and Ω6 = 0.99 × 10−20 cm2. The radiative life of the 5F5 state has been calculated to be 5.39 ms. The experimental lifetime of the 5F5 state is much lower than the value calculated from the theory. This is owing to the nonradiative relaxation proceeding in the holmium complex doped gel in the presence of the O–H and C–H groups in the sol–gel-derived materials. NIR luminescent properties of the dysprosium complex doped xerogels with the same acetylacetone (acac) ligand and different neutral ligands (TPPO = triphenyl phosphine oxide; and phen = 1,10-phenanthroline) were also studied by our group.53 In the NIR region, the dysprosium complex doped xerogels show multiband emission of the Dy3+ ion (Fig. 2). Herein, the 1300 nm and 1520 nm emissions from the dysprosium complex doped xerogels match the working window of telecommunications. It is observed that TPPO could increase the emission intensity of the dysprosium complex, while phen gives a negative effect. This is caused by different energy transfer processes from different neutral ligands (TPPO and phen) to the Dy3+ ion (Fig. 3). The S1 energy level of TPPO (36400 cm−1) matches that of acac (∼29000 cm−1) and the 4F9/2 energy level of the Dy3+ ion (20830 cm−1), so the energy absorbed by TPPO could be transferred to acac or the Dy3+ ion directly and have a positive effect on the Dy3+ luminescence. However, the S1 (26320 cm−1) and T1 (∼23000 cm−1) energy levels of phen are lower than those of acac (T1 = 25300 cm−1). Thus, the energy transfer from phen to acac is impossible, and the reverse energy transfer occurred. Moreover, the energy difference between the T1 energy level of phen and the 4F9/2 energy level of the Dy3+ ion is not suitable for the efficient energy transfer from phen to Dy3+, resulting in a negative effect on the luminescence of the xerogel.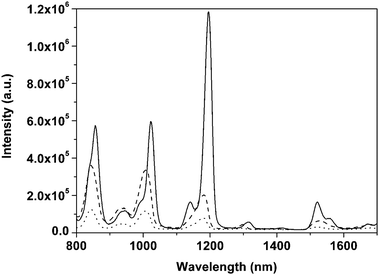 |
| | Fig. 2 NIR emission spectra of Dy(acac)3TPPO2 (solid line), Dy(acac)3phen (dashed line), and Dy(acac)3(H2O)2 (dotted line) doped xerogels. Reprinted with permission from ref. 53. Copyright 2009, Royal Society of Chemistry Publishing Company. | |
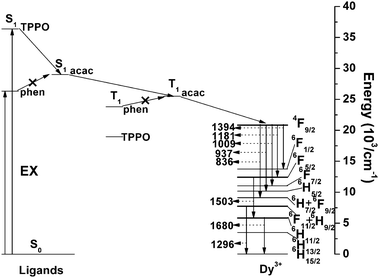 |
| | Fig. 3 Schematic energy level diagram and energy transfer process of the Dy–ligand system. EX, excitation; S1, first excited singlet state; T1, first excited triplet state. Reprinted with permission from ref. 53. Copyright 2009, Royal Society of Chemistry Publishing Company. | |
3.2 Chemical immobilization
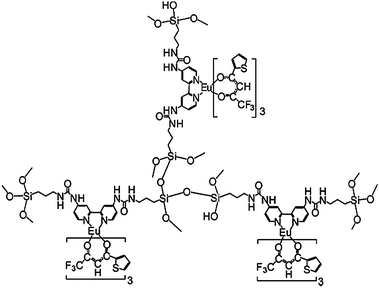
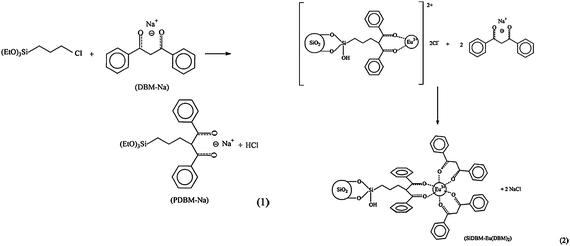
A major improvement in the luminescence behavior can be expected by immobilizing lanthanide complexes to the silica matrix. The previous work (LnDP gel) was continued by our group. The Ln(DBM)3phen (Ln = Er, Nd, Yb) complexes were covalently linked to the xerogels by a chelate ligand 5-(N,N-bis-3-(triethoxysilyl)propyl)ureyl-1,10-phenanthroline (phen-Si) in situ via a sol–gel method.54 In this case, no quenching effects were detected as the concentration of the Ln3+ ion (Ln/Si molar ratio) increased from 1/27 to 1/11. The radiative properties of the Nd3+ ion in the host were predicted from optical absorption measurements and by using Judd–Ofelt theory. The parameters Ωt (t = 2, 4 and 6) in this work have been calculated to be 94.88 × 10−20, 3.771 × 10−20 and 2.818 × 10−20 cm2, respectively. It is suggested that upon covalent bonding to the xerogel, the Ω2-parameter of the xerogel-bonded Nd complex(1/11) increased, when compared with the doped material of the Nd complex. The larger value for Ω2 in this work suggests that the DBM and phen-Si ligands bind more strongly with the Nd3+ ion and shield the Nd3+ ion from the water molecules.55 The Ω6 value decreases in this case, compared with that of the doped material of the Nd complex, suggesting that a more rigid material was obtained. The stimulated emission cross-section σp (one of the most important parameters for laser design) of the 4F3/2 → 4I11/2 fluorescence transition of the Nd3+ ion is 1.16 × 10−20 cm2, which is comparable to those shown by glasses used in solid state laser applications. Lin et al. prepared luminescent lanthanide-complex-bridged polysilsesquioxanes by sol–gel processing of a monomer with a large heterocyclic bridging group in the presence of lanthanide ions under acidic conditions, and further investigated the sensitization of Ln3+ ions by bridged polysilsequioxanes (Fig. 4).56 The monomer containing large organic bridging groups of a complex chemical structure, consisting of a heterocyclic ring integrated through flexible alkyl chains with three trimethoxysilyl groups. They found a relatively high quantum yield (30%) for Eu-containing material. In the above two works, one used two kinds of ligands (DBM and phen), and the other used only one kind of ligand (the large heterocyclic group). For the two kinds of ligands system, generally, the first ligand (such as DBM) is more important than the second ligand (the neutral ligand) to the luminescence of Ln3+. According to the literature,11,57–65 replacement of C–H bonds in a β-diketone ligand (the first ligand) with lower-energy C–F oscillators could decrease the vibration energy of the ligand and the energy loss caused by ligand vibration, thus enhancing the emission intensity of the lanthanide ion. Our group synthesized a fluorinated β-diketone ligand 4,4,5,5,5-pentafluoro-1-(2-naphthyl)-1,3-butanedione to further prepare the luminescent sol–gel hybrid materials.66 It is observed that the corresponding sol–gel hybrids show intense NIR luminescence. He et al. also suggested that the halogenation of ligands (porphyrin, acetate, phenanthroline) will further eliminate the C–H bonds in the surrounding environment and so enhance the emission efficiency.67 One of the prominent advantages of the chemical immobilization approach is increasing the concentration of the lanthanide complex in the silica matrix. To further elevate the concentration, a feasible method, using silylated organic ligands as the only siloxane network precursor could be performed. Li and co-workers reported organic–inorganic hybrid materials with a high loading of lanthanide 2,2′-bipyridine moieties, by using the silylated bipyridine as the only siloxane network precursor in the presence of Ln3+ ions or lanthanide complexes (Ln = Eu, Tb) (Fig. 5).68 They found a higher monochromatic purity upon the incorporation of Eu(TTA)3·2H2O instead of Eu3+ ions. The radiative lifetime of Eu3+ in the hybrid material incorporating Eu(TTA)3·2H2O (0.540 ms) is much longer than that in the pure complex Eu(TTA)3·2H2O (0.368 ms) and the hybrid material incorporating Eu3+ ions (0.459 ms). This suggests that the tris(2-thenoyltrifluoroacetonato)europium(III) dihydrate complex was bound to the 2,2-bipyridine groups on the silica gel and the coordinated water was expelled. When the europium ions in the hybrid material were replaced by terbium ions, the intense green photoluminescence can be observed. |
| | Fig. 4 Predicted structure of the bridged polysilsesquioxane material (Ln = Eu, Tb). Reprinted with permission from ref. 56. Copyright 2008, Wiley Publishing Company. | |
 |
| | Fig. 5 Predicted structure of the organic–inorganic hybrid material derived from a urea-based bis-silylated bipyridine. Reprinted with permission from ref. 68. Copyright 2009, Wiley Publishing Company. | |
Lanthanide complex grafted sol–gel hybrid materials could also be achieved by functionalized β-diketonate ligands. o-Hydroxydibenzoylmethane (DBM-OH) and 3-isocyanatepropyltriethoxysilane were used to synthesize the functionalized β-diketonate ligand (DBM-Si).69,70 A series of transparent, homogeneous, and colorless hybrid materials were obtained by covalent bonding with the binary lanthanide complex Ln(DBM-OH)3 (Ln = Nd, Er, Yb, Sm, and Eu) via DBM-Si (Fig. 6). The calculated radiative lifetimes for Nd (1.63 ms) and Er hybrids (14.56 ms) are comparable to those of Nd-doped or Er-doped glass systems, which have been used to make lasers and optical amplifiers. Also, the values of radiative lifetimes are much higher than those in previous work, which indicates that Nd and Er hybrids are efficient for laser pumping. Wang et al. grafted the tetrakis complex [(C2H5)4N][Eu(DBM)4] instead of the tris complex Eu(DBM)3 to the silica matrix through the modified DBM-OH.71 In this case, Eu3+ ions are well protected by the DBM ligands. Thus, the vibrational coupling from the coordinated H2O molecules could be avoided and the luminescence properties of the resulting material could be distinctly improved. Moreover, the vinyltriethoxysilane (VTES)/tetraethoxysilane (TEOS) composite system was considered, but not the pure TEOS system. At the optimal molar ratio of VTES to TEOS, the luminescence intensity of the VTES/TEOS composite hybrid material is enhanced by 3.3 and 2.4 times compared with the TEOS-derived hybrid material and pure [(C2H5)4N][Eu(DBM)4], respectively. It indicated that using vinyl-modified silicates as matrices could be a promising method to improve the luminescence properties of lanthanide complexes. The other reactive β-diketonate bridge (trifluoroacetylacetone) has been achieved through the extraction reaction of hydrogen atoms of the methylene group by Yan et al.72 They also developed a series of sol–gel hybrids by means of the functionalized 1-(2-naphthoyl)-3,3,3-trifluoroacetonate as the linkage. Besides, the other precursors, which were synthesized through the addition polymerization of the monomer 4-vinylpyridine and methacrylic acid, were added. Luminescent rare earth molecular-based polymeric hybrid materials with chemical bonds, containing the long organic carbon chains and organic network (Si–O–Si) through the sol–gel process, were obtained. In this case, lanthanide ions could coordinate with the β-diketonate ligand and the carboxyl group or the N atom from the polymer precursors.
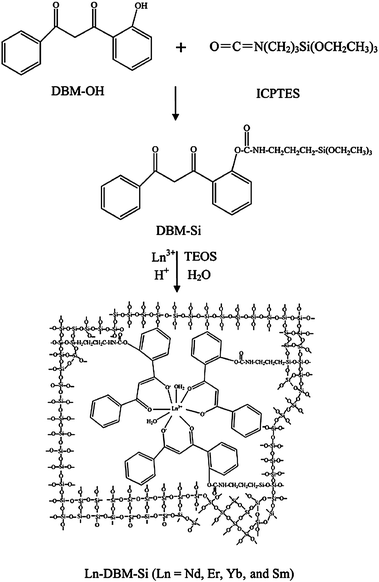 |
| | Fig. 6 Schematic diagram of the synthetic procedure and the possible structure for the hybrid materials Ln-DBM-Si. Reprinted with permission from ref. 69. Copyright 2009, American Chemical Society. | |
Lanthanide complex-based sol–gel hybrids could also be obtained by means of other various functionalized alkoxysilane ligands, such as modified calix[4]arene derivatives,73,74 8-hydroxyquinolinate,75 terpyridine,76 thiosalicylic acids,77 triazine,78 2-pyrrolidinone-5-carboxylic acid,79 and so on. Among these functionalized ligands, the interesting macrocyclic precursors that derive from p-tert-butylcalix[4]arene and calix[4]arene grafted by 3-(triethoxysilyl)propyl isocyanate through base-initiated nucleophilic addition, were reported by Lu et al.73 Then, the two novel macrocyclic bridge molecules were engaged in the construction of three series of novel luminescent (Eu3+, Tb3+) chemically bonded hybrid material systems (Fig. 7). They also performed other hybrid material systems based on calix[4]arene derivatives.74 In fact, they did not obtain satisfactory luminescent behavior (luminescence spectra and quantum yields) for this kind of hybrid material. Ligand emission can be seen in combination with lanthanide-centered emission. It is claimed that the energy level differences between the lowest triplet state energy of calix[4]arene derivatives and the resonant emitting levels of Tb3+ or Eu3+ are not appropriate for producing efficient energy transfer. The other possible reason is due to the fact that the lanthanide center is not well protected by the phenolic group of the calix[4]arene derivatives, which results in luminescence quenching by the hydroxyl groups (water, solvent, and silanol groups). However, in this case, the specific energy transfer mechanism is more favorable for Tb3+ than for Eu3+ ions. Our group developed visible-light-sensitized (>400 nm) hybrid materials (LnQSi-Gel), covalently linked with tris(8-hydroxyquinolinate)-lanthanide [Er(III), Nd(III), and Yb(III)] derivatives (Fig. 8 and 9).75 Normally, the hybrid materials containing the lanthanide complexes can be excited by ultraviolet light. However, materials excited by visible light could strongly reduce the background fluorescence and interference by the inner filter effects, due to the absorption of light. For some applications, visible light-excitable hybrid materials are desirable.
![The proposed structure of the obtained hybrid materials: p-tert-butylcalix[4]arene systems (top); calix[4]arene systems (bottom). Adapted with permission from ref. 73. Copyright 2009, American Chemical Society.](/image/article/2013/CS/c2cs35069f/c2cs35069f-f7.gif) |
| | Fig. 7 The proposed structure of the obtained hybrid materials: p-tert-butylcalix[4]arene systems (top); calix[4]arene systems (bottom). Adapted with permission from ref. 73. Copyright 2009, American Chemical Society. | |
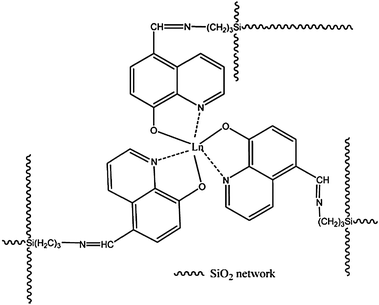 |
| | Fig. 8 Proposed Structure of the LnQSi-Gel, Ln = Er, Nd, Yb. Reprinted with permission from ref. 75. Copyright 2010, American Chemical Society. | |
In the above materials, only one kind of lanthanide complex is grafted onto the silica host. Nevertheless, more than one kind linked to the silica matrix could receive some significant results. Armelao and co-workers synthesized the tunable colour changes of silica single films grafted with Eu(III), Tb(III) complexes, and the mixed complexes (Tb/Eu molar ratios of 0.4, 1.0 and 2.2, yielding Eu2–Tb1, Eu1–Tb1 and Eu1–Tb2 films) respectively, by the sol–gel dip-coating method.80 In this case, a macrocycle was used as a coordinating unit with an appended chromophore as the light harvesting functionality (Fig. 10). The change in relative intensities between the Eu(III) and Tb(III) bands (Fig. 11) appears to be related to the molar ratios of the metal complexes and, in turn, to the colour output. The obtained films are optically homogeneous, resulting from the statistical and homogeneous composition of the red and green emitters. One more co-luminescence system, inert rare earth (RE) ion (Y3+, La3+, Gd3+) and active rare earth ion (Tb3+, Eu3+) co-fabricated organic–inorganic hybrid materials were developed by Wang and Yan.81 They took modified 5-hydroxyisophthalic acid as the bridge and one of the sol–gel precursors (the other is TEOS). Different ratios of active rare earth ions (Eu3+ or Tb3+) and inert rare earth ions (La3+, Ga3+, or Y3+) were set into the hybrid molecular materials. At the appropriate ratio [Y3+ (Gd3+or La3+):Eu3+ (Tb3+) = 1:1], the characteristic luminescence intensity of the active rare earth ions (Eu3+, Tb3+) was strongest for the 5D4–7F5 (Tb3+) and 5D0–7F2 (Eu3+) emissions. Besides, the luminescence lifetimes of the cohybrids fabricated with inert rare earth ions are longer than those of pure Eu or Tb hybrids. They suggested that the introduction of inert rare earth ions (Y3+, Gd3+, La3+) decreased the non-radiative deactivation energy transfer process, resulting in enhancement of the luminescent lifetime. In this case, they considered that active rare earth ions and inert rare earth ions coexist in the same molecular hybrids on the molecular scale and the distance between the inert rare earth complexes and the active rare earth complexes is very small. Thus, the inert RE ions can act as an energy bridge through which the energy of the modified 5-hydroxyisophthalic acid in the inert rare earth complexes can be transferred to the active Tb3+ ions through intramolecular energy transfer, which leads to the enhanced luminescence of Tb3+ in the hybrid. In addition, the concentration of inert rare earth ions is large enough, and each of the Eu-complex molecular fragments is surrounded by many Gd-complex molecular fragments. These Gd-complex molecular fragments can form a cage as an energy-insulating sheath to prevent collision with water molecules and decrease the energy loss of the Eu-complex; thus luminescence quantum efficiency and luminescence intensity are improved. This technology can be expected in the assembly of other luminescent molecular-based hybrid materials.
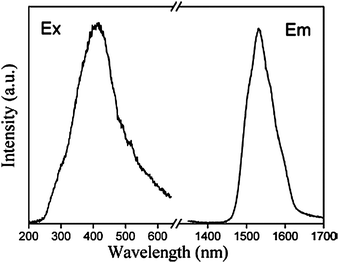 |
| | Fig. 9 Excitation (λem = 1531 nm) and emission (λex = 415 nm) spectra for the ErQSi-Gel material. Reprinted with permission from ref. 75. Copyright 2010, American Chemical Society. | |
 |
| | Fig. 10 Chemical structure of the trivalent lanthanide complexes linked to the silica matrix. Reprinted with permission from ref. 80. Copyright 2007, Royal Society of Chemistry Publishing Company. | |
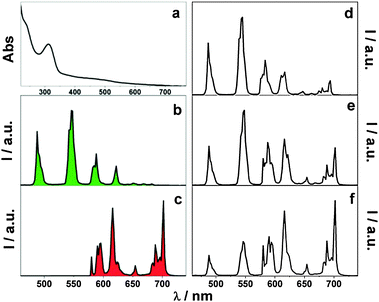 |
| | Fig. 11 Absorption (a), (Tb1–Eu1) and corrected emission spectra (λexc = 300 nm) of (b) Tb, (c) Eu, (d) Tb2–Eu1, (e) Tb1–Eu1 and (f) Tb1–Eu2 in SiO2 matrices. Reprinted with permission from ref. 80. Copyright 2007, Royal Society of Chemistry Publishing Company. | |
4. Hybrid mesoporous materials based on the lanthanide organic complex
Since mesoporous silica materials were first introduced in 1992,82,83 the development of ordered mesoporous molecular sieves has been of widespread interest in materials science. Ordered mesoporous materials with unique properties (high surface area, ordered pore structure of varying morphology, and controllable pore size over wide ranges) continue to be widely investigated because of their many potential applications in catalysis, adsorption and sensing, etc.84–89 Recently, a particular interest is the use of the ordered mesoporous silica material as a support for lanthanide complexes. Up to now, lanthanide complexes could be incorporated into mesoporous matrices, such as MCM-41 (2D hexagonal, space group p6mm), MCM-48 (cubic, space group Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d), FDU-1 (cubic structure with a 3D hexagonal intergrowth, space group Fm3m), SBA-15 (2D hexagonal, space group p6mm) or SBA-16 (cubic, space group Imm) either by a simple doping method (no strong bonding between the phases; only weak physical interactions, such as hydrogen bonding, van der Waals force, or weak static effects, exist between the mesoporous materials and the lanthanide complexes) or by the covalent bond grafting technique. The hybrid mesoporous materials are mainly synthesized via a sol–gel process by the hydrolysis and condensation of tetraalkoxysilane and/or organosilicon compound in the presence of a surfactant, such as hexadecyltrimethylammonium bromide (C16TABr), octadecyltrimethylammonium bromide (C18TABr), EO20PO70EO20 (P123), EO106PO70EO106 (F127), n-dodecylamine, supramolecular polyoxyethylene (10) stearyl ether (Brij 76), and so on. Compared to the pure lanthanide complexes, the obtained hybrid mesoporous materials usually show stronger luminescence intensity and higher quantum efficiency.90–92
d), FDU-1 (cubic structure with a 3D hexagonal intergrowth, space group Fm3m), SBA-15 (2D hexagonal, space group p6mm) or SBA-16 (cubic, space group Imm) either by a simple doping method (no strong bonding between the phases; only weak physical interactions, such as hydrogen bonding, van der Waals force, or weak static effects, exist between the mesoporous materials and the lanthanide complexes) or by the covalent bond grafting technique. The hybrid mesoporous materials are mainly synthesized via a sol–gel process by the hydrolysis and condensation of tetraalkoxysilane and/or organosilicon compound in the presence of a surfactant, such as hexadecyltrimethylammonium bromide (C16TABr), octadecyltrimethylammonium bromide (C18TABr), EO20PO70EO20 (P123), EO106PO70EO106 (F127), n-dodecylamine, supramolecular polyoxyethylene (10) stearyl ether (Brij 76), and so on. Compared to the pure lanthanide complexes, the obtained hybrid mesoporous materials usually show stronger luminescence intensity and higher quantum efficiency.90–924.1 Physical doping method
Originally, lanthanide complexes were introduced into mesoporous materials by a simple doping method, because it is easy to achieve. In order to improve the properties of the resultant materials, the researchers usually modify the mesoporous matrices. According to the reports, the luminescence intensity and lifetime of encapsulation of the lanthanide complex into a modified mesoporous matrix are much stronger and longer than those of the lanthanide complex into an unmodified mesoporous matrix. Usually, N-(3-trimethoxysilyl)ethylethylenediamine, (3-chloropropyl)triethoxysilane, and phenyltriethoxysilane molecules are used to modify the mesoporous matrices. Our group developed a simple ion exchange method incorporating a luminescent lanthanide complex [Eu(phen)2Cl3·2H2O] inside the channels of mesoporous silica MCM-41 with its external surface modified by phenyltriethoxysilane [Ph-Si(OEt)3] (Fig. 12).93 The preparation process involves the passivation of silanol groups on the external surface of as-synthesized MCM-41 with Ph-Si(OEt)3 and ion-exchange of the surfactant cations by [Eu(phen)2]3+. It is found that the resultant hybrid mesoporous material exhibits the characteristic emission of Eu3+ ions under UV irradiation with a higher 5D0 luminescence quantum efficiency, longer lifetime and better thermal stability than the corresponding pure complex. Li et al. encapsulated the Tb(acac)3phen (acac = acetylacetone; phen = 1,10-phenanthroline) complex in unmodified and 3-aminopropyl1)triethoxysilane (APTES) modified mesoporous molecular sieves, SBA-15.94 By comparing the intensity and the concentration of Tb3+ in different samples, they conclude that the unit mass of Tb(acac)3phen in the unmodified and modified samples gives a stronger luminescence than Tb(acac)3phen, implying improvement of the outer luminescence efficiency. Previously, Xu et al. also observed a similar phenomenon in the encapsulated Eu3+ complexes.95,96 In Tb(acac)3phen, a large part of the excited-state energy is transferred into vibration transition energy because of strong electron–phonon coupling. For the encapsulated complexes, vibration transition is suppressed because of the existence of a solid bone with a boundary of pores. As a consequence, more energy is transferred to Tb3+, leading to improvement of the photoluminescence. The authors pointed out that more excited energy in the modified sample is adsorbed by the modified SBA-15 instead of the acac complexes, which cannot be effectively transferred to Tb3+. The temperature dependence of the fluorescence lifetime indicated that the radiative transition rate of 5D4–7FJ in the unmodified and modified samples decreased in comparison to the pure complexes, which originated from the influence of the refractive index of the surrounding media (SBA-15). In the unmodified sample, the nonradiative transition rate for the 5D4 state became larger at room temperature, leading the inner quantum efficiency to decrease. Anyway, the outer quantum efficiency of photoluminescence in the unmodified sample became much larger than the pure complexes as well as the modified sample, which was attributed to more efficient energy transfer from the ligands to Tb3+. In the SBA-15, the photostability of the Tb complexes was improved considerably, in comparison to the pure complexes. Interestingly, in the modified sample, it is observed that the emission intensity increased with increasing irradiation time, which was attributed to the modification of surface defects under the exposure of ultraviolet light. The thermal stability for the samples encapsulated in SBA-15 was also improved considerably.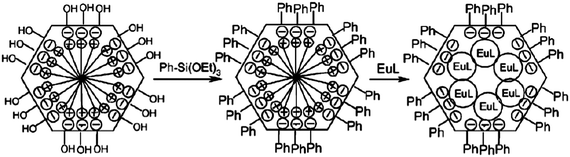 |
| | Fig. 12 Schematic diagram of incorporation of the lanthanide complex into the channels of functionalized mesoporous material MCM-41 with L = phen. The charge in Eu-phen was omitted for simplicity. Reprinted with permission from ref. 93. Copyright 2005, Royal Society of Chemistry Publishing Company. | |
Although the luminescence properties of the encapsulated complexes are somewhat improved, there are some intrinsic shortcomings, such as nonhomogeneous distribution and the aggregation of the lanthanide complex in the mesoporous matrix, and ease of separation out from the matrix. These could be resolved by a method called “complexes covalently bonded to the mesoporous matrix”.
4.2 Complexes covalently bonded to the mesoporous matrix
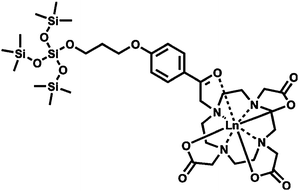
Generally, a lanthanide complex covalently bonded to mesoporous materials could be achieved through postsynthetic functionalization (grafting) or direct synthesis (co-condensation). Both methods need a functional ligand to be the ligand for the lanthanide ions and an organosilane precursor to synthesize the hybrid mesoporous materials. However, the nonhomogeneous distribution of the complexes is still a problem when using the grafting method, and is often encountered upon embedding the luminescent complexes in the mesoporous material, leading to the clustering of lanthanide ions and a decrease in luminescence intensity. In fact, compared with the physical doping method and postsynthetic functionalization (grafting), the co-condensation method enables a higher and more homogeneous surface coverage of organosilane functionalities and can effectively prevent the aggregation of emitting centers and the leaching of the photoactive molecules in the resultant materials. Because of the advantages, the co-condensation method is very appropriate for the preparation of lanthanide complex anchored mesoporous materials. Researchers began with europium and terbium complexes, which covalently bonded to the mesoporous matrix. Then, the other lanthanide complexes that emit in the NIR region were considered. Through 5-[N,N-bis-3-(triethoxysilyl)propyl]ureyl-1,10-phenanthroline (phen-Si), Eu(TTA)3phen was covalently bonded to SBA-15 mesoporous material by our group.92 The luminescence spectra and theoretical studies of all the materials showed that, compared with the doping sample SBA-15/Eu(TTA)3phen and the binary europium complex functionalized sample Eu-phen-SBA-15, the Eu(TTA)3phen-SBA-15 mesoporous hybrid material exhibited a higher luminescence intensity and emission quantum efficiency (37.61%). Also, the thermal stability of the Eu(TTA)3phen complex was evidently improved as it was covalently bonded to the mesoporous SBA-15 matrix. Successively, erbium, neodymium, ytterbium, samarium, praseodymium, and thulium complexes were covalently bonded to the MCM-41 and SBA-15 mesoporous materials in the follow-up work.97–100 Particularly, the luminescence spectra of the Pr3+ ion are more complicated in comparison with those of other lanthanide systems, as the Pr3+ ion can show emission lines originating from three different levels (3P0, 1D2 and 1G4) upon excitation of the organic ligand absorption. It is suggested that the NIR emission of the Pr3+ ion results from the 1D2 state to the nether states in that case, not from the 3P0 or the 1G4 state. The luminescence behaviors of thulium complexes covalently bonded to mesoporous MCM-41 with different β-diketones [4,4,4-trifluoro-1-2-thenoyl-1,3-butanedione (Htta) and 4,4,4-trifluoro-1-(2-naphthyl)-1,3-butanedione (Htfnb)] were compared.99 The results show that the material containing Htfnb is somewhat superior to the material with the Htta ligand as an optical amplifier, because the triplet state energy level of Htfnb matches better with the 3F2 level of the Tm3+ ion in comparison with that of Htta. Besides 5-[N,N-bis-3-(triethoxysilyl)propyl]ureyl-1,10-phenanthroline, another modified neutral ligand could be used as the linkage between the mesoporous matrix and the lanthanide complexes. Carlos and co-workers immobilized the lanthanide complexes with the modified neutral ligand {(3-triethoxysilylpropyl)[3-(2-pyridyl)-1-pyrazolyl]acetamide} (Fig. 13).101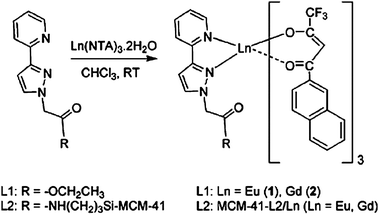 |
| | Fig. 13 Synthesis of the model complexes Ln(NTA)3·L1 and the supported materials MCM-41-L2/Ln. Reprinted with permission from ref. 101. Copyright 2005, American Chemical Society. | |
Besides the neutral ligands, the functionalized β-diketones were also used as bridges to synthesize lanthanide complex anchored mesoporous materials. A route for coordination of Eu3+ by a functionalized dibenzoylmethane (DBM) and two free DBM molecules covalently bonded inside hexagonal mesoporous silica was established by Serra et al. (Fig. 14).102 Yan et al. also linked the lanthanide complexes to the functionalized ordered mesoporous materials (SBA-15, MCM-41, SBA-16) with the modified β-diketones, such as 1-(2-naphthoyl)-3,3,3-trifluoroacetonate (NTA),103 2-thenoyltrifluoroacetone (TTA),104–108 1,3-diphenyl-1,3-propanepione (DBM),109–111 and acetylacetone (ACAC).109,112 Meanwhile, they selected 2,2′-bipyridine, 1,10-phenanthroline, polymethyl methacrylate (PMMA), poly(methacrylic acid) (PMAA), and polyvinylpyrrolidone (PVP) as the additive ligands to satisfy the coordination number of the central lanthanide ion. Also, the polymers were used as second matrices in such materials. This will be discussed in the Lanthanide organic complex-based polymers section. Other functionalized ligands, such as 8-hydroxyquinoline and macrocyclic calix[4]arene derivatives, were also used as bridges. Alkoxysilane modified 8-hydroxyquinoline (Q-Si) was used as one of the precursors for the preparation of the Q-SBA-15 or Q-MCM-41 material due to its excellent complexation properties.113–115 Followed by a ligand exchange reaction, LnQ2Cl(H2O)2 (Ln = Er, Nd, Yb) complexes were linked to the Q-SBA-15 or Q-MCM-41 material (denoted as LnQ3-SBA-15 or LnQ3-MCM-41) (Fig. 15). In this case, the Q-Si ligand efficiently replaces the water molecules in the first coordination sphere of the introduced [LnQ2Cl(H2O)2] complexes and protects the Ln3+ ions from the residual water molecules and silanol groups in the mesoporous materials. Differently from this work, Yan and co-workers directly reacted 3-(triethoxysilyl)-propyl isocyanate with the hydroxyl group (–OH) of 8-hydroxyquinoline.116 Then they introduced the lanthanide ions (Eu, Tb, Er, Nd) to coordinate with the functionalized 8-hydroxyquinoline. In this case, the ligands cannot coordinate with the Ln3+ ions and protect the Ln3+ ions efficiently. As a result, they did not observe the characteristic emissions of the corresponding lanthanide ions. Yan et al. also used modified macrocylic calix[4]arene derivatives as bridge molecules to prepare lanthanide organic–inorganic SBA-15 hybrid materials.117 Additionally, carboxylic acids are a class of commonly used ligands for the lanthanide ions. Lanthanide (Eu, Tb) complexes have been immobilized to the mesoporous materials by using the modified carboxylic acids (such as salicylic acid,118 2-hydroxyl-3-methylbenzoic acid,118 5-sulfosalicylic acid,119meta-aminobenzoic acid,120 or dipicolinic acid,121 and so on) as the linkages. Actually, because of the relatively high energy level of carboxylic acids, they are usually suitable for Ln3+ ions with relatively high excited 4f states, such as Tb3+ ions.
 |
| | Fig. 14 Synthetic procedure and schematic diagram of the obtained material. Reprinted with permission from ref. 102. Copyright 2007, American Chemical Society. | |
![Synthetic procedure of LnQ3-MCM-41 (Ln = Er, Nd, Yb). Reagents and experimental conditions: (a) TEOS, CTAB, H2O, surfactant removal; (b) [LnQ2Cl–(H2O)2], reflux. Reprinted with permission from ref. 115. Copyright 2008, Elsevier Ltd.](/image/article/2013/CS/c2cs35069f/c2cs35069f-f15.gif) |
| | Fig. 15 Synthetic procedure of LnQ3-MCM-41 (Ln = Er, Nd, Yb). Reagents and experimental conditions: (a) TEOS, CTAB, H2O, surfactant removal; (b) [LnQ2Cl–(H2O)2], reflux. Reprinted with permission from ref. 115. Copyright 2008, Elsevier Ltd. | |
4.3 Periodic mesoporous organosilica materials (PMOs) as the hosts
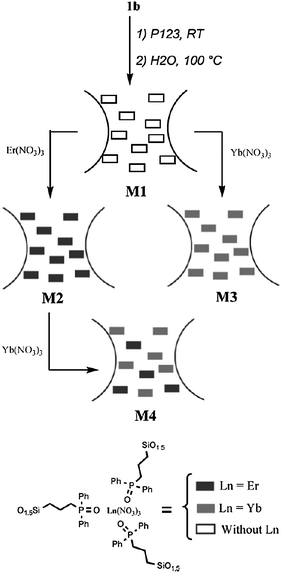
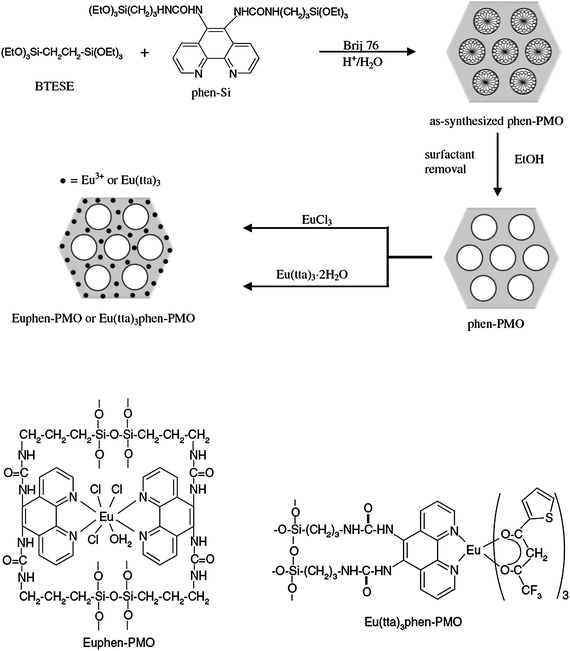
Besides the traditional mesoporous silica, a new class of mesostructured silica materials, designated as the periodic mesoporous organosilica materials (PMOs) were reported by three different independently working research groups in 1999.122–124 PMOs are formed by hydrolytic polycondensation of bridged organosilica precursors, such as (R′O)3–Si–R–Si–(OR′)3 with two trialkoxysilyl groups bridged by an organic group (R), in the presence of a structure-directing agent. The resulting materials were novel in that the organic fragments were located within the framework, leaving the channel pores unoccupied. This route confirms that PMO possesses a uniform distribution of organic and inorganic moieties at the molecular level within the framework.125–132 Moreover, compared with the mesoporous silica materials, PMO presents better aging, hydrothermal and mechanical stabilities, as well as unique physical and chemical properties.133–135 In recent years, many organic groups have been covalently grafted into the network of PMO silica matrices in the presence of aliphatic, aromatic, and heterocyclic surfactants.136 However, the incorporation of lanthanide complexes into the silica network of PMOs is rarely reported. Corriu et al. reported an original route allowing the introduction of Er3+ and/or Yb3+ ions encapsulated by the silylated phosphine oxide as the ligand (Fig. 16) into the framework of a mesoporous organosilica.137 First, they obtained the erbium complex 1b (Fig. 16). Then, the hybrid material was prepared by co-hydrolysis and polycondensation of 1b with tetraethylorthosilicate (TEOS) in a lyotropic liquid crystal medium. They observed that the erbium salt is lost after hydrothermal treatment. Subsequently, recomplexation of Er(NO3)3 into M1 was attempted. It is observed that 70% of the total erbium content was incorporated into the framework. Using the same method, the ytterbium(III) complex was introduced into the framework of PMOs (M3, Fig. 17). The material M2 (containing erbium(III)) was treated with an ethanolic solution of Yb(NO3)3. The resulting material M4 revealed that 60% of erbium(III) was displaced by ytterbium(III), giving rise to a material containing both Er3+ and Yb3+ ions. This method could control the lanthanide content. It is demonstrated that such materials are of importance as the further functionalisation of the channel pores by either a post-synthetic treatment or a one-step synthesis. Our group succeeded in incorporating the europium complexes inside the framework of PMOs by co-condensation of 1,2-bis(triethoxysilyl)ethane (BTESE) and 5,6-bis(N-3-(triethoxysilyl)propyl)ureyl-1,10-phenanthroline (phen-Si) using polyoxyethylene (10) stearyl ether (Brij 76) surfactant as a template under acidic conditions (Fig. 18).138 The corresponding pore sizes of phen-functionalized PMOs before [phen(0.10)-PMO] and after [Eu(tta)3phen(0.10)-PMO] introducing the Eu(tta)3·2H2O complex show scarcely any change, supporting the fact that the europium complexes are covalently coordinated with phen into the wall of the PMOs. Compared to the pure complex Eu(tta)3phen (30.5%),92 Eu(tta)3phen(0.10)-PMO (27.9%) also displays a similar emission quantum efficiency and better thermal stability. In a follow-up study, terbium complexes were incorporated inside the framework of PMOs by using 3,5-bis(N,N-(triethoxysilyl)propyl)ureyl-benzoic acid (BA-Si) as a precursor for the synthesis of PMOs and as an organic ligand for the terbium ions (Fig. 19).139 The Tb-BA-PMO hybrid material displays the characteristic emission of Tb3+ ions under UV and better thermal stability compared to the pure complex. Interestingly, the microscopic morphologies of the samples transfer slightly from the curved cylinder forms to the spheres with increasing BA-Si concentration. Moreover, it is suggested that more ureylene groups were introduced into the pore walls and stronger hydrogen bond interaction occurs with increasing content of the organosilane BA-Si, which contributes to the organization of the PMOs into a more regular morphology.140,141 More recently, Yan's group incorporated lanthanide complexes covalently bonded to PMO by a functionalized calix[4]arene derivative.142 Despite the above achievements, it is still necessary to design more new lanthanide complex-based PMO materials for different desirable targets.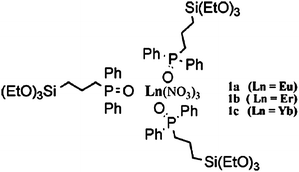 |
| | Fig. 16 Mesoporous hybrid material containing free phosphine oxide ligands located in the framework. Free phosphine oxide ligands operate as templates for further lanthanide(III) ion complexation. Reprinted with permission from ref. 137. Copyright 2006, Royal Society of Chemistry Publishing Company. | |
 |
| | Fig. 17 Incorporation of lanthanide salts in the framework. Reprinted with permission from ref. 137. Copyright 2006, Royal Society of Chemistry Publishing Company. | |
 |
| | Fig. 18 Schematic diagram of incorporation of the europium complex into the framework of PMOs via a co-condensation of 1,2-bis(triethoxysilyl)ethane (BTESE) and 5,6-bis(N-3-(triethoxysilyl)propyl-1,10-phenanthroline (phen-Si) using covalent bonds. Reprinted with permission from ref. 138. Copyright 2008, Elsevier Ltd. | |
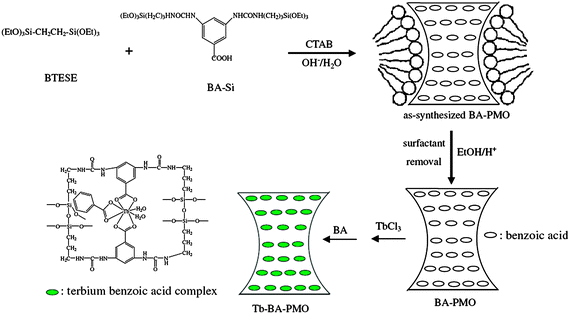 |
| | Fig. 19 Schematic diagram of synthesis of the novel PMOs via co-condensation of 1,2-bis(triethoxysilyl)ethane (BTESE) and benzoic acid-functionalized organosilane (BA-Si) and its application in the incorporation of the Tb3+ ions into the matrix. Reprinted with permission from ref. 139. Copyright 2009, Elsevier Ltd. | |
Out of the lanthanide complex supporting materials, which is the most suitable one, MCM-41, SBA-15, or PMO? Comparative studies were carried out. First, the luminescent properties of MCM-41 and SBA-15 covalently bonded with Ln(DBM)3phen (Ln = Er, Nd, Yb) complexes by a modified 1,10-phenanthroline ligand were compared.143 All the results revealed that SBA-15 is somewhat superior to MCM-41 as the parent of the Ln(DBM)3phen complexes. The SBA-15-supported materials show an overall increase in relative luminescence intensity and lifetime, despite the lower lanthanide ion content, as compared to that of the MCM-41-supported materials. The luminescence behavior of the lanthanide complex functionalized mesoporous materials may depend on the lanthanide ion content and the mesoporous structure of the materials. As we know, the mesoporous structure of SBA-15 is different to that of MCM-41. It consists not only of large, uniform, and ordered mesopores, but also of much smaller complementary micropores that provide connectivity between the ordered mesopores. On the one hand, the micropores may too small for the Ln(dbm)3(H2O)2 (around 1.6 nm) complexes to enter. On the other hand, some phen-Si groups may block the pore mouth of the micropores. Thus, lanthanide ion content was reduced in the SBA-15-supported hybrid materials. The relatively small pore size, higher lanthanide ion loading and locally high lanthanide concentration of the Ln(dbm)3phen-MCM-41 materials may result in a self-quenching effect. Energy transfer from one Ln3+ ion to another can occur and the photon can migrate among the clustered regions, followed by quenching at a defect site in the Ln(dbm)3phen-MCM-41 materials. In succession, the luminescence properties of Eu(tta)3phen complex linked SBA-15 and PMO materials were compared.144 Compared to Eu(tta)3phen linked PMO, Eu(tta)3phen linked SBA-15 exhibited a longer luminescence decay time and higher luminescence intensity, emission quantum efficiency (q), and absolute quantum yield (Φ). Meanwhile, it is found that the europium complex in SBA-15-supported material possessed a better thermal stability than that in PMO-supported material. The photoluminescence and thermal stability features indicated that SBA-15 is a more suitable host material for a lanthanide complex than MCM-41 and PMOs. However, the effect of the microenvironment between the lanthanide complex and these three kinds of silica matrices on the luminescence properties still needs further fundamental investigation.
From the above examples, it can be seen that various mesoporous hybrid materials with lanthanide luminescence properties have been reported. As the methods of synthesis can be easily applied to other complexes and different modified organic ligands, the desired properties of mesoporous hybrids can be tailored by an appropriate choice of precursors and lanthanide ions. However, for such kinds of hybrids, how to decrease the quenching of lanthanide luminescence from the microenvironment (Si–OH group and H2O molecule) and increase the quantum yield is still an unsolved problem.
5. Titania materials based on lanthanide organic complex
To date, lanthanide organic complexes containing hybrid materials were mainly developed for silicate systems. The extension of this chemistry to other oxides than silica would allow new options for the development of hybrid organic–inorganic materials based on lanthanide complexes. Among the oxides, titania is a very interesting material in view of photocatalytic and energy conversion applications.145,146 In this section, we will provide some advances on titania materials based on lanthanide organic complexes.5.1 Embedding lanthanide organic complex into titania
Recently, the sensitization of lanthanide ions by TiO2 has been described.147–149 Generally speaking, the energy transfer process is as follows. UV light is absorbed in the band gap of the titania with subsequent relaxation to the defect states, followed by energy transfer to the crystal field states of the lanthanide ion. Encapsulation of lanthanide complexes into the titania host has also been reported.150–152 The emission of the hybrid materials is mainly from energy absorbed by the ligands, not the titania host. Sasaki and co-workers encapsulated lanthanide complexes [Eu(phen)2Cl32H2O and Tb(phen)2Cl32H2O, phen = 1,10-phenanthroline] in the host titania nanosheets by the restacking method.150 They found the composite ex-Ti0.91O2/Eu(phen)2 exhibited characteristic red emission from the europium complex, while ex-Ti0.91O2/Tb(phen)2 only gave featureless emission originating from the ligand. The quantum yield of the europium complex in the composite is estimated to be 60.2%, which is much higher than that of the free complex. In addition, the unit emission efficiency also enhanced in the composite benefited from the improvement in absorption coefficient and quantum yield. Li's group reported an organic–inorganic transparent titania thin film, in which a highly luminescent Eu3+ or Tb3+ complex was formed in situ, by employing a TTA stabilized titania precursor.1535.2 Grafting lanthanide organic complex into titania
Besides embedding as exemplified above, the lanthanide organic complex could also be attached to titania. A representative example is that Li's group has successfully immobilized lanthanide complexes on titania via modification of the titanium alkoxide by iso-nicotinic acid, not by the Ti–C bonds as with silicon (Fig. 20).154 The carboxylic acid group can react with titanium alkoxide to moderate its reactivity toward hydrolysis and condensation, while the heterocyclic group can coordinate lanthanide ions as well as sensitize their luminescence. Later, they use an Eu(TTA)3(H2O)2 complex instead of EuCl3 (Fig. 21).155 β-Diketonate ligands can act both as an antenna to absorb and transfer energy to the lanthanide ions and to expel water molecules from the first coordination sphere.156 Great improvements in luminescence behavior have been achieved by comparing the luminescence data of hybrid material based on the Eu(TTA)3(H2O)2 complex with those of hybrid material based on EuCl3. Otherwise, the europium(III) ions can be replaced in this system by other lanthanide ions that show luminescence in near-infrared regions (Ln = Nd, Er).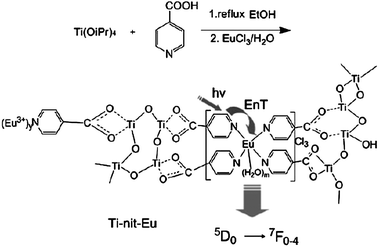 |
| | Fig. 20 Procedure and proposed structure of materials Ti-nit-Eu. Reprinted with permission from ref. 154. Copyright 2008, Royal Society of Chemistry Publishing Company. | |
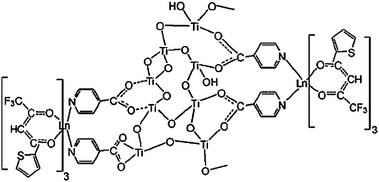 |
| | Fig. 21 Proposed structure of materials Ti-nit-Ln(tta)3 (Ln = Eu, Nd, Er). Reprinted with permission from ref. 155. Copyright 2009, American Chemical Society. | |
Similar to the silica-polymer system, Yan et al. developed a titania-polymer mixed system.157 They achieved the functional precursor by grafting nicotinic acid to titanium alkoxide, which is then coordinated to lanthanide ions (Tb3+/Eu3+) to prepare the titania hybrid materials via a sol–gel process in the presence of water. Then, the mixed systems are assembled by the introduction of the organic polymers polymethacrylic acid (PMAA)/polyvinylpyrrolidone (PVP) via coordination reactions with lanthanide ions. It is observed that the luminescence properties of the hybrids based on titania-polymer exhibit stronger luminescence intensities, longer lifetimes, and higher luminescence quantum efficiencies than the hybrid based on titania, indicating that their introduction is of benefit to the photoluminescence of titania hybrid materials. Moreover, the obtained luminescence data indicate that PVP is more efficient to sensitize the luminescence of titania hybrid materials compared to PMAA in this case. The examples of hybrid materials based on the silica–titania mixed matrix were also presented by Yan's group.158–161 In their work, a derivative of nicotinic acid (2-sulfanylpyridine-3-carboxylic acid) was selected due to its three potential binding sites: pyridine N, sulfhydryl S and the carboxylic O atom.158 In this case, the sulfhydryl S could link with the silane crosslinking reagent, the carboxylic O could link with titanium alkoxide and the pyridine N could coordinate with the lanthanide ions to form lanthanide organic complexes. Then, lanthanide organic complexes are covalently grafted to the silica–titania mixed matrix. They found that the silica–titania mixed system exhibits a much higher luminescence intensity and quantum efficiency than the single titania system. Another example reported by Yan's group is the lanthanide organic complex covalently bonded to an ordered mesoporous Si–O network and an amorphous Ti–O network simultaneously.160 First, a β-diketonate ligand reacts with 3-(triethoxysilyl)-propyl isocyanate to form the ordered SBA-15 Si–O network. Second, nicotinic acid reacts with tetraisopropyl titanate to construct the Ti–O network. Finally, the mesoporous hybrid materials were obtained by introducing Ln3+ (Ln = Eu, Tb) ions, which could coordinate with β-diketonate and the N atoms of nicotinic acid. They also selected 4-mercaptobenzoic acid as a molecular bridge to construct the linkage between inorganic and organic components.161 Similarly to 2-sulfanylpyridine-3-carboxylic acid,158 the S atom linked to the Si–O network, and the –COOH connected with the Ti–O network. In this case, they introduced phen as the second ligand. Then, the lanthanide organic complex was formed in the silica–titania mixed matrix. Ranyuk et al. elaborated on new hybrid materials hosting covalently bonded lanthanide organic complexes in the titania matrix.162 The reaction of Ti(O-i-Pr)4 with [Eu.L](X)3 [L = (diethoxyphosphoryl)naphthyl-substituted tetramide cyclone, X = SO3CF3, NO3] could form the P–O–Ti bond. Then, the [Eu.L](X)3 complexes were incorporated into the titania matrix to form the hybrid materials. It is suggested that attaching lanthanide organic complexes to mixed-matrix hybrid materials can be conveniently applied to other hybrid material systems and the desired properties can be tailored by the appropriate choice of precursors and lanthanide ions. In this way, more hybrid luminescent materials could be expected.
6. Lanthanide organic complex-containing ionic liquid systems
Typically, ionic liquids are built up by a large organic cation such as imidazolium, pyridinium, or quaternary ammonium ions and anionic counterions such as halides or BF4−, PF6−, or CF3SO3−. By a common definition, the melting point of ionic liquids is below 100 °C. Many ionic liquids are even liquid at room temperature (room-temperature ionic liquids) and below.163–165 The physical and chemical properties of ionic liquids such as miscibility with water and other solvents, dissolving ability, polarity, viscosity, and density, could be fine-tuned by choosing the appropriate anion/cation combination. Recently, it is an interesting topic to investigate the spectroscopic behavior of lanthanide complex-doped ionic liquids, which can be regarded as new luminescent ‘soft' materials.6.1 Lanthanide complex-doped ionic liquids
Binnemans et al. dissolved neodymium(III) tosylate, bromide, triflate and sulfonylimide complexes in 1-alkyl-3-methylimidazolium ionic liquids containing the same anion as the neodymium(III) complexes.166 It is demonstrated that ionic liquids are promising solvents for NIR emitting lanthanide compounds, because ionic liquids are polar non-coordinating solvents that can solubilize lanthanide complexes. The other reason is that many ionic liquids are transparent through almost the whole of the visible and NIR spectral regions. Ionic liquids doped with luminescent lanthanide complexes would be an active component in, for instance, liquid chelate lasers, which operate in a similar way as dye lasers do. They found a 1-hexyl-3-methylimidazolium tetrakis(2-thenoyltrifluoroacetonato)europate(III) complex {[HMIM][Eu(tta)4]} (Fig. 22) that combines good shielding of the lanthanide ion with photosensitization by an efficient light-harvesting antenna such as 2-thenoyltrifluoroacetonate, and which is soluble in an ionic liquid [HMIM][Tf2N] (HMIM = 1-hexyl-3-methylimidazolium; Tf2N = bis(trifluoromethanesulfonyl)imide).167 By introducing the [HMIM]+ cation in the ionic complex, the solubility in the corresponding ionic liquid was enhanced in comparison with alkali counter ions. They observed that the photochemical stability of the [HMIM][Eu(tta)4] complex was obviously enhanced after dissolving in the ionic liquid [HMIM][Tf2N]. Bünzli et al. doped the lanthanide ternary complexes [Ln(tta)3(phen)] (tta = thenoyltrifluoroacetonate, phen = 1,10-phenanthroline, Ln = Eu, Er, Nd, Yb) into 1-dodecyl-3-methylimidazole chloride ([C12-mim]Cl).168 It is observed that all the materials exhibit relatively intense luminescence, although the concentration of the doped ternary complex (1 mol%) in [C12-mim]Cl is low (Fig. 23 and 24). They determined the quantum yields of Yb(tta)3(phen) in [C12-mim]Cl, [Yb(tta)3(phen)] in the solid state and [Yb(tta)3(phen)] in toluene to be 2.1% 1.6%, and 1.1%, respectively, by taking [Yb(tta)3(H2O)2] as standard. Puntus et al. studied the influence of their ionic liquid anion nature on the properties of Eu-containing luminescent materials. They doped the [EuCl2phen2(H2O)2]Cl·H2O complex (1 mol%) into the 1-methyl-3-tetradecyl imidazolium ionic liquid with N(CN)2−, CF3SO3− and N(CF3SO2)2− anions.169 They suggested that the properties of IL anions, such as the strength of the anion–cation interaction, the molecular volume and their coordination ability towards lanthanide ions, determine the formation of the solution or suspension of the europium complex. The results demonstrated that a relatively large IL anion molecular volume promotes the formation of micro particles of the europium complex, and the high coordinating ability of IL anions towards the europium ion, along with the high temperature for obtaining Eu-containing ILs, produces the improvement in luminescence. Mudring et al. reported new Eu-based ionic liquid systems, [C4mim][DTSA]:[Eu(DTSA)3] and 2[C4mim][DTSA]:[Eu(DTSA)3] (C4mim = 1-butyl-3-methylimidazolium, DTSA = ditoluenesulfonylamide), which exhibit strong red luminescence of a high color purity and have the potential to be used for optical applications such as in emission displays.170 The reported work aimed to pursue new lanthanide complex-containing ionic liquids and the new approaches to prepare such materials. As described above, most of the lanthanide complex-doped ionic liquids to date were prepared by dissolving the as-synthesized lanthanide complexes in ionic liquids. More recently, Li and co-workers reported an original method of introducing a highly luminescent lanthanide complex into an ionic liquid by directly dissolving Eu2O3 and the organic ligands 2-thenoyltrifluoroacetone (TTA) and 1,10-phenanthroline (phen) into the task-specific ionic liquid 3-(5-carboxypropyl)-1-methylimidazolium bromide (Fig. 25).171 It is demonstrated that the europium complex is synthesized in situ in the ionic liquid. The number of water molecules (nw) coordinated to the europium ions in the material was estimated to be 0.5. This means that most of the water molecules have been expelled from the first coordination sphere of the Eu3+ ions. In this case, the key point is the task-specific ionic liquid (containing a carboxylate group), which can dissolve metal oxide and organic ligands without the addition of extra solvent. They did not observe the broad emission band resulting from organic ligand molecules in the blue region. The obtained soft material shows a high red luminescence (Fig. 26) (experimental quantum yield 39%) and has potential applications in emission displays and laser dyes, despite the low concentration of lanthanide complexes in the ionic liquid (ca. 3 mol%). The extension of this method to lanthanide complexes other than those of europium was also done by them.172 Interestingly, the ionic liquids have a selective solubilizing ability for lanthanide oxides under the reaction conditions used in this study. They have tried Eu2O3, Tb4O7, Nd2O3, Er2O3, Dy2O3, and Pr6O11. Among them, Eu2O3, Nd2O3, Er2O3 and Dy2O3 were found to be soluble, whereas Tb4O7 and Pr6O11 were insoluble or very poorly soluble in the ionic liquids. They selected 3-(5-carboxypropyl)-1-methylimidazolium bromide and 3-(5-carboxypropyl)-1-butylimidazolium bromide as the task-specific ionic liquids. It is found that 3-(5-carboxypropyl)-1-butylimidazolium bromide can sensitize the luminescence of Eu3+ ions, and the fine-tuning of the emission characteristics is easily achieved through adjustment of the Eu3+ ion concentration. The quantum yield has been increased from 48% for Eu-TTA-phen/3-(5-carboxypropyl)-1-methylimidazolium bromide to 55% for Eu-TTA-phen/3-(5-carboxypropyl)-1-butylimidazolium bromide. Lately, they synthesized propanoic acid functionalized ILs, which could dissolve Eu2O3 or Tb4O7, followed by the formation of europium(III) or terbium(III) carboxylate and lanthanide complex-containing ionic liquids.173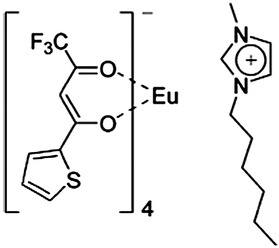 |
| | Fig. 22 1-Hexyl-3-methylimidazolium tetrakis(2-thenoyltrifluoro-acetonato)europate(III) complex. Reprinted with permission from ref. 167. Copyright 2005, Royal Society of Chemistry Publishing Company. | |
![Luminescence spectra of [Eu(tta)3(phen)] (top) and Eu1 (bottom) at 295 K obtained under ligand excitation at 385 nm. Reprinted with permission from ref. 168. Copyright 2005, Wiley Publishing Company.](/image/article/2013/CS/c2cs35069f/c2cs35069f-f23.gif) |
| | Fig. 23 Luminescence spectra of [Eu(tta)3(phen)] (top) and Eu1 (bottom) at 295 K obtained under ligand excitation at 385 nm. Reprinted with permission from ref. 168. Copyright 2005, Wiley Publishing Company. | |
![Near-infrared photoluminescence of (a) [Er(tta)3(phen)], (b) Er1, (c) [Nd(tta)3(phen)], (d) Nd1, (e) [Yb(tta)3(phen)], and (f) Yb1 at 295 K. The excitation wavelength is 385 nm. Reprinted with permission from ref. 168. Copyright 2005, Wiley Publishing Company.](/image/article/2013/CS/c2cs35069f/c2cs35069f-f24.gif) |
| | Fig. 24 Near-infrared photoluminescence of (a) [Er(tta)3(phen)], (b) Er1, (c) [Nd(tta)3(phen)], (d) Nd1, (e) [Yb(tta)3(phen)], and (f) Yb1 at 295 K. The excitation wavelength is 385 nm. Reprinted with permission from ref. 168. Copyright 2005, Wiley Publishing Company. | |
 |
| | Fig. 25 Ionic liquid and organic ligand used (top) and the procedure for obtaining the lanthanide complex in ionic liquid (bottom). Reprinted with permission from ref. 171. Copyright 2008, Royal Society of Chemistry Publishing Company. | |
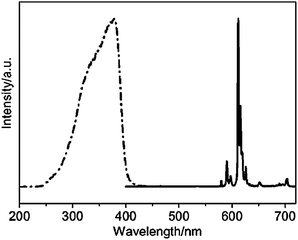 |
| | Fig. 26 Excitation (dot-dash) and emission (solid) spectra of IL-Eu-TTA-phen. Reprinted with permission from ref. 171. Copyright 2008, Royal Society of Chemistry Publishing Company. | |
Generally, lanthanide complex-containing ionic liquids can be obtained mainly through two methods. One is to dissolve a lanthanide complex or lanthanide oxide and organic ligands in an ionic liquid. The merit of this method is its easy feasibility. However, the structure of the optically active component, especially the local environment of the Ln3+ ion, is most of the time unknown and makes a strategic improvement of the material difficult. The other is to construct a lanthanide salt with an ionic liquid that shares the same counter anion as the used ionic liquid, leading to the formation of a compound or solution where the Ln3+ ion is solely coordinated by the common anion. The target compound can be tuned by using the salt reacted with the ionic liquid at different molar ratios or by selecting different cations and anions.
6.2 Lanthanide organic complex-containing ionogels
Recently, a new type of luminescent material has emerged through the incorporation of lanthanide complexes into ionogels, which is endowed with the mechanical, electrical and luminescent properties of all of the components (silica or polymer, lanthanide complex and ionic liquid).174,175 The novel ionogels offer several advantages, including transparency, outstanding ionic conductivity performance, stability in water and various organic solvents, and the ability to be easily shaped into coatings, rods or pellets, making them excellent candidates for use in dye-sensitized solar cells, fuel cell lithium batteries, and electroluminescent devices. Feng et al. present a facile method to prepare a transparent and luminescent ionogel monolith by hydrolysis and condensation of the silylated bipyridine, which can sensitize the luminescence of europium(III) in the presence of a carboxyl-functionalized ionic liquid where Eu3+ ions coordinate the oxygen atoms of carboxylate groups from the ionic liquid (Fig. 27).176 Such materials consist of an ionic liquid confined inside the nanosized pores of a silica matrix.177–179 Wang et al. succeeded in preparing the luminescent organic–inorganic hybrid materials by hydrolysis and condensation of silylated β-diketone under acidic conditions in the presence of a carboxyl-functionalized ionic liquid, in which Eu3+ ions are coordinated to the oxygen atoms of carboxylate groups from the ionic liquids.180 Lately, Binnemans et al. used ionic liquid 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide [C6mim][Tf2N] as a plasticizer to prepare europium(III)-doped luminescent poly(methyl methacrylate) films.181 The ionic liquid also enhances the solubility of the europium(III) complexes in the PMMA matrix. Different europium(III) complexes, such as [C6mim][Eu(nta)4], [C6mim][Eu(tta)4], [Eu(tta)3(phen)] and [choline]3[Eu(dpa)3] (where nta is 2-naphthoyltrifluoroacetonate, tta is 2-thenoyltrifluoroacetonate, phen is 1,10-phenanthroline, dpa is 2,6-pyridinedicarboxylate (dipicolinate) and choline is the 2-hydroxyethyltrimethyl ammonium cation), have been incorporated in the polymer/ionic liquid matrix. Bright red photoluminescence was observed for all the films upon irradiation with ultraviolet radiation. As an extension of this work, the luminescence color of the films could be tuned by a suitable choice of lanthanide ion, ranging from blue for Tm3+, green for Tb3+ and near-infrared emission for ions such as Nd3+, Er3+ and Yb3+.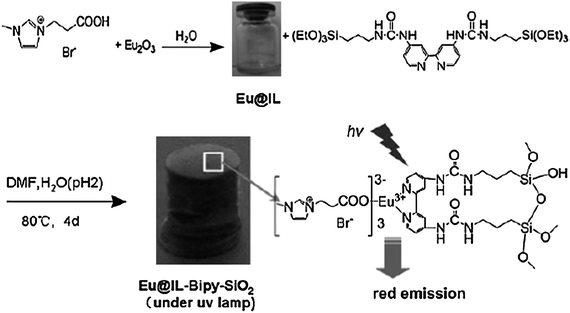 |
| | Fig. 27 Illustration of the procedure used to obtain the transparent and luminescent ionogel. Reprinted with permission from ref. 176. Copyright 2010, Royal Society of Chemistry Publishing Company. | |
7. Lanthanide organic complex-based polymers
Polymers are ideal matrices for lanthanide complexes, due to several attractive features including mechanical strength, flexibility, ease of processing, and controllable cost. Similarly to the other lanthanide complex-based organic–inorganic hybrid materials, lanthanide complex-based polymers could be obtained through two main routes: (1) dispersing lanthanide complexes in the polymer; (2) immobilizing lanthanide complexes to the polymer. The development of lanthanide complex-based polymers keeps advancing, from the design of the lanthanide complexes to the selection of the polymer matrix. Lanthanide complex-based polymers include the single systems (single lanthanide complex and single polymer), and the mixed systems (mixed-lanthanide complexes or mixed-matrices).7.1 Single systems
It has always been a challenge to synthesize lanthanide complex-containing polymers with high quantum yields. Biju et al. reported that the 3-phenyl-4-acyl-5-isoxazolonate complex of Tb3+ exhibited high green luminescence efficiency in the solid state, with quantum yields between 59–72%.182 Then, poly-β-hydroxybutyrate (PHB) polymer films doped with Tb3+-3-phenyl-4-acyl-5-isoxazolonate complexes to different extents are prepared. It is noted that the luminescence efficiency of doped films is enhanced (quantum yields between 74–86%) compared with the pure complexes, revealing that the polymer matrix acts as a co-sensitizer for Tb3+ centers. Richards and co-workers reported complexes of formula [Eu(β-diketonate)3(DPEPO)] (β-diketonate = hexafluoroacetylacetonate, tetradecafluorononadione, and 2,2-dimethyl-6,6,7,7,8,8,8-heptafluoro-3,5-octadione; DPEPO = bis(2-(diphenylphosphino)phenyl)ether oxide) displaying a total photoluminescence quantum yield up to 0.76 in solution and comparable values in polymethylmethacrylate (PMMA).183 At that time, such a high total photoluminescence quantum yield in PMMA was unprecedented. Then, Raj et al. designed a complex [Eu(CPFHP)3(DDXPO)][CPFHP = 1-(9H-carbazol-2-yl)-4,4,5,5,5-pentafluoro-3-hydroxypent-2-en-1-one, DDXPO = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene oxide] and incorporated it into PMMA polymer films with different content, which were shown to exhibit exceptionally high PL photoluminescence quantum yields (79–84%).184 This range is comparable to that reported by Richards et al. for luminescent europium β-diketonate complexes doped in PMMA matrix. They suggested that the PMMA with high molecular weight enwraps the Eu3+ complex and keeps the donor and acceptor close, which results in the effective intermolecular energy transfer and, consequently, the high sensitization efficiency. The PMMA films doped with [Eu(CPFHP)3(DDXPO)] show a promising PL efficiency and therefore have potential applications as polymer light-emitting diodes and as active polymer optical fibers.Besides pursuing the high luminescence quantum yield of the hybrid polymers, other investigations have also been studied. The properties of the Eu(DBM)3phenx complex doped in PMMA with different molar ratios of phen (x = 0.0, 0.5, 1.0, and 1.5 mol%) were unraveled by Mishra and co-workers.185 The maximum fluorescence is observed for Eu(DBM)3phen1.5, which is new as compared to the usual composition. They observed a systematic and significant improvement in the thermal stability and fluorescence properties with increasing molar concentration of phen. Meanwhile, the glass transition temperature decreases with increasing phen concentration. It is suggested that this study may help in finding suitable materials for luminescence solar collectors, as well as other photonic devices. Lanthanide complexes have been introduced into the hybrid polymer-based oxo-hydroxo butyltin cluster to prepare novel hybrid materials (Fig. 28).186,187 The PMMA-co-Sn12Cluster matrix exhibited a high physical doping quantity of [Ln(TTA)3phen], which can be attributed to the relatively flexible framework and nanospace supported by the Sn12clusters of this kind of hybrid material. It is worth noting that the luminescent hybrid materials could be easily redissolved in organic solvent and repeatedly gelled, which endows the luminescent hybrid materials with shape controllability. Chen et al. demonstrated the synthesis of the first photoluminescent lanthanide conducting metallopolymer.188 The ligand, 3,8-bis(3,4-(ethylenedioxy)thien-2-yl)-1,10-phenanthroline, in which the 1,10-phenanthroline (phen) moiety serves as a metal binding group and 3,4-(ethylenedioxy)thiophene as a polymerizable group, was prepared. The structure is assembled from a well-characterized europium-containing monomer via controlled electropolymerization (Fig. 29). This approach represents a novel perspective on the use of luminescent conducting metallopolymers for a wide range of light-emitting applications.
 |
| | Fig. 28 The proposed mechanisms for the formation of PMMA-co-Sn12Cluster/Ln(TTA)3phen and PMMA-co-Sn12Cluster-co-LnAA(TTA)2phen hybrid materials (Ln =Eu, Er, Yb, Nd, and Sm). Reprinted with permission from ref. 187. Copyright 2010, Royal Society of Chemistry Publishing Company. | |
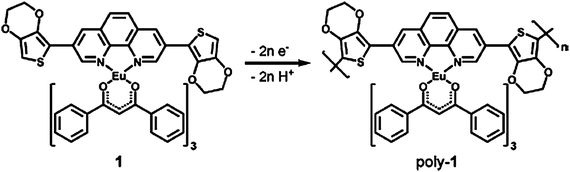 |
| | Fig. 29 Electrochemical polymerization of the europium-containing monomer to the conducting metallopolymer. Reprinted with permission from ref. 188. Copyright 2008, American Chemical Society. | |
7.2 Mixed systems
In order to obtain multicoloured materials, researchers mixed lanthanide complexes with different emission colours into the polymer matrices. Luo et al. co-doped the terbium and europium β-diketonate complex in the poly(N-vinylcarbazole) (PVK) matrix to construct white LEDs.189 In this case, PVK is not only used as the blue emitter but also as a matrix dispersing the green and red emitter. White light could be obtained by optimizing the concentration of the dopants and the excitation of different wavelengths. In this system, energy transfer from PVK to the terbium and europium complex and from the terbium to europium complex exists. Kai et al. reported the preparation and photoluminescence study of poly(methylmethacrylate) (PMMA) thin films co-doped with [Eu(tta)3(H2O)2] and [Tb(acac)3(H2O)2] complexes.190 In this work, the PMMA polymer matrix acts as a co-sensitizer and enhances the overall luminescence intensity of the polymer films. The emission properties of these multicoloured films can be fine-tuned by both the adjustment of the composition and the excitation wavelength, exhibiting two individual primary colours, green and red, as well as the intermediate emission colours (Fig. 30). Moreover, the phenomenon of an intermolecular energy transfer from the 5D4 level of the Tb3+ ion to the T1 state of the tta ligands is observed in polymer-based luminescent materials for the first time.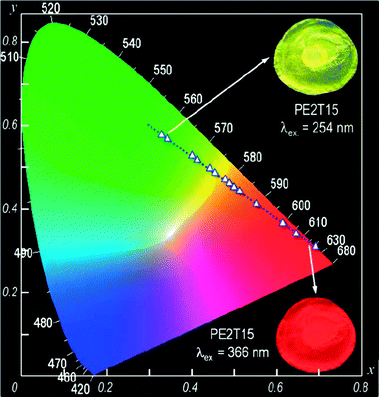 |
| | Fig. 30 CIE chromaticity diagram showing the x,y emission color coordinates for PMMA:Eu(tta)3:Tb(acac)3 films irradiated at different wavelengths. The inset figures are photographs of the film taken with a digital camera displaying the green and red emissions under UV irradiation at 254 and 366 nm, respectively. Reprinted with permission from ref. 190. Copyright 2011, Royal Society of Chemistry Publishing Company. | |
Recently, researchers recognized that the mixed matrices beyond the single polymer matrix could endow the hybrid materials with different microstructures and novel properties. Yan et al. prepared a series of organic–inorganic luminescent polymeric hybrid materials containing lanthanide complexes covalently bonded to silica (mesoporous or amorphous) and the polymers.191–195 In such materials, the functionalized organic ligands (e.g. TTA, DBM, ACAC, calix[4]arene, 4-mercaptobenzoic acid, and phenylphenacyl sulfoxide) are used as the first precursor, and the other precursor is the polymer (synthesized through the addition polymerization reaction of the corresponding monomer). It is concluded that the introduction of the polymer chain is favorable for the thermal stability, regular microstructure and luminescence of the overall hybrid systems. Moreover, the different structures of the polymers [polyacrylamide (PAM), polyvinylpyrrolidone (PVP), polymethyl methacrylate (PMMA) and polyethyl methacrylate (PEMA)] induce different microstructures and different photoluminescence behaviors (lifetime and quantum efficiency) for the hybrid systems. Tang's group successfully assembled a series of transparent homogeneous hybrid materials based on the silica-polymer mixed matrix embedded with the europium ternary complex.196 It is suggested that the hybrid materials based on the mixed matrix in comparison with the single silica matrix show a more efficient emission and a much higher thermal stability and exposure durability since the polymers interacted with europium complexes.
8. Bifunctional magnetic-optical composites
To give the materials more interesting character, more than one functional component could be introduced into a single micro- or nanocomposite system, such as magnetic nanoparticles and luminescent materials. Such materials would be very useful in the biomedical and biopharmaceutical fields. Bifunctional materials with both magnetic and optical properties have recently become a class of intriguing materials. Much work has been focused on fabricating such nanoarchitectures by combining the magnetic nanoparticles with the luminescent reagents.197–200 For applications involving magnetic delivery or separation, superparamagnetic properties are more desirable than ferromagnetism, because there could be no residual magnetism after the removal of the magnetic field. Thus, γ-Fe2O3 and Fe3O4 nanoparticles were selected as the magnetic component because of their superparamagnetic properties. For luminescent materials, organic fluorescent compounds and quantum dots (QDs) have been used as fluorescent labels. However, rapid photobleaching of organic fluorescent compounds and the potential toxicity of QDs would hinder them from biological detection and medical diagnosis. In other cases, ruthenium(II) complexes [Ru(bpy)32+] were used as luminescent markers in bioanalysis and biodetection or in electrochemiluminescence (ECL) fields.201,202 As we know, lanthanide complexes show narrow band photoluminescence and have a high luminescence quantum yield, which makes them interesting candidates for luminescence applications such as biochemical sensors and fluoroimmunoassays.For instance, our group reported a simple and straightforward synthetic method by a modified Stöber method combined with the layer-by-layer assembly technique to prepare the bifunctional materials.203 The steps involve covering the Fe3O4 nanoparticles with silica layers (Fe3O4@SiO2), grafting the terbium complex to silica through N-(4-benzoic acid-yl),N′-(propyltriethoxysilyl) urea (Fe3O4@SiO2@PABI-Tb), and coating an extra silica shell on the bifunctional nanoparticles (Fig. 31).
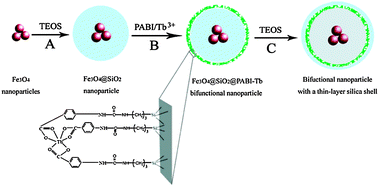 |
| | Fig. 31 Synthetic procedure to obtain bifunctional magnetic–optical nanoparticles. Reprinted with permission from ref. 203. Copyright 2007, American Chemical Society. | |
Moreover, suitable structures of the bifunctional composites for special applications, such as mesoporous structures, are designed.204 Our group reported a new kind of bifunctional nanocomposite with Fe3O4 magnetic particles encapsulated in the cores of mesoporous silica nanospheres, following by bonding neodymium/ytterbium complexes to the framework of mesoporous silica based on functionalized 1,10-phenanthroline. In a successive study, Eu(TTA)3phen was used instead of Ln(DBM)3phen (Ln = Nd, Yb).205 The detailed steps are as follows: (1) introducing the oleic acid stabilized Fe3O4 magnetic particles and functionalized 1,10-phenanthroline synchronously at the time of forming the mesoporous silica nanospheres; (2) introducing the lanthanide complex [Ln(DBM)3(H2O)2 (Ln = Nd, Yb), Eu(TTA)3(H2O)2] into the channels of the mesoporous silica nanospheres by a simple ligand exchange reaction (Fig. 32). Thus, the mesoporous nanocomposites with unique luminescent (Fig. 33) and magnetic properties (Fig. 34) were obtained. Such nanocomposites are potential materials for applications in drug delivery, optical imaging or sensors, which have the following advantages. (a) Mesoporous silica with a nontoxic and biocompatible nature, a large surface area, a large pore volume, and a tunable pore diameter with abundant Si–OH bonds on the pore surface is a promising candidate for use as a carrier in drug delivery systems. (b) The emissions from NIR luminescent nanocomposites are critical for in vivo optical imaging, since the blood and tissues are relatively transparent in the NIR region, especially in the range of 700–1000 nm, thereby minimizing complications resulting from intrinsic background interference. (c) The europium ion is one of the most intense emitting ions among the lanthanide ions. The Yb3+ ion emits relatively intense NIR luminescence compared to the other lanthanide ions. (d) It can be guided to target sites by means of an external magnetic field.
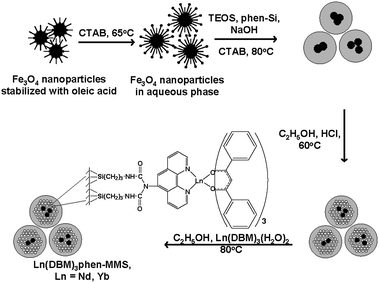 |
| | Fig. 32 Synthesis of Ln(DBM)3phen-MMS (Ln = Nd, Yb) nanospheres. Reprinted with permission from ref. 204. Copyright 2010, American Chemical Society. | |
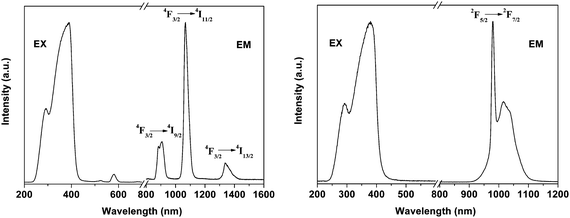 |
| | Fig. 33 Excitation (EX) and emission (EM) spectra of Nd(DBM)3phen-MMS and Yb(DBM)3phen-MMS. Reprinted with permission from ref. 204. Copyright 2010, American Chemical Society. | |
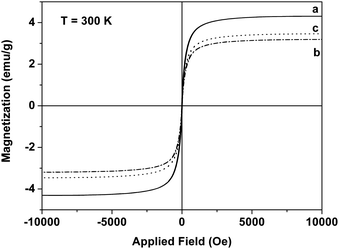 |
| | Fig. 34 Magnetization curves of surfactant-extracted phen-MMS (a), Nd(DBM)3phen-MMS (b), and Yb(DBM)3phen-MMS (c) at 300 K. Reprinted with permission from ref. 204. Copyright 2010, American Chemical Society. | |
Hybrid macroporous materials doped with lanthanide complexes and Fe3O4 magnetic particles have also been investigated.206 The macroporous structure could accommodate bigger chemical species than mesoporous materials do. Also, 3D-ordered macroporous materials have photonic stop bands and have potential applications in optical waveguides, optical circuits, and low-threshold telecommunications lasers.207–212 Our group synthesized a near-infrared luminescent/magnetic bifunctional macroporous material by integrating Fe3O4@polystyrene core–shell nanoparticles (Fe3O4@PS) and erbium complexes into a macroporous construction (Fig. 35).206 In such material, the erbium complex could be replaced by other lanthanide complexes.213 Thus, a series of bifunctional macroporous materials, which emit in the visible or near-IR region for different applications, could be obtained (Fig. 36). One could select different structures and porous sizes of the host functional material for special applications.
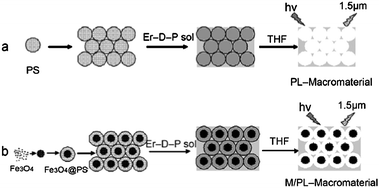 |
| | Fig. 35 Synthetic procedures for the PL-Macromaterial (a) and the M/PL-Macromaterial (b). Reprinted with permission from ref. 206. Copyright 2008, Wiley Publishing Company. | |
 |
| | Fig. 36 SEM images: PS (a), PL-Macromaterial film (b), magnified view of PL-Macromaterial (c), M/PL-Macromaterial film (e), and magnified view of M/PL-Macromaterial (f). TEM image of Fe3O4@PS (d). Reprinted with permission from ref. 206. Copyright 2008, Wiley Publishing Company. | |
9. Conclusions and outlook
Generally, hybrid materials based on lanthanide organic complexes can be grouped into two subclasses on the basis of the type of interaction established between the organic and inorganic components. While in class I the organic component is bonded physically to the inorganic phase (i.e., via hydrogen bonding or van der Waals forces), class II includes all those hybrids in which the organic and inorganic components are bonded through strong chemical bonds, in particular covalent bonds. However, the basic problem is how to enhance the luminescence intensities and further improve the luminescence properties of such hybrids. To achieve this target, one route is to select and design the organic ligand correctly. The relevant aspects are as follows: (i) the ligand should contain a number of properly organized binding sites providing complete saturation of the Ln3+ sphere coordination sites, in order to minimize the nonradiative deactivation by vibronic quenchers; and (ii) the energy gap between the triplet state of the ligand and the resonance level of the Ln3+ cation should be proper, to ensure the efficient energy transfer and to avoid possible back energy transfer deactivation processes. The other route is to incorporate lanthanide organic complexes into a stable matrix. Various hosts enrich the family of hybrid materials based on lanthanide organic complexes. At present, research into hybrid materials based on lanthanide organic complexes are devoted to class II hybrids because, compared with class I hybrids, many problems, such as clustering of the emitters, inhomogeneous dispersion of components, leaching of dopants, and limited concentration of organic species, could be resolved. Furthermore, multi-functionalities introduced into one system rather than luminescent properties is a trend. To researchers, it is also a big challenge to put these hybrids into practical applications, but they will not stop pursuing this goal, especially with hybrids that could be used in the field of biology.Acknowledgements
The authors are grateful for the financial aid from the National Natural Science Foundation of China (Grant Nos. 21071140, 21001100), National Natural Science Foundation of China Major Project (Grant Nos. 91122030), ‘863’ Plan-National High Technology Research and Development Program of China (Grant Nos. 2011AA03A407) and National Natural Science Foundation for Creative Research Group (Grant Nos. 20921002).References
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214 CrossRef CAS.
- L. R. Melby, N. J. Rose, E. Abramson and J. C. Caris, J. Am. Chem. Soc., 1964, 86, 5117 CrossRef CAS.
- K. Binnemans, P. Lenaerts, K. Driesen and C. Görller-Walrand, J. Mater. Chem., 2004, 14, 191 RSC.
- L. D. Carlos, R. A. Sá Ferreira, J. P. Rainho and V. de Zea Bermudez, Adv. Funct. Mater., 2002, 12, 819 CrossRef CAS.
- V. Bekiari and P. Lianos, Adv. Mater., 1998, 10, 1455 CrossRef CAS.
- N. Sabbatini, A. Mecati, M. Guardigli, V. Balazani, J. M. Lehn, R. Zeissel and R. Ungaro, J. Lumin., 1991, 48–49, 463 CrossRef CAS.
- N. Sabbatini, M. Guardigli and J. M. Lehn, Coord. Chem. Rev., 1993, 123, 201 CrossRef CAS.
- K. Driesen, R. V. Deun, C. Görller-Walrand and K. Binnemans, Chem. Mater., 2004, 16, 1531 CrossRef CAS.
- T. Jin, S. Tsutsumi, Y. Deguchi, K. Machida and G. Adachi, J. Electrochem. Soc., 1995, 142, 195 CrossRef.
- D. W. Dong, S. C. Jiang, Y. F. Men, X. L. Ji and B. Z. Jiang, Adv. Mater., 2000, 12, 646 CrossRef CAS.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2007, 107, 2592 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2005, 105, 4148 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2002, 102, 2303 CrossRef CAS.
- L. D. Carlos, R. A. S. Ferreira, V. de Zea Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509 CrossRef CAS.
- C. Sanchez, B. Lebeau, F. Chaput and J.-P. Boilot, Adv. Mater., 2003, 15, 1969 CrossRef CAS.
- P. Escribano, B. Julián-López, J. Planelles-Aragó, E. Cordoncillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23 RSC.
- C. Sanchez, P. Belleville, M. Popalld and L. Nicole, Chem. Soc. Rev., 2011, 40, 696 RSC.
- K. Kuriki, Y. Koike and Y. Okamoto, Chem. Rev., 2002, 102, 2347 CrossRef CAS.
- S. I. Klink, G. A. Hebbink, L. Grave, F. C. J. M. van Veggel, D. N. Reinhoudt, L. H. Slooff, A. Polman and J. W. Hofstraat, J. Appl. Phys., 1999, 86, 1181 CrossRef CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS.
- A. Beeby, S. Faulkner, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 2001, 1268 RSC.
- R. Van Deun, P. Fias, K. Binnemans and C. Görller-Walrand, Phys. Chem. Chem. Phys., 2003, 5, 2754 RSC.
- G. A. Crosby, R. E. Whan and R. M. Alire, J. Chem. Phys., 1961, 34, 743 CrossRef CAS.
- N. Filipescu, W. F. Sager and F. A. Serafin, J. Phys. Chem., 1964, 68, 3324 CrossRef CAS.
- K. Binnemans, Rare-Earth Beta-Diketonates. in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr., J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, Amsterdam, 2005, vol. 35, ch. 225, p. 107 Search PubMed.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef.
- K. Kojima, K. Tsuchiya and N. Wada, J. Sol-Gel Sci. Technol., 2000, 19, 511 CrossRef CAS.
- L. H. Slooff, M. J. A. de Dood, A. van Blaaderen and A. Polman, J. Non-Cryst. Solids, 2001, 296, 158 CrossRef CAS.
- T. Ishizaka and Y. Kurokawa, J. Appl. Phys., 2001, 90, 243 CrossRef CAS.
- S. Y. Chen, C. C. Ting and W. F. Hsieh, Thin Solid Films, 2003, 434, 171 CrossRef CAS.
- Z. Elalamy, E. Drouard, T. Mcgovern, L. Escoubas, J. J. Simon and F. Flory, Opt. Commun., 2004, 235, 365 CrossRef CAS.
- G. D. Qian, M. Q. Wang, M. Wang, X. P. Fan and Z. L. Hong, J. Mater. Sci. Lett., 1997, 16, 322 CrossRef CAS.
- G. D. Qian and M. Q. Wang, Mater. Res. Bull., 2001, 36, 2289 CrossRef CAS.
- G. D. Qian, Z. Yang and M. Q. Wang, J. Lumin., 2002, 96, 211 CrossRef CAS.
- G. D. Qian, M. Q. Wang and Z. Yang, J. Phys. Chem. Solids, 2002, 63, 1829 CrossRef CAS.
- L. S. Fu, R. A. S. Ferreira, S. S. Nobre, L. D. Carlos and J. Rocha, J. Lumin., 2007, 122, 265 CrossRef.
- L. N. Sun, H. J. Zhang, Q. G. Meng, F. Y. Liu, L. N. Fu, C. Y. Peng, J. B. Yu, G. L. Zheng and S. B. Wang, J. Phys. Chem. B, 2005, 109, 6174 CrossRef CAS.
- K. Binnemans, P. Lenaerts, K. Driesen and C. Görller-Walrand, J. Mater. Chem., 2004, 14, 191 RSC.
- P. Lenaerts, A. Storms, J. Mullens, J. D'Haen, C. Görller-Walrand, K. Binnemans and K. Driesen, Chem. Mater., 2005, 17, 5194 CrossRef CAS.
- K. Driesen, R. Van Deun, C. Görller-Walrand and K. Binnemans, Chem. Mater., 2004, 16, 1531 CrossRef CAS.
- H. R. Li, J. Lin, H. J. Zhang, H. C. Li, L. S. Fu and Q. G. Meng, Chem. Commun., 2001, 1212 RSC.
- H. R. Li, J. Lin, H. J. Zhang, L. S. Fu, Q. G. Meng and S. B. Wang, Chem. Mater., 2002, 14, 3651 CrossRef CAS.
- F. Y. Liu, L. S. Fu, J. Wang, Q. G. Meng, H. R. Li, J. F. Guo and H. J. Zhang, New J. Chem., 2003, 27, 233 RSC.
- H. R. Li, J. B. Yu, F. Y. Liu, H. J. Zhang, L. S. Fu, Q. G. Meng, C. Y. Peng and J. Lin, New J. Chem., 2004, 28, 1137 RSC.
- S. Quici, C. Scalera, M. Cavazzini, G. Accorsi, M. Bolognesi, L. Armelao and G. Bottaro, Chem. Mater., 2009, 21, 2941 CrossRef CAS.
- L. N. Sun, H. J. Zhang, L. S. Fu, F. Y. Liu, Q. G. Meng, C. Y. Peng and J. B. Yu, Adv. Funct. Mater., 2005, 15, 1041 CrossRef CAS.
- B. R. Judd, Phys. Rev., 1962, 127, 750 CrossRef CAS.
- G. S. Oflet, J. Chem. Phys., 1962, 37, 511 CrossRef.
- S. Dang, L. N. Sun, H. J. Zhang, X. M. Guo, Z. F. Li, J. Feng, H. D. Guo and Z. Y. Guo, J. Phys. Chem. C, 2008, 112, 13240 CAS.
- J. Feng, L. Zhou, S. Y. Song, Z. F. Li, W. Q. Fan, L. N. Sun, Y. N. Yu and H. J. Zhang, Dalton Trans., 2009, 6593 RSC.
- L. N. Sun, H. J. Zhang, J. B. Yu, Q. G. Meng, F. Y. Liu and C. Y. Peng, J. Photochem. Photobiol., A, 2008, 193, 153 CrossRef CAS.
- C. K. Jørgensen and R. Reisfeld, J. Less Common Met., 1983, 93, 107 CrossRef.
- N. N. Lin, H. R. Li, Y. G. Wang, Y. Feng, D. S. Qin, Q. Y. Gan and S. D. Chen, Eur. J. Inorg. Chem., 2008, 4781 CrossRef CAS.
- Y. X. Zheng, J. Lin, Y. J. Liang, Q. Lin, Y. N. Yu, Q. G. Meng, Y. H. Zhou, S. B. Wang, H. Y. Wang and H. J. Zhang, J. Mater. Chem., 2001, 11, 2615 RSC.
- Y. Hasegawa, Y. Kimura, K. Murakoshi, Y. Wada, J. H. Kim, N. Nakashima, T. Yamanaka and S. Yanagida, J. Phys. Chem., 1996, 100, 10201 CrossRef CAS.
- J. L. Yuan and K. Matsumoto, Anal. Sci., 1996, 12, 31 CrossRef CAS.
- G. Mancino, A. J. Ferguson, A. Beeby, N. J. Long and T. S. Jones, J. Am. Chem. Soc., 2005, 127, 524 CrossRef CAS.
- G. E. Buono-Core, H. Li and B. Marciniak, Coord. Chem. Rev., 1990, 99, 55 CrossRef CAS.
- J. H. Melman, T. J. Emge and J. G. Brennan, Inorg. Chem., 2001, 40, 1078 CrossRef CAS.
- A.-S. Chauvin, F. Gumy, I. Matsubayashi, Y. Hasegawa and J.-C. G. Bünzli, Eur. J. Inorg. Chem., 2006, 473 CrossRef CAS.
- S. Yanagida, Y. Hasegawa, K. Murakoshi, Y. Wada, N. Nakashima and T. Yamanaka, Coord. Chem. Rev., 1998, 171, 461 CrossRef CAS.
- L. D. Carlos, C. De Mello Donegá, R. Q. Albuquerque, S. Jr. Alves, J. F. S. Menezes and O. L. Malta, Mol. Phys., 2003, 101, 1037 CrossRef CAS.
- J. Feng, J. B. Yu, S. Y. Song, L. N. Sun, W. Q. Fan, X. M. Guo, S. Dang and H. J. Zhang, Dalton Trans., 2009, 2406 RSC.
- H. S. He, M. Dubey, A. G. Sykes and P. Stanley May, Dalton Trans., 2010, 39, 6466 RSC.
- H. R. Li, N. N. Lin, Y. G. Wang, Y. Feng, Q. Y. Gan, H. J. Zhang, Q. L. Dong and Y. H. Chen, Eur. J. Inorg. Chem., 2009, 519 CrossRef CAS.
- X. M. Guo, H. D. Guo, L. S. Fu, L. D. Carlos, R. A. S. Ferreira, L. N. Sun, R. P. Deng and H. J. Zhang, J. Phys. Chem. C, 2009, 113, 12538 CAS.
- X. M. Guo, H. D. Guo, L. S. Fu, H. J. Zhang, L. D. Carlos, R. P. Deng and J. B. Yu, J. Photochem. Photobiol., A, 2008, 200, 318 CrossRef CAS.
- Y. H. Wang, B. Li, L. M. Zhang, Q. H. Zuo, L. N. Liu and P. Li, J. Colloid Interface Sci., 2010, 349, 505 CrossRef CAS.
- B. Yan, L. L. Kong and B. Zhou, J. Non-Cryst. Solids, 2009, 355, 1281 CrossRef CAS.
- H. F. Lu, B. Yan and J. L. Liu, Inorg. Chem., 2009, 48, 3966 CrossRef CAS.
- B. Yan, Q. M. Wang and D. J. Ma, Inorg. Chem., 2009, 48, 36 CrossRef CAS.
- L. N. Sun, S. Dang, J. B. Yu, J. Feng, L. Y. Shi and H. J. Zhang, J. Phys. Chem. B, 2010, 114, 16393 CrossRef CAS.
- B. H. Tong, S. J. Wang, J. Jiao, F. R. Ling, Y. Z. Meng and B. Wang, J. Photochem. Photobiol., A, 2007, 191, 74 CrossRef CAS.
- B. Yan and H. F. Lu, Inorg. Chem., 2008, 47, 5601 CrossRef CAS.
- N. N. Lin, H. R. Li, Y. G. Wang, Y. Feng, D. S. Qin, Q. Y. Gan and S. D. Chen, Eur. J. Inorg. Chem., 2008, 4781 CrossRef CAS.
- B. Yan and H. F. Lu, J. Organomet. Chem., 2009, 694, 2597 CrossRef CAS.
- L. Armelao, G. Bottaro, S. Quici, M. Cavazzini, M. C. Raffo, F. Barigelletti and G. Accorsi, Chem. Commun., 2007, 2911 RSC.
- F. F. Wang and B. Yan, J. Photochem. Photobiol., A, 2008, 194, 238 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615 CrossRef CAS.
- B. J. Scott, G. Wirnsberger and G. D. Stucky, Chem. Mater., 2001, 13, 3140 CrossRef CAS.
- A. Stein, Adv. Mater., 2003, 15, 763 CrossRef CAS.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821 CrossRef CAS.
- P. Selvam, S. K. Bhatia and C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237 CrossRef CAS.
- M. Fernandes, V. de Zea Bermudez, R. A. Sá Ferreira, L. D. Carlos, A. Charas, J. Morgado, M. M. Silva and M. J. Smith, Chem. Mater., 2007, 19, 3892 CrossRef CAS.
- C. Molina, K. Dahmouche, Y. Messadeq, S. J. L. Ribeiro, M. A. P. Silva, V. de Zea Bermudez and L. D. Carlos, J. Lumin., 2003, 104, 93 CrossRef CAS.
- C. Y. Peng, H. J. Zhang, J. B. Yu, Q. G. Meng, L. S. Fu, H. R. Li, L. N. Sun and X. M. Guo, J. Phys. Chem. B, 2005, 109, 15278 CrossRef CAS.
- X. M. Guo, L. S. Fu, H. J. Zhang, L. D. Carlos, C. Y. Peng, J. F. Guo, J. B. Yu, R. P. Deng and L. N. Sun, New J. Chem., 2005, 29, 1351 RSC.
- S. W. Li, H. W. Song, W. L. Li, X. G. Ren, S. Z. Lu, G. H. Pan, L. B. Fan, H. Q. Yu, H. Zhang, R. F. Qin, Q. L. Dai and T. Wang, J. Phys. Chem. B, 2006, 110, 23164 CrossRef CAS.
- Q. H. Xu, L. S. Li, B. Li and R. R. Xu, Microporous Mesoporous Mater., 2000, 38, 351 CrossRef CAS.
- Q. H. Xu, L. S. Li, X. S. Liu and R. R. Xu, Chem. Mater., 2002, 14, 549 CrossRef CAS.
- L. N. Sun, J. B. Yu, H. J. Zhang, Q. G. Meng, E. Ma, C. Y. Peng and K. Y. Yang, Microporous Mesoporous Mater., 2007, 98, 156 CrossRef CAS.
- L. N. Sun, Y. Zhang, J. B. Yu, C. Y. Peng and H. J. Zhang, J. Photochem. Photobiol., A, 2008, 199, 57 CrossRef CAS.
- J. Feng, S. Y. Song, W. Q. Fan, L. N. Sun, X. M. Guo, C. Y. Peng, J. B. Yu, Y. N. Yu and H. J. Zhang, Microporous Mesoporous Mater., 2009, 117, 278 CrossRef CAS.
- J. Feng, S. Y. Song, Y. Xing, H. J. Zhang, Z. F. Li, L. N. Sun, X. M. Guo and W. Q. Fan, J. Solid State Chem., 2009, 182, 435 CrossRef CAS.
- S. Gago, J. A. Fernandes, J. P. Rainho, R. A. Sá Ferreira, M. Pillinger, A. A. Valente, T. M. Santos, L. D. Carlos, P. J. A. Ribeiro-Claro and I. S. Gonçalves, Chem. Mater., 2005, 17, 5077 CrossRef CAS.
- E. DeOliveira, C. R. Neri, O. A. Serra and A. G. S. Prado, Chem. Mater., 2007, 19, 5437 CrossRef CAS.
- Y. Li, B. Yan and H. Yang, J. Phys. Chem. C, 2008, 112, 3959 CAS.
- Y. J. Li and B. Yan, Inorg. Chem., 2009, 48, 8276 CrossRef CAS.
- B. Yan and Y. Li, Dalton Trans., 2010, 39, 1480 RSC.
- Y. J. Li and B. Yan, Dalton Trans., 2010, 39, 2554 RSC.
- Y. J. Li, B. Yan and Y. Li, J. Solid State Chem., 2010, 183, 871 CrossRef CAS.
- Y. J. Li, B. Yan and Y. Li, Microporous Mesoporous Mater., 2010, 131, 82 CrossRef CAS.
- B. Yan and B. Zhou, J. Photochem. Photobiol., A, 2008, 195, 314 CrossRef CAS.
- L. L. Kong, B. Yan and Y. Li, J. Solid State Chem., 2009, 182, 1631 CrossRef.
- Y. J. Li, B. Yan and Y. Li, Chem.–Asian J., 2010, 5, 1642 CrossRef CAS.
- L. L. Kong, B. Yan and Y. Li, J. Alloys Compd., 2009, 481, 549 CrossRef CAS.
- S. Torelli, D. Imbert, M. Cantuel, G. Bernardinelli, S. Delahaye, A. Hauser, J.-C. G. Bünzli and C. Piguet, Chem.–Eur. J., 2005, 11, 3228 CrossRef CAS.
- L. N. Sun, H. J. Zhang, J. B. Yu, S. Y. Yu, C. Y. Peng, S. Dang, X. M. Guo and J. Feng, Langmuir, 2008, 24, 5500 CrossRef CAS.
- L. N. Sun, Y. Zhang, J. B. Yu, S. Y. Yu, S. Dang, C. Y. Peng and H. J. Zhang, Microporous Mesoporous Mater., 2008, 115, 535 CrossRef CAS.
- L. L. Kong, B. Yan, Y. J. Li and Y. Li, Microporous Mesoporous Mater., 2010, 135, 45 CrossRef CAS.
- Y. J. Li, B. Yan and L. Wang, Dalton Trans., 2011, 40, 6722 RSC.
- Y. Li and B. Yan, J. Fluoresc., 2009, 19, 191 CrossRef CAS.
- B. Yan and L. L. Kong, Nanoscale Res. Lett., 2010, 5, 1195 CrossRef CAS.
- Y. Li and B. Yan, Microporous Mesoporous Mater., 2010, 128, 62 CrossRef CAS.
- D. J. Zhang, D. H. Tang, X. M. Wang, Z. A. Qiao, Y. T. Li, Y. L. Liu and Q. S. Huo, Dalton Trans., 2011, 40, 9313 RSC.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS.
- B. J. Melde, B. T. Hollande, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867 CAS.
- T. Asefa, G. A. Ozin, H. Grondey, M. Kruk and M. Jaroniec, Stud. Surf. Sci. Catal., 2002, 141, 1 CrossRef CAS.
- S. Inagaki, Nanotechnology in Mesostructured Materials, Proceedings of the 3rd International Mesostructured Materials Symposium, Stud. Surf. Sci. Catal., ed. S. E. Park, R. Ryoo, W. S. Ahn, C. W. Lee and J. S. Chang, Elsevier, Amsterdam, 2003, vol. 146, p. 1 Search PubMed.
- M. C. Burleigh, S. Jayasundera, C. W. Thomas, M. S. Spector, M. A. Markowitz and B. P. Gaber, Colloid Polym. Sci., 2004, 282, 728 CAS.
- J. Morell, G. Wolter and M. Fröba, Chem. Mater., 2005, 17, 804 CrossRef CAS.
- W. H. Zhang, B. Daly, J. O'Callaghan, L. Zhang, J. L. Shi, C. Li, M. A. Morris and J. D. Holmes, Chem. Mater., 2005, 17, 6407 CrossRef CAS.
- M. A. Wahab, I. Imae, Y. Kawakami and C. S Ha, Chem. Mater., 2005, 17, 2165 CrossRef CAS.
- B. Hatton, K. Landskron, W. Whitnall, D. Perovic and G. A. Ozin, Acc. Chem. Res., 2005, 38, 305 CrossRef CAS.
- S. Z. Qiao, C. Z. Yu, Q. H. Hu, Y. G. Jin, X. F. Zhou, X. S. Zhao and G. Q. Lu, Microporous Mesoporous Mater., 2006, 91, 59 CrossRef CAS.
- M. C. Burleigh, M. A. Markowitz, S. Jayasundera, M. S. Spector, C. W. Thomas and B. P. Gaber, J. Phys. Chem. B, 2003, 107, 12628 CrossRef CAS.
- Q. H. Yang, J. Liu, J. Yang, L. Zhang, Z. C. Feng, J. Zhang and C. Li, Microporous Mesoporous Mater., 2005, 77, 257 CrossRef CAS.
- T. Asefa, M. Kruk, M. J. MacLachlan, N. Coombs, H. Grondey, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2001, 123, 8520 CrossRef CAS.
- D. M. Jiang, Q. H. Yang, H. Wang, G. R. Zhu, J. Yang and C. Li, J. Catal., 2006, 239, 65 CrossRef CAS.
- E. Besson, A. Mehdi, C. Reyé and R. J. P. Corriu, J. Mater. Chem., 2006, 16, 246 RSC.
- X. M. Guo, X. M. Wang, H. J. Zhang, L. S. Fu, H. D. Guo, J. B. Yu, L. D. Carlos and K. Y. Yang, Microporous Mesoporous Mater., 2008, 116, 28 CrossRef CAS.
- X. M. Guo, H. D. Guo, L. S. Fu, H. J. Zhang, R. P. Deng, L. N. Sun, J. Feng and S. Dang, Microporous Mesoporous Mater., 2009, 119, 252 CrossRef CAS.
- J. J. E. Moreau, B. P. Pichon, M. W. C. Man, C. Bied, H. Pritzkow, J. L. Bantignies, P. Dieudonné and J.-L. Sauvajol, Angew. Chem., Int. Ed., 2004, 43, 203 CrossRef CAS.
- J. N. Li, T. Qi, L. N. Wang, Y. Zhou, C. H. Liu and Y. Zhang, Microporous Mesoporous Mater., 2007, 103, 184 CrossRef CAS.
- Y. J. Li, L. Wang and B. Yan, J. Mater. Chem., 2011, 21, 1130 RSC.
- L. N. Sun, H. J. Zhang, C. Y. Peng, J. B. Yu, Q. G. Meng, L. S. Fu, F. Y. Liu and X. M. Guo, J. Phys. Chem. B, 2006, 110, 7249 CrossRef CAS.
- X. M. Guo, H. D. Guo, L. S. Fu, R. P. Deng, W. Chen, J. Feng, S. Dang and H. J. Zhang, J. Phys. Chem. C, 2009, 113, 2603 CAS.
- M. H. Bartil, S. W. Boettcher, K. F. Frindell and G. D. Stucky, Acc. Chem. Res., 2005, 38, 263 CrossRef.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef.
- A. Conde-Gallardo, M. Garcia-Rocha, I. Hernández-Calderón and R. Palomino-Merino, Appl. Phys. Lett., 2001, 78, 3436 CrossRef CAS.
- K. L. Frindell, M. H. Bartl, A. Popitsch and G. D. Stucky, Angew. Chem., Int. Ed., 2002, 41, 960 CrossRef.
- K. L. Frindell, M. H. Bartl, M. R. Robinson, G. C. Bazan, A. Popitsch and G. D. Stucky, J. Solid State Chem., 2003, 172, 81 CrossRef CAS.
- H. Xin, Y. Ebina, R. Ma, K. Takada and T. Sasaki, J. Phys. Chem. B, 2006, 110, 9863 CrossRef CAS.
- Y. G. Wang, L. Wang, H. R. Li, P. Liu, D. S. Qin, B. Y. Liu, W. J. Zhang, R. P. Deng and H. J. Zhang, J. Solid State Chem., 2008, 181, 562 CrossRef CAS.
- F. Xie, H. Yang, C. J. Xu and X. H. Zhang, J. Zhejiang Univ., Sci., 2005, 6A, 619 CrossRef.
- Y. G. Wang, L. Wang, H. R. Li, P. Liu, D. S. Qin, B. Y. Liu, W. J. Zhang, R. P. Deng and H. J. Zhang, J. Solid State Chem., 2008, 181, 562 CrossRef CAS.
- P. Liu, H. R. Li, Y. G. Wang, B. Y. Liu, W. J. Zhang, Y. J. Wang, W. D. Yan, H. J. Zhang and U. Schubert, J. Mater. Chem., 2008, 18, 735 RSC.
- H. R. Li, P. Liu, Y. G. Wang, L. Zhang, J. B. Yu, H. J. Zhang, B. Y. Liu and U. Schubert, J. Phys. Chem. C, 2009, 113, 3945 CAS.
- K. Binnemans, P. Lenaerts, K. Driesen and C. Görller-Walrand, J. Mater. Chem., 2004, 14, 191 RSC.
- X. L. Wang and B. Yan, Colloid Polym. Sci., 2011, 289, 423 CAS.
- L. Guo, L. S. Fu, R. A. S. Ferreira, L. D. Carlos, Q. P. Li and B. Yan, J. Mater. Chem., 2011, 21, 15600 RSC.
- C. Wang and B. Yan, Mater. Res. Bull., 2011, 46, 2515 CrossRef CAS.
- B. Yan and Y. J. Li, J. Mater. Chem., 2011, 21, 18454 RSC.
- Y. Zhao and B. Yan, Photochem. Photobiol., 2012, 88, 21 CrossRef CAS.
- E. Ranyuk, C. M. Douaihy, A. B. Lemeune and R. Guilard, New J. Chem., 2011, 35, 1189 RSC.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef CAS.
- Ionic Liquids in Synthesis, ed. P.Wasserscheid and T.Welton, VCH-Wiley, Weinheim, 2003 Search PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- K. Driesen, P. Nockemann and K. Binnemans, Chem. Phys. Lett., 2004, 395, 306 CrossRef CAS.
- P. Nockemann, E. Beurer, K. Driesen, R. Van Deun, K. VanHecke, L. Van Meervelt and K. Binnemans, Chem. Commun., 2005, 4354 RSC.
- L. N. Puntus, K. J. Schenk and J.-C. G. Bünzli, Eur. J. Inorg. Chem., 2005, 4739 CrossRef CAS.
- L. N. Puntus, I. S. Pekareva, K. A. Lyssenko, A. S. Shaplov, E. I. Lozinskaya, A. T. Zdvizhkov, M. I. Buzin and Y. S. Vygodskii, Opt. Mater., 2010, 32, 707 CrossRef CAS.
- S. F. Tang, J. Cybinska and A.-V. Mudring, Helv. Chim. Acta, 2009, 92, 2375 CrossRef CAS.
- H. R. Li, H. F. Shao, Y. G. Wang, D. S. Qin, B. Y. Liu, W. J. Zhang and W. D. Yan, Chem. Commun., 2008, 5209 RSC.
- H. R. Li, P. Liu, H. F. Shao, Y. G. Wang, Y. X. Zheng, Z. Sun and Y. H. Chen, J. Mater. Chem., 2009, 19, 5533 RSC.
- H. R. Li, D. Li, Y. G. Wang and Q. R. Ru, Chem.–Asian. J., 2011, 6, 1443 CrossRef CAS.
- K. Lunstroot, K. Driesen, P. Nockemann, C. Görller-Walrand, K. Binnemans, S. Bellayer, J. Le Bideau and A. Vioux, Chem. Mater., 2006, 18, 5711 CrossRef CAS.
- K. Lunstroot, K. Driesen, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Görller-Walrand, K. Binnemans, S. Bellayer, L. Viau, J. Le Bideau and A. Vioux, Dalton Trans., 2009, 298 RSC.
- Y. Feng, H. R. Li, Q. Y. Gan, Y. G. Wang, B. Y. Liu and H. J. Zhang, J. Mater. Chem., 2010, 20, 972 RSC.
- M. A. Neouze, J. Le Bideau, F. Lerroux and A. Vioux, Chem. Commun., 2005, 1082 RSC.
- M. A. Neouze, J. Le Bideau and A. Vioux, Prog. Solid State Chem., 2005, 33, 217 CrossRef CAS.
- M. A. Neouze, J. Le Bideau, P. Gaveau, S. Bellayer and A. Vioux, Chem. Mater., 2006, 18, 3931 CrossRef CAS.
- Y. G. Wang, Y. Feng, H. S. Zhao, Q. Y. Gan and X. Y. Yu, J. Sol-Gel Sci. Technol., 2011, 58, 711 CrossRef CAS.
- K. Lunstroot, K. Driesen, P. Nockemann, L. Viau, P. H. Mutin, A. Viouxb and K. Binnemans, Phys. Chem. Chem. Phys., 2010, 12, 1879 RSC.
- S. Biju, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, J. Mater. Chem., 2009, 19, 5179 RSC.
- O. Moudam, B. C. Rowan, M. Alamiry, P. Richardson, B. S. Richards, A. C. Jones and N. Robertson, Chem. Commun., 2009, 6649 RSC.
- D. B. A. Raj, B. Francis, M. L. P. Reddy, R. R. Butorac, V. M. Lynch and A. H. Cowley, Inorg. Chem., 2010, 49, 9055 CrossRef CAS.
- A. K. Singh, S. K. Singh, H. Mishra, R. Prakash and S. B. Rai, J. Phys. Chem. B, 2010, 114, 13042 CrossRef CAS.
- W. Q. Fan, J. Feng, S. Y. Song, Y. Q. Lei, G. L. Zheng and H. J. Zhang, Chem.–Eur. J., 2010, 16, 1903 CrossRef CAS.
- W. Q. Fan, J. Feng, S. Y. Song, Y. Q. Lei, L. Zhou, G. L. Zheng, S. Dang, S. Wang and H. J. Zhang, Nanoscale, 2010, 2, 2096 RSC.
- X. Y. Chen, X. P. Yang and B. J. Holliday, J. Am. Chem. Soc., 2008, 130, 1546 CrossRef CAS.
- Y. H. Luo, Q. Yan, Z. S. Zhang, X. W. Yu, W. X. Wu, W. Su and Q. J. Zhang, J. Photochem. Photobiol., A, 2009, 206, 102 CrossRef CAS.
- J. Kai, M. C. F. C. Felinto, L. A. O. Nunes, O. L. Malta and H. F. Brito, J. Mater. Chem., 2011, 21, 3796 RSC.
- K. Sheng and B. Yan, J. Photochem. Photobiol., A, 2009, 206, 140 CrossRef CAS.
- X. F. Qiao, H. Y. Zhang and B. Yan, Dalton Trans., 2010, 39, 8882 RSC.
- K. Sheng and B. Yan, J. Mater. Sci.: Mater. Electron., 2010, 21, 65 CrossRef CAS.
- K. Sheng, B. Yan, H. F. Lu and L. Guo, Eur. J. Inorg. Chem., 2010, 3498 CrossRef CAS.
- L. Guo, B. Yan, J. L. Liu, K. Sheng and X. L. Wang, Dalton Trans., 2011, 40, 632 RSC.
- X. G. Huang, Q. Wang, X. H. Yan, J. Xu, W. S. Liu, Q. Wang and Y. Tang, J. Phys. Chem. C, 2011, 115, 2332 CAS.
- J. Kin, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C.-H. Shin, J.-G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688 CrossRef.
- Y.-S. Lin, S.-H. Wu, Y. Hung, Y.-H. Chou, C. Chang, M.-L. Lin, C.-P. Tsai and C.-Y. Mou, Chem. Mater., 2006, 18, 5170 CrossRef CAS.
- L. H. Zhang, B. F. Liu and S. J. Dong, J. Phys. Chem. B, 2007, 111, 10448 CrossRef CAS.
- M. J. Li, Z. F. Chen, V. W.-W. Yam and Y. B. Zu, ACS Nano, 2008, 2, 905 CrossRef CAS.
- L. H. Zhang, B. F. Liu and S. J. Dong, J. Phys. Chem. B, 2007, 111, 10448 CrossRef CAS.
- M. J. Li, Z. F. Chen, V. W.-W. Yam and Y. B. Zu, ACS Nano, 2008, 2, 905 CrossRef CAS.
- S. Y. Yu, H. J. Zhang, J. B. Yu, C. Wang, L. N. Sun and W. D. Shi, Langmuir, 2007, 23, 7836 CrossRef CAS.
- J. Feng, H. J. Zhang, J. B. Yu, C. Wang, L. N. Sun and W. D. Shi, Langmuir, 2010, 26, 3596 CrossRef CAS.
- J. Feng, W. Q. Fan, S. Y. Song, Y. N. Yu, R. P. Deng and H. J. Zhang, Dalton Trans., 2010, 39, 5166 RSC.
- W. Q. Fan, J. Feng, S. Y. Song, Y. Q. Lei, Y. Xing, R. P. Deng, S. Dang and H. J. Zhang, Eur. J. Inorg. Chem., 2008, 5513 CrossRef CAS.
- R. C. Schroden and A. Stein, “3D Ordered Macroporous Materials”, in Colloids and Colloid Assemblies, ed. F. Caruso, Wiley-VCH, Weinheim, Germany, 2004, pp. 465–493 Search PubMed.
- E. Yablonovitch, J. Opt. Soc. Am. B, 1993, 10, 283 CrossRef CAS.
- J. D. Joannopoulos, P. R. Villeneuve and S. Fan, Nature, 1997, 386, 143 CrossRef CAS.
- S. Y. Lin, E. Chow, V. Hietala, P. R. Villeneuve and J. D. Joannopoulos, Science, 1998, 282, 274 CrossRef CAS.
- Y. Lu, Y. Yin and Y. Xia, Nano Lett., 2002, 2, 785 CrossRef CAS.
- B. Rodriguez-Gonzalez, V. Salgueirino-Maceria, F. Garcia-Santamaria and L. M. Liz-Marzan, Nano Lett., 2002, 2, 471 CrossRef CAS.
- W. Q. Fan, J. Feng, S. Y. Song, Y. Q. Lei, S. Dang and H. J. Zhang, Opt. Mater., 2011, 33, 582 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.