 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton transfer and hydrogen bonding in the organic solid state: a combined XRD/XPS/ssNMR study of 17 organic acid–base complexes†
Joanna S.
Stevens
*a,
Stephen J.
Byard
b,
Colin C.
Seaton
a,
Ghazala
Sadiq
a,
Roger J.
Davey
a and
Sven L. M.
Schroeder
*ac
aSchool of Chemical Engineering and Analytical Science, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: joanna.stevens@manchester.ac.uk; s.schroeder@manchester.ac.uk
bCovance Laboratories, Willowburn Avenue, Alnwick, Northumberland NE66 2JH, UK
cSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 5th November 2013
Abstract
The properties of nitrogen centres acting either as hydrogen-bond or Brønsted acceptors in solid molecular acid–base complexes have been probed by N 1s X-ray photoelectron spectroscopy (XPS) as well as 15N solid-state nuclear magnetic resonance (ssNMR) spectroscopy and are interpreted with reference to local crystallographic structure information provided by X-ray diffraction (XRD). We have previously shown that the strong chemical shift of the N 1s binding energy associated with the protonation of nitrogen centres unequivocally distinguishes protonated (salt) from hydrogen-bonded (co-crystal) nitrogen species. This result is further supported by significant ssNMR shifts to low frequency, which occur with proton transfer from the acid to the base component. Generally, only minor chemical shifts occur upon co-crystal formation, unless a strong hydrogen bond is formed. CASTEP density functional theory (DFT) calculations of 15N ssNMR isotropic chemical shifts correlate well with the experimental data, confirming that computational predictions of H-bond strengths and associated ssNMR chemical shifts allow the identification of salt and co-crystal structures (NMR crystallography). The excellent agreement between the conclusions drawn by XPS and the combined CASTEP/ssNMR investigations opens up a reliable avenue for local structure characterization in molecular systems even in the absence of crystal structure information, for example for non-crystalline or amorphous matter. The range of 17 different systems investigated in this study demonstrates the generic nature of this approach, which will be applicable to many other molecular materials in organic, physical, and materials chemistry.
Introduction
The crystallization of solid state acid–base donor–acceptor systems with more than one component is a commonly used route to tailor the physical and chemical properties of solid organic products and their formulations. For example, during the development of active pharmaceutical ingredients (APIs), the physicochemical properties of the pure API may be unsuitable for a chosen drug delivery route. Combining with a Brønsted donor or acceptor can result in intermolecular bonding, either by forming an ionic (salt) or hydrogen-bonded (co-crystal1–3) complex with superior properties,1–4 which may include solubility,5 bioavailability,5,6 chemical and physical stability,7,8 hygroscopicity,9 and mechanical properties.10,11Single crystal X-ray diffraction (XRD) is most commonly used for determining whether proton transfer or hydrogen bonding takes place between acid and base components,3,5,12–18 often in conjunction with an analysis of structural indicators such as bond angles and bond lengths.3,12,16,19 However, the unequivocal determination of hydrogen positions is not always straightforward, particularly with systems exhibiting proton disorder, temperature-dependent migration, or other unusual behaviour.18 Analysis by XRD is also constrained by a requirement for suitable single crystals, which may not be available when solid-state preparation techniques such as milling are used to form materials with more than one component.20,21 Where single crystal XRD fails other commonly available laboratory techniques such as vibrational spectroscopies can sometimes provide the required information.12,13 Often, however, the determination of hydrogen and proton positions has to rely on more advanced techniques such as neutron diffraction,12,18,19as demonstrated for, e.g., urea/phosphoric acid,17,22,23 4,4′-bipyridyl/benzene-1,2,4,5-tetracarboxylic acid,24 4-methylpyridine/pentachlorophenol,25 and benzoic acid.26,27
Over the last decade, solid-state NMR (ssNMR) methods combined with computational chemical shift analysis5,28,29 have been added to the inventory of techniques for the characterisation of hydrogen bonding and Brønsted interactions in the organic solid state. Studies have shown that H-bonding and proton transfer in two-component systems can be distinguished using 15N ssNMR,5,21,29–32 as for example shown for co-crystals and salts of a cancer-treatment API5 and for theophylline (1,3-dimethyl-7H-purine-2,6-dione) systems.21,28,30 Density functional theory (DFT) can be applied to correlate 15N ssNMR parameters with crystal structures. For example, calculations for the co-crystal form of a pharmaceutical API correctly predicted chemical shifts (to low frequency) relative to the free base on formation of strong H-bonds, in agreement with experimental data.5 Similarly, comparison of predicted 15N chemical shift values for complexes of dabco (1,4-diazabicyclo[2.2.2]-octane) and dicarboxylic acids with experimental data confirmed Brønsted and H-bonding interactions in these systems.29
Recently, we reported that X-ray photoelectron spectroscopy (XPS) also reliably detects proton transfer: a strong positive N 1s binding energy shift occurs due to protonation and identifies the formation of a salt.21,28,30,33 The information of this chemical shift analysis is conceptually very similar to that used in the ssNMR approach,28 opening up an avenue to mutual validation of results obtained with either technique. The principle of a large N 1s shift with protonation was applied to identify the nature of meta-aminobenzoic acid polymorphs (non-ionic vs. zwitterionic),34 and an O 1s shift to higher binding energy was also observed for an oxygen proton acceptor with formation of a H-bond on moving from the gas to condensed phase.35 Very importantly, long-range order is not a pre-requisite for the application of XPS and ssNMR; both techniques are also applicable to systems for which single crystal XRD would be unfeasible, for example when a suitable single crystal cannot be obtained, or when non-crystalline or amorphous samples are analysed.36
The feasibility of a combined ssNMR/XPS approach to local structure characterization in the organic solid state was introduced for theophylline co-crystal/salt systems,28 but to explore it more systematically we report here the examination of the nature of the intermolecular interactions between components of 17 acid–base complexes (Table 1 and Fig. 1), comparing XPS results with experimental and calculated 15N ssNMR data. As in our previous XPS study,33 we incorporate several development API substances alongside a range of non-development acid–base complexes including theophylline, aminobenzoic, and isonicotinamide base components with different acid co-formers and thus differing acid strength.
| Salt or co-crystal | ΔpKa | |
|---|---|---|
| 4-Aminobenzoic acid/4-hydroxy-3-nitrobenzoic acid | C | −3.9 |
| Isonicotinamide/4-hydroxy-3-nitrobenzoic acid | C | −3.0 |
| Theophylline/glutaric acid | C | −2.6 |
| Theophylline/citric acid | C | −1.4 |
| Theophylline/malonic acid | C | −1.1 |
| 4-Aminobenzoic acid/3,5-dinitrobenzoic acid | C | −0.3 |
| Theophylline/maleic acid | C | −0.2 |
| Theophylline/oxalic acid | C | 0.3 |
| Fumaric acid co-crystal 1c | C | 0.3 |
| Isonicotinamide/2,4,6-trihydroxybenzoic acid | S | 1.8 |
| Theophylline/5-sulfosalicylic acid dihydrate | S | 2.3 |
| Theophylline/5-sulfosalicylic acid monohydrate | S | 2.3 |
| 3,5-Diaminobenzoic acid/3,5-dinitrobenzoic acid | S | 2.5 |
| Difumarate salt 2 | S | 7.2, 4.6 |
| Di-HCl salt 1s | S | 11.3, 3.3 |
| HCl salt 3 | S | 15.4 |
| HCl salt 4 | S | 17.7 |
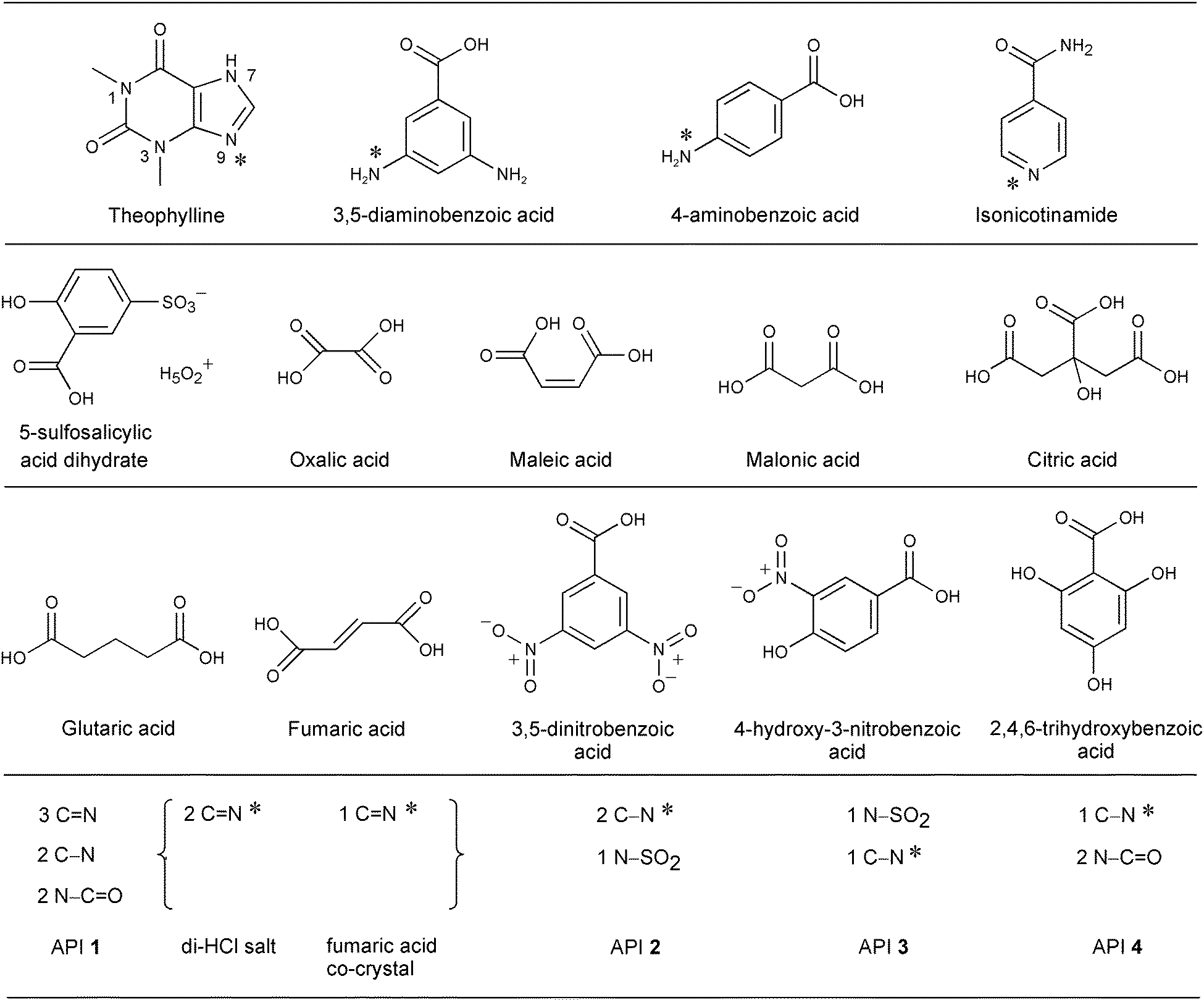 | ||
| Fig. 1 Chemical structures of the individual components (the nitrogen moieties are listed for the development drug substances). The asterisks (*) indicate the nitrogen atoms sensitive to protonation (Brønsted donation) or intermolecular H-bonding in this study. | ||
These systems cover a wide range of pKa differences (ΔpKa), from −3.9 to +17.7, between the acceptor and donor functional groups, which will allow us to examine correlations between ΔpKa values and the chemical shifts observable by XPS and ssNMR in a systematic manner. Of the 17 complexes, 6 (4 salts, 2 co-crystals) are within the range 0 < ΔpKa < 3 (Table 1), for which it is generally unpredictable whether H-bonding or proton transfer takes place.12,37
Experimental
Starting materials
All non-development starting materials were obtained with >99% purity (Sigma-Aldrich, UK); co-crystal or salt formation was verified by comparison of X-ray powder diffraction (XRPD) patterns to known structures7,21,28,41–43 or XPS/ssNMR/XRD for new complexes. The five development complexes were used as supplied (Sanofi-Aventis, Alnwick) and comprise the dihydrochloride salt (1s) and fumaric acid co-crystal (1c) of an API 1, the difumarate salt 2, and the hydrochloride salts 3 and 4.Preparation of salts and co-crystals
The non-development co-crystals and salts were prepared by solid-state-grinding (milling) or solution crystallization. Milling was performed with a Retsch MM200 mixer mill at a rate of 30 Hz, using two 5 mL stainless steel jars, each containing one 7 mm-diameter stainless steel ball. The complexes were prepared as follows:(i) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Theophyllinium salicylic-5-sulfonate dihydrate salt was prepared by both milling and solution crystallization as described previously.28
:1 Theophyllinium salicylic-5-sulfonate dihydrate salt was prepared by both milling and solution crystallization as described previously.28
(ii) 1:1 Theophyllinium salicylic-5-sulfonate monohydrate salt was prepared by both milling and solution crystallization as described previously.30
(iii) 2:1 Theophylline/oxalic acid co-crystal was prepared by both milling and solution crystallization as described previously.30
(iv) 1:1 Theophylline/maleic acid co-crystal. Anhydrous theophylline (2.023 g, 11.23 mmol) and maleic acid (1.303 g, 1 equiv.) were dissolved in 35 mL of a 6:1 chloroform:methanol solvent mixture under reflux (53 °C). The solution was removed from heat and allowed to cool to ambient temperature while stirring. The resulting precipitate was removed by filtration under vacuum to give co-crystals with an excess of anhydrous theophylline. The filtrate was then allowed to stand overnight, affording the physically pure 1:1 co-crystal, which was isolated by filtration under vacuum. The 1:1 co-crystal was also formed by milling theophylline (109.99 mg, 0.61 mmol) and maleic acid (70.80 mg, 1 eq.) for 20 minutes.
(v) 1:1 Theophylline/malonic acid co-crystal. Milling anhydrous theophylline (111.70 mg, 0.62 mmol) and malonic acid (64.52 mg, 1 eq.) for 20 minutes formed the 1:1 co-crystal. Anhydrous theophylline (0.5873 g, 3.26 mmol) and malonic acid (0.3392 g, 1.0 equiv.) were also dissolved in 40 mL chloroform and 2 mL methanol under reflux (55 °C). The solution was removed from heat and seeded with ∼45 mg of co-crystal formed by milling. The solution was allowed to evaporate until ∼15 mL remained, and then filtered under vacuum to give the 1:1 co-crystal.
(vi) 1:1 Theophylline/citric acid co-crystal was prepared by milling as described previously.21
(vii) 1:1 Theophylline/glutaric acid co-crystal. Milling anhydrous theophylline (93.7 mg, 0.52 mmol) and glutaric acid (68.7 mg, 1 eq.) for 60 minutes formed the 1:1 co-crystal, which was physically pure as determined by X-ray powder diffraction. Theophylline (0.4972 g, 2.76 mmol) and glutaric acid (0.3646 g, 1 equiv.) were also dissolved in 35 mL chloroform under reflux (59 °C). The solution was removed from heat and seeded with ∼35 mg of co-crystal formed by milling. The solution was allowed to evaporate until ∼20 mL volume remained, and the precipitate (present at the surface) was filtered under vacuum to give the 1:1 co-crystal.
(viii) 1:1 3,5-Diaminobenzoic acid/3,5-dinitrobenzoic acid salt. 3,5-Dinitrobenzoic acid (0.4176 g, 2 mmol) and 3,5-diaminobenzoic acid (0.3148 g, 2 mmol) were dissolved in 95% ethanol (25 mL), heated to ensure complete dissolution and left to slowly cool. Dark brown needle crystals were obtained.
(ix) 1:1 4-Aminobenzoic acid/3,5-dinitrobenzoic acid co-crystal. 4-Aminobenzoic acid (0.3477 g, 2.5 mmol) and 3,5-dinitrobenzoic acid (0.5596 g, 2.6 mmol) were dissolved in methanol (20 mL). The resulting yellow solution was left to slowly evaporate and yellow crystals were obtained.
(x) 1:1 4-Aminobenzoic acid/4-hydroxy-3-nitrobenzoic acid co-crystal. 4-Aminobenzoic acid (0.1372 g, 1 mmol) and 4-hydroxy-3-nitrobenzoic acid (0.1359 g, 0.7 mmol) were mixed in methanol (1 mL) and heated to 50 °C, and subsequently cooled to 10 °C at a rate of 0.5 °C min−1 using an Avantium Crystal16.44 Orange-yellow block crystals were obtained.
(xi) 1:1 Isonicotinamide/2,4,6-trihydroxybenzoic acid salt. Isonicotinamide (0.0619 g, 5 mmol) and 2,4,6-trihydroxybenzoic acid monohydrate (0.1227 g, 7 mmol) were dissolved in methanol (7.5 mL). The sample was heated to ensure complete dissolution and left to cool to room temperature. On slow evaporation, light brown, blocky crystals were produced.
(xii) 1:1 Isonicotinamide/4-hydroxy-3-nitrobenzoic acid co-crystal. Isonicotinamide (0.666 g, 6 mmol) and 4-hydroxy 3-nitrobenzoic acid (0.9309 g, 5 mmol) were dissolved in hot methanol (35 mL). Upon cooling the solution was left to slowly evaporate until yellow crystals were obtained.
X-ray diffraction (XRD)
Powder XRD patterns were collected using a Rigaku Miniflex instrument utilizing Cu Kα radiation (λ = 1.5406 Å), operating over 5–40° 2θ at 1.5° min−1 with a 0.03° step, 30 kV voltage, and 15 mA current. Typically 5 mg of the sample was placed on a small sample attachment and smoothed to achieve a level surface.Single crystal XRD data were collected at 100 K using an Oxford Diffraction X-Calibur 2 diffractometer utilizing Mo Kα radiation (λ = 0.71073 Å) and an Oxford CryoSystems Cryostream Controller 700. Data reduction, cell refinement, and multi-scan absorption corrections were carried out using the program CrysAlis RED (Oxford Diffraction Ltd., version 1.171.32.24, 2008). The isonicotinamide/4-hydroxybenzoic acid co-crystal structure was solved with SHELXS-97 and refined on F2 against all reflections with SHELXL-97.45 The isonicotinamide/2,4,6-trihydroxybenzoic acid salt structure was solved with SIR9246 and refined on F2 against all reflections using SHELXL-97.45 All non-hydrogen atoms were refined by direct methods anisotropically; hydrogen atoms were located in difference Fourier maps and refined isotropically, or placed in geometric positions and refined as riding atoms. CCDC 960081 and 960082.†
X-ray photoelectron spectroscopy (XPS)
XP spectra were recorded using a Kratos Axis Ultra instrument employing a monochromatic Al Kα source (1486.69 eV), a hemispherical analyser with a hybrid (electrostatic and magnetic) lens system, charge neutralization by filament-generated, magnetically channeled low-energy electrons, and a delay line detector (DLD). Samples were fixed using double-sided tape. Experiments were performed while operating the X-ray source with a power of 180 W (15 kV and 12 mA), with the pressure below 10−8 mbar during analysis. The instrument was operated in CAE (constant analysis energy) mode, with a pass energy of 20 eV for high resolution scans of the photoemission from individual core levels, with a calibrated intensity/energy response and transmission function.36 High resolution spectra were measured within the spectral range of interest (ca. ±20 eV around the core level emission peaks) with 0.1 eV steps and 300–500 ms dwell time per data point. Repeats were carried out to check for radiation damage.Analysis of the data was carried out using Casa XPS software.47 A linear background was used in all curve-fitting (minimizing χ2)47,48 along with a GL(30) lineshape (70% Gaussian, 30% Lorentzian using the Gaussian/Lorentzian product form).47 For the remainder of this paper, when referring to spectral contributions of specific atoms in the molecular structure in the text, the atom of interest will be indicated by being underlined. Samples naturally containing aliphatic ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –C environments were referenced to adventitious hydrocarbon contamination at 285 eV.48 Samples without C–C environments were referenced to the lowest EB photoemission; those containing citric acid were referenced to –COOH (β-COOH) at 285.2 eV49 due to the absence of a clear hydrocarbon shoulder; samples containing 5-sulfosalicylic acid were referenced to
–C environments were referenced to adventitious hydrocarbon contamination at 285 eV.48 Samples without C–C environments were referenced to the lowest EB photoemission; those containing citric acid were referenced to –COOH (β-COOH) at 285.2 eV49 due to the absence of a clear hydrocarbon shoulder; samples containing 5-sulfosalicylic acid were referenced to ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 284.8 eV.50 Individual chemical environments/functional groups often exhibit similar ranges of binding energy values where the bonding or electronegativity is similar, thus where peaks arising from photoemission from different chemical environments occurred at the same position or within ±0.1 eV, a single peak was used to represent both environments for clarity and ease of discussion. Repeatability of the peak positions was within 0.1 eV.
C at 284.8 eV.50 Individual chemical environments/functional groups often exhibit similar ranges of binding energy values where the bonding or electronegativity is similar, thus where peaks arising from photoemission from different chemical environments occurred at the same position or within ±0.1 eV, a single peak was used to represent both environments for clarity and ease of discussion. Repeatability of the peak positions was within 0.1 eV.
Solid-state nuclear magnetic resonance (ssNMR)
Solid-state 15N cross-polarisation magic angle spinning (CP-MAS) data were acquired using a Bruker DRX500 spectrometer operating at 50.69 MHz, equipped with a standard bore magnet and a 4 mm CP-MAS probe. The magic angle adjustment was optimized using KBr and field homogeneity optimized using adamantane. Data were collected using a spectral width of 30.3 kHz, 3072 complex data points, and an acquisition time of 50.8 ms. The relaxation delay and contact time were optimized for each individual sample. Sample rotation rates of 5.0–8.0 kHz were employed and typically greater than 2048 transients acquired. 1H decoupling was achieved using a TPPM-15 sequence (86.2 kHz). Chemical shifts (δN) were referenced indirectly with respect to nitromethane, a standard recommended by IUPAC,51,52via the high frequency signal of a traceable standard of ammonium nitrate (δN = −5.1 ppm). Chemical shifts for the theophylline and API 1 salts and co-crystals are compared with the values for the free base forms by the change in chemical shift (Δδ = δcomplex − δfree base).Isotropic chemical shift differences53 were calculated using the CASTEP density functional theory (DFT) code,54–56 which employs the gauge including projector augmented wave (GIPAW) algorithm. The generalised gradient approximation (GGA) PBE functional was used with core-valence interactions described by ultrasoft pseudo-potentials generated on-the-fly. Integrals over the Brillouin zone were completed using a Monkhorst–Pack grid with a reciprocal space k-point spacing of 0.04 Å−1. A cut-off energy of 610 eV was defined for kinetic energy of planewaves used to provide a basis for wavefunctions. For each crystal structure, full geometry optimization was performed using an energy cut-off of 300 eV and a k-point spacing of 0.05 Å−1, with fixed lattice parameters. Calculations were performed using Materials Studio version 4.4, as provided by Accelrys.57 CASTEP DFT chemical shielding values (σ) were obtained for the nitrogen atoms of the theophylline, 4-aminobenzoic acid, 3,5-diaminobenzoic acid, and isonicotinamide complexes using the previously reported7,21,28,41–43 and new isonicotinamide co-crystal/salt crystal structures as input. Shielding values for the co-crystal and salts are compared with chemical shifts by plotting calculated chemical shieldings (σ) versus experimental chemical shifts (δN). A plot of calculated chemical shieldings (σ) versus experimental chemical shifts (δN) for all of the CASTEP data gives a linear correlation with y = −0.9525x − 153.61 (R2 = 0.9893). Predicted chemical shift differences use the free base as a reference [Δδcalc = −(σcomplex − σfree base)].
Results and discussion
XPS
The four nitrogen environments (Fig. 1) in the theophylline complexes result in two X-ray photoemission peak components.21,28,30 The peak arising from the C–![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) /–CO nitrogens occurs at 401.0 ± 0.1 eV for both the co-crystal and salt forms (Fig. 2, Table 2). In the five theophylline co-crystals, the C photoemission occurs at 399.6 ± 0.1 eV. However, for the two theophylline salts, this second photoemission peak is at 401.9 eV (Fig. 2b, Table 2), with protonation (CH+) shifting the CN peak by +2.3 eV.28,30
/–CO nitrogens occurs at 401.0 ± 0.1 eV for both the co-crystal and salt forms (Fig. 2, Table 2). In the five theophylline co-crystals, the C photoemission occurs at 399.6 ± 0.1 eV. However, for the two theophylline salts, this second photoemission peak is at 401.9 eV (Fig. 2b, Table 2), with protonation (CH+) shifting the CN peak by +2.3 eV.28,30
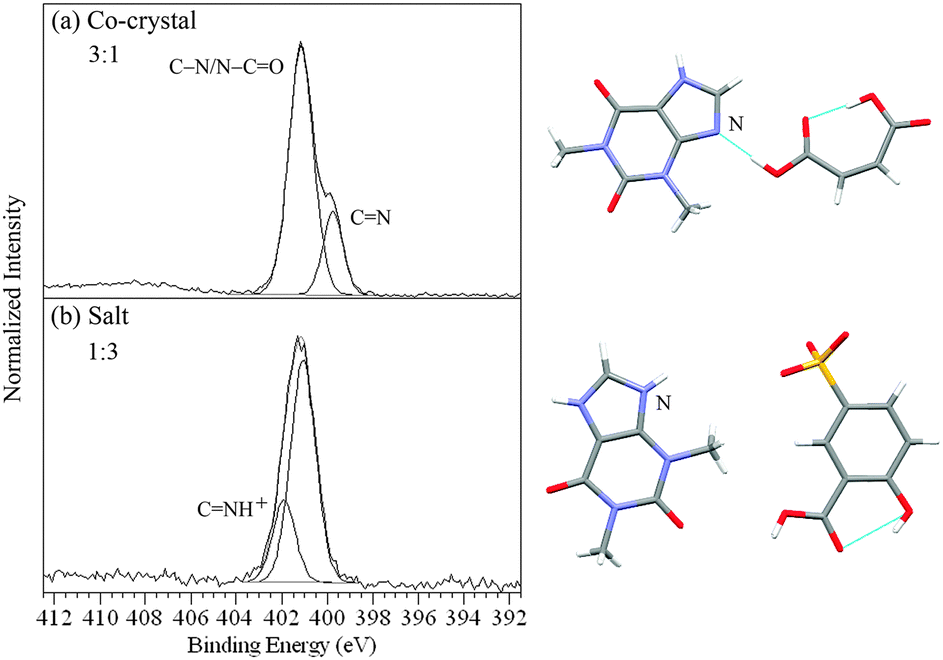 | ||
| Fig. 2 N 1s XPS spectra of (a) the theophylline/maleic acid co-crystal and (b) the theophylline/5-sulfosalicylic acid dihydrate salt (CNH+, SO3−)28 showing the ratios for types of nitrogen and the corresponding intermolecular H-bonding/protonation from the single crystal XRD structures7,28 with the nitrogen sensitive to H-bonding/protonation marked. | ||
| Complex (base/acid) | Binding energy/eV | |||||||
|---|---|---|---|---|---|---|---|---|
| –SO2 |
C– |
C |
C⋯HO |
–CO |
C–H+ |
CH+ |
O2 |
|
| Theophylline/5-sulfosalicylic acid dihydrate salt | 401.0 | 401.0 | 401.9 | |||||
| Theophylline/5-sulfosalicylic acid monohydrate salt | 401.0 | 401.0 | 401.9 | |||||
| Theophylline/oxalic acid co-crystal | 401.0 | 399.6 | 401.0 | |||||
| Theophylline/maleic acid co-crystal | 401.1 | 399.7 | 401.1 | |||||
| Theophylline/malonic acid co-crystal | 401.1 | 399.7 | 401.1 | |||||
| Theophylline/citric acid co-crystal | 400.9 | 399.5 | 400.9 | |||||
| Theophylline/glutaric acid co-crystal | 401.0 | 399.5 | 401.0 | |||||
| 4-Aminobenzoic acid/3,5-dinitrobenzoic acid co-crystal | 399.4 | 406.3 | ||||||
| 4-Aminobenzoic acid/4-hydroxy-3-nitrobenzoic acid co-crystal | 399.6 | 406.3 | ||||||
| 3,5-Diaminobenzoic acid/3,5-dinitrobenzoic acid salt | 399.5 | 401.9 | 406.3 | |||||
| Isonicotinamide/4-hydroxy-3-nitrobenzoic acid co-crystal | 399.6 | 400.1 | 406.3 | |||||
| Isonicotinamide/2,4,6-trihydroxybenzoic acid salt | 400.1 | 401.6 | ||||||
| Di-HCl salt 1s | 399.1 | 399.1 | 400.3 | 401.2 | ||||
| Fumaric acid co-crystal 1c | 399.2 | 399.2 | 399.8 | 400.6 | ||||
| Difumarate salt 2 | 399.5 | 401.7 | ||||||
| HCl salt 3 | 399.6 | 401.7 | ||||||
| HCl salt 4 | 399.9 | 401.5 | ||||||
The 4-aminobenzoic and 3,5-diaminobenzoic complexes all have at least one primary amine nitrogen C–H2 on the base component and a nitro group O2 on the acid component (Fig. 1 and 3). For the 4-aminobenzoic acid/3,5-dinitrobenzoic acid and 4-aminobenzoic acid/4-hydroxy-3-nitrobenzoic acid co-crystals (heterodimer bonding) this gives rise to two peaks at 399.5 ± 0.1 eV and 406.3 eV, from C–H2 and O2 respectively (Fig. 3a and b, Table 2).40 When a salt is formed, as in the case of the 3,5-diaminobenzoic acid/3,5-dinitrobenzoic acid complex, one of the two amine nitrogens of 3,5-diaminobenzoic acid is protonated (C–H3+), shifting its peak position to a higher binding energy of 401.9 eV, a shift of +2.4 eV (Fig. 3c, Table 2).40
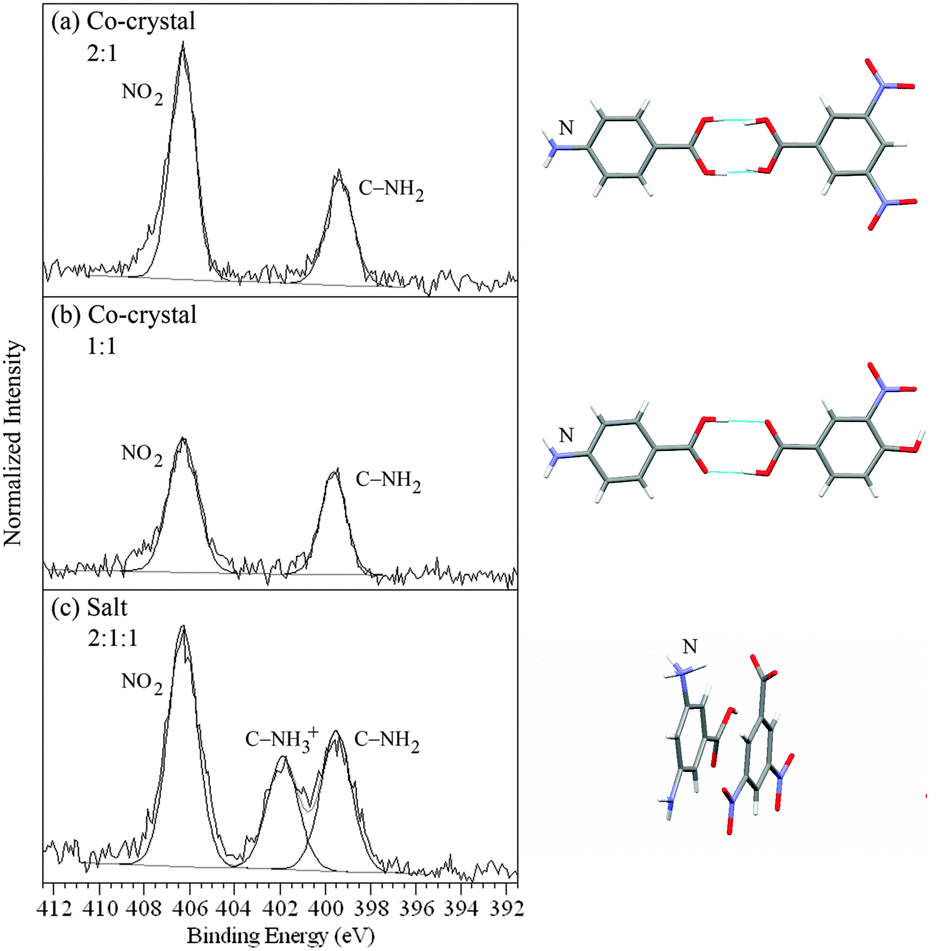 | ||
| Fig. 3 N 1s XPS spectra of (a) the 4-aminobenzoic acid/3,5-dinitrobenzoic acid co-crystal, (b) the 4-aminobenzoic acid/4-hydroxy-3-nitrobenzoic acid co-crystal, and (c) the 3,5-diaminobenzoic acid/3,5-dinitrobenzoic acid salt (NH3+, COO−), with the corresponding intermolecular H-bonding/protonation from the single crystal XRD structures.42,43 | ||
There are three different types of nitrogen atom in isonicotinamide/4-hydroxy-3-nitrobenzoic acid – pyridine C and amide –CO of isonicotinamide and nitro O2 of the acid molecule (Fig. 1). The nitrogen 1s XPS (Fig. 4a) shows two maxima, with the high binding energy peak at 406.3 eV arising from the O2 photoemsission, while the lower energy peak can be fit with two components at 399.6 and 400.1 eV representing the C and amide –CO nitrogen functionalities respectively. With 2,4,6-trihydroxybenzoic acid, there are no nitrogens in the acid molecule (no O2 photoemission), leaving just the C and –CO from isonicotinamide. Aside from the isonicotinamide –CO peak at 400.1 eV, the C peak is shifted by 2.0 eV compared to that of isonicotinamide/4-hydroxy-3-nitrobenzoic acid and is observed at 401.6 eV, representing the change in chemical state from pyridine C to the pyridinium nitrogen CH+ and evidence of salt formation (Fig. 4b, Table 2). Subsequent acquisition of the single crystal structure confirms the XPS-assignment of isonicotinamide/4-hydroxy-3-nitrobenzoic acid as a co-crystal and isonicotinamide/2,4,6-trihydroxybenzoic acid as a salt (Fig. 4).
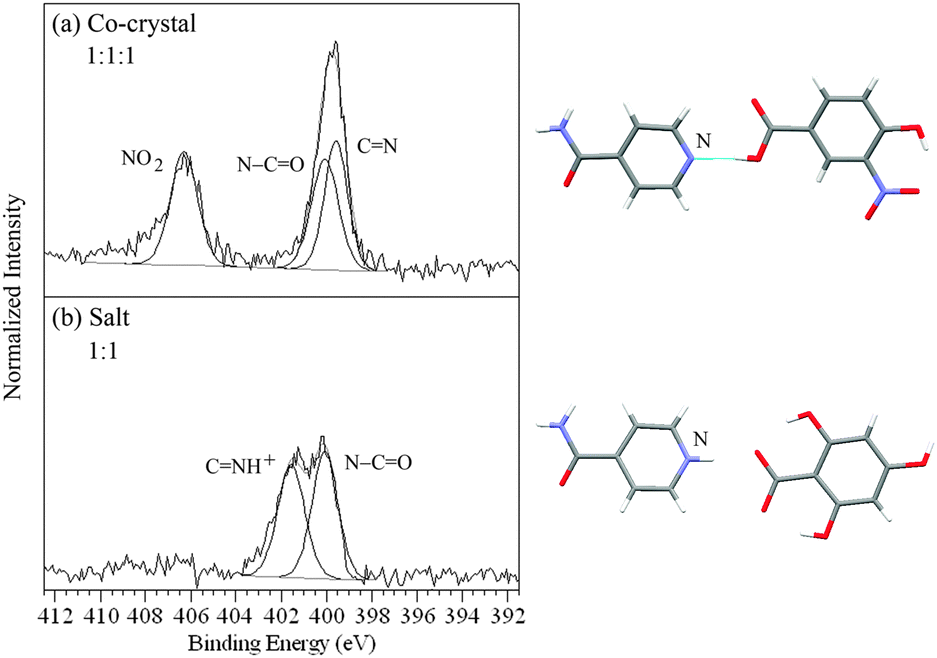 | ||
| Fig. 4 N 1s XPS spectra of (a) the isonicotinamide/4-hydroxy-3-nitrobenzoic acid co-crystal and (b) the isonicotinamide/2,4,6-trihydroxybenzoic acid salt (NH3+, COO−), with the corresponding intermolecular H-bonding/protonation from the single crystal XRD structures. | ||
The API 1 provides a variety of nitrogen environments (Fig. 1), which can be grouped into pyridine derivatives (C × 3), amines (C– × 2), and amides (–CO × 2). For the free base form (API 1), the photoemission by the C– and C nitrogens appears around the same binding energy position at 399.1 ± 0.1 eV, while that of –CO ranges from 400.3 to 400.6 eV (Fig. 5, Table 2). Differences with respect to the free base are clearly evident for the salt 1s, with the presence of a pyridinium CH+ peak component at 401.2 eV shifted by +2.1 eV compared to unprotonated, pyridine nitrogen (Fig. 5, Table 2). The relative intensity of this peak is in accord with the protonation of two of the pyridine nitrogens in the di-HCl salt 1s. There is also an observable difference for the co-crystal 1c, with a decrease in the intensity of the C–/CN peak relative to the free base and a broadening between the two free base peak components. This is accounted for by a third peak at 399.8 eV, of a relative intensity indicative of one of the C–/C nitrogens (Fig. 5, Table 2). A shift of +0.6 eV from the C–/CN photoemissions is too small a magnitude for complete proton transfer, so it appears likely that formation of a new hydrogen bond results in a high binding energy shift for the nitrogen acceptor atom.
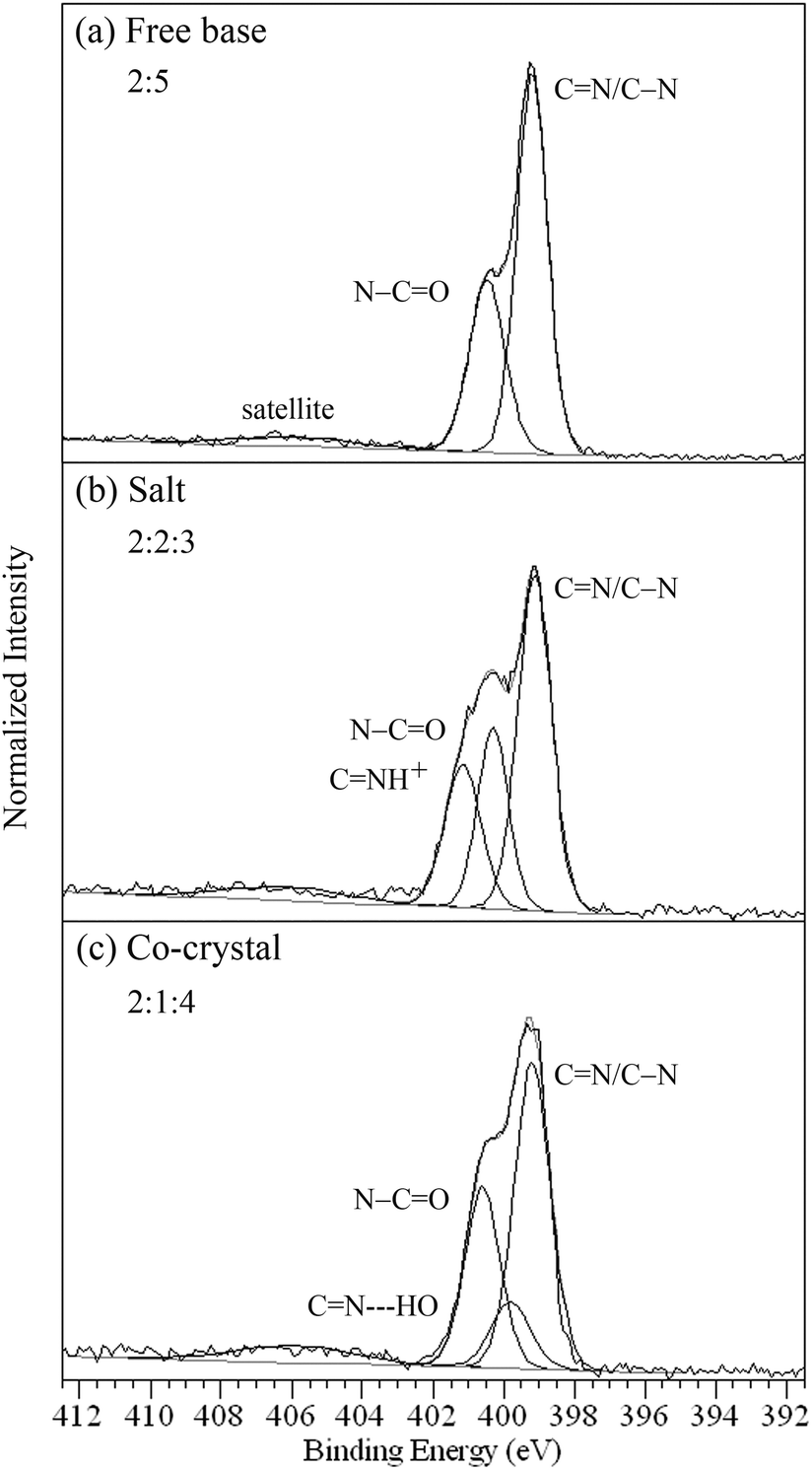 | ||
| Fig. 5 N 1s XPS spectra of (a) the free base 1, (b) di-HCl salt 1s, and (c) fumaric acid co-crystal 1c showing the ratios for the types of nitrogen. | ||
The difumarate salt 2 has three nitrogen environments (Fig. 1), comprising two heterocyclic amines (C– × 2) and one sulfonamide (–SO2 × 1). The sulfonamide nitrogen gives rise to the peak at 399.5 eV, while protonation of the amine nitrogens results in a peak at 401.7 eV (Fig. 6a, Table 2), although this API is particularly sensitive to X-ray induced reduction of NH3+ to NH2 (occurring at coinciding binding energy with –SO2), leading to a departure from the expected 2:1 ratio.58 The HCl salts 3 and 4 have two and three nitrogens respectively (Fig. 1), with 3 including an amine (C– × 1) and sulfonamide (–SO2 × 1), while 4 has a heterocyclic amine (C– × 1) and two amides (–CO × 2). The sulfonamide peak of 3 and amide peak of 4 occur at 399.6 and 399.9 eV, respectively, while the protonated amine C–H+ nitrogen photoemissions occur at higher binding energy, 401.7 and 401.5 eV, respectively (Fig. 6b and c, Table 2).
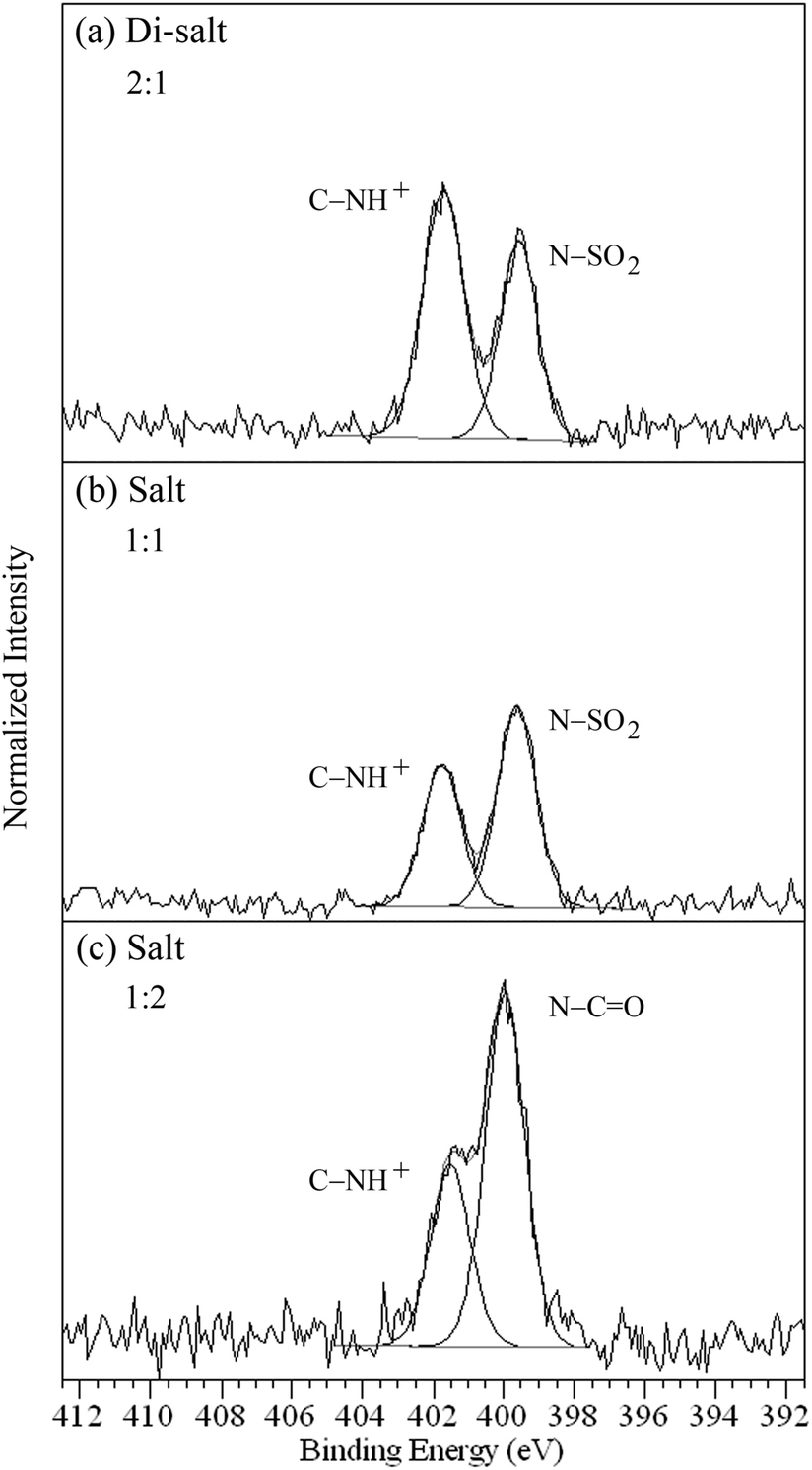 | ||
| Fig. 6 N 1s photoemission of (a) difumarate salt 2,58 (b) HCl salt 3, and (c) HCl salt 4 showing the ratios for the types of nitrogen. | ||
15N ssNMR
The spectra of the theophylline complexes,21,28,301s, and 1c, were compared with the free base form of each in order to investigate the changes in chemical shifts occurring with formation of the complexes. 50–100 ppm shifts relative to the starting components (chemical shift difference Δδ) are typical for protonation of aromatic, heterocyclic nitrogen,5,28–31 and shifts of 47–118 ppm to low frequency confirm salt formation (protonated nitrogen, CNH+) for the two theophylline/5-sulfosalicylic acid salts28,30 (Table 3), the isonicotinamide/2,4,6-trihydroxybenzoic acid salt (Fig. 7, Table 5) and the di-HCl salt 1s (Table 4). The theophylline salts are protonated at C (N9, Fig. 1, Table 3), with ca. −50 ppm shifts (Δδ),28,30 and the isonicotinamide salt has a −68.76 ppm shift on protonation (Fig. 7, Table 5), while di-HCl salt 1s is protonated in two pyridine positions, N5 and N7, with shifts of −108.0 and −117.8 ppm respectively (Table 4). In contrast, the fumaric acid co-crystal 1c (Table 4) and the theophylline co-crystal resonances21,30 (Table 3) show only minor changes in 15N chemical shifts compared to the base component (Δδ), with the exception of the pyridine N5 resonance in 1c. A shift of 32.6 ppm to low frequency occurs for N5 of 1c (Table 4), indicating formation of a strong hydrogen bond that is not present in the free base form. A component of this change in chemical shift may be ascribed to conformational/crystallographic packing differences induced by the hydrogen bonding, which is observed on comparing different polymorphic forms of the free base.
| Compound | δ/ppm | |||
|---|---|---|---|---|
| N1 | N3 | N7 | N9 | |
| OC–N–CO |
N–CO |
C–N | CN |
|
| Theophylline | −227.04 | −269.41 | −218.47 | −162.74 |
| Complex (base/acid) | Δδ = δ (complex) − δ (theophylline) | |||
|---|---|---|---|---|
| N1 | N3 | N7 | N9 | |
| OC–N–CO |
N–CO |
C–N | CN |
|
| Theophylline/5-sulfosalicylic acid dihydrate salt | −0.60 | −1.11 | −2.44 | −47.59 |
| Theophylline/5-sulfosalicylic acid monohydrate salt | −1.34 | −0.41 | −5.33 | −50.48 |
| Theophylline/oxalic co-crystal | −0.44 | 1.99 | −3.79 | −1.82 |
| Theophylline/maleic co-crystal | −0.81 | 1.96 | −4.89 | −5.39 |
| Theophylline/malonic co-crystal | −3.85 | 3.31 | −6.08 | −4.07 |
| Theophylline/citric co-crystal | −2.32 | 3.96 | −8.16 | 3.72 |
| Theophylline/glutaric co-crystal | −2.53 | 2.95 | −4.16 | 1.01 |
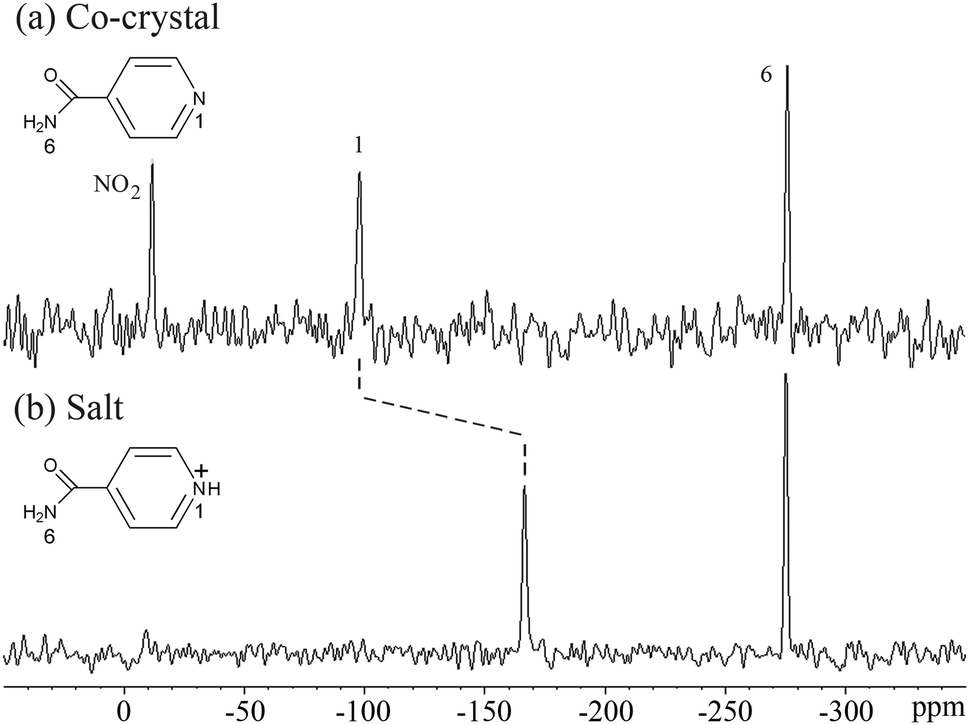 | ||
| Fig. 7 15N ssNMR spectra of (a) the isonicotinamide/4-hydroxy-3-nitrobenzoic acid co-crystal and (b) the isonicotinamide/2,4,6-trihydroxybenzoic acid salt, showing the shift with proton transfer. | ||
| Compound | δ/ppm | ||||||
|---|---|---|---|---|---|---|---|
| N1 amine | N2 amide | N3 amine | N4 amide | N5 pyridine | N6 pyridine | N7 pyridine | |
| Free base (API 1) | −298.04 | −275.91 | −249.82 | −240.45 | −93.20 | −117.03 | −70.04 |
| Complex (base/acid) | Δδ = δ (complex) − δ (free base) | ||||||
|---|---|---|---|---|---|---|---|
| N1 amine | N2 amide | N3 amine | N4 amide | N5 pyridine | N6 pyridine | N7 pyridine | |
| Free base form I | 0.91 | −2.31 | −0.58 | 1.93 | −13.95 | 2.49 | −4.30 |
| Fumaric acid co-crystal 1c | 9.57 | 2.96 | 3.83 | 2.14 | −32.60 | 2.82 | 1.12 |
| Di-HCl salt 1s | 15.83 | 0.37 | 4.21 | 2.24 | −108.02 | −3.13 | −117.79 |
Aliphatic nitrogen often exhibits much smaller shifts with protonation than its heterocyclic counterparts (e.g. amines vs. pyridines).59 The aliphatic nature of the protonated nitrogen (C–H3+) in the 3,5-diaminobenzoic acid/3,5-dinitrobenzoic acid salt results in a smaller magnitude shift of −14.11 ppm relative to the unprotonated amine, but this is still sufficient to distinguish formation of a salt from a co-crystal. There are no significant shifts indicating protonation of the amine in the 4-aminobenzoic acid co-crystals (Table 5), confirming they are not salts.
| Complex (base/acid) | δ/ppm | |||||
|---|---|---|---|---|---|---|
| C |
C– |
–CO |
CH+ |
C–H+ |
O2 |
|
| 4-Aminobenzoic acid/3,5-dinitrobenzoic acid co-crystal | −311.04 | −16.28 | ||||
| 4-Aminobenzoic acid/4-hydroxy-3-nitrobenzoic acid co-crystal | −318.15 | −8.64 | ||||
| 3,5-Diaminobenzoic acid/3,5-dinitrobenzoic acid salt | −314.94 | −329.05 | −14.13 | |||
| −18.53 | ||||||
| Isonicotinamide/4-hydroxy-3-nitrobenzoic acid co-crystal | −97.87 | −275.69 | −11.61 | |||
| Isonicotinamide/2,4,6-trihydroxybenzoic acid salt | −275.31 | −166.63 | ||||
Density functional theory (DFT) calculations (chemical shielding values, σ) were performed for the nitrogens of the theophylline, aminobenzoic, and isonicotinamide complexes in order to compare with experimental data (chemical shifts, δ). The plot of σ versus δ (Fig. 8) shows a linear correlation with y = −0.9525x − 153.61 (R2 = 0.9893). This demonstrates the robustness of using CASTEP for relating 15N ssNMR chemical shifts to molecular structure and, therefore, providing NMR based evidence for proof of co-crystal or salt formation.
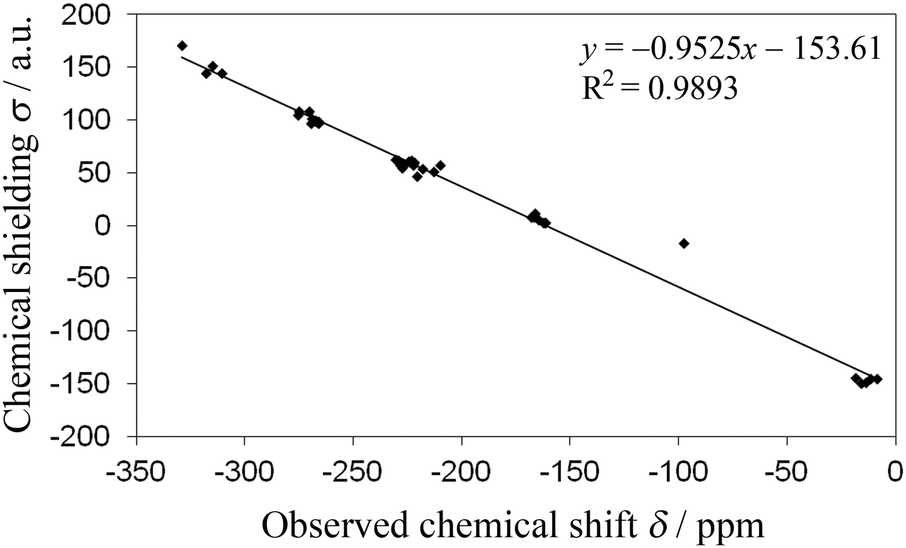 | ||
| Fig. 8 CASTEP DFT nitrogen chemical shieldings (σ) vs. experimental 15N ssNMR chemical shifts (δ). | ||
The predicted chemical shift differences can be related to the H-bond length, giving a measure of H-bond strength, and providing an example of how NMR crystallography can be used in a complementary way with respect to X-ray crystallography.60 For example, the predicted values as a function of distance between N of the base and H of the acid component show a clear separation between unprotonated (co-crystals, d ≥ 1.53 Å) and protonated nitrogen (salts, d ≤ 1.11 Å) for the theophylline complexes (Fig. 9). Additional multi-dimensional solid-state NMR studies are in progress to further characterize the nature of the hydrogen bonding in these co-crystals and salts.
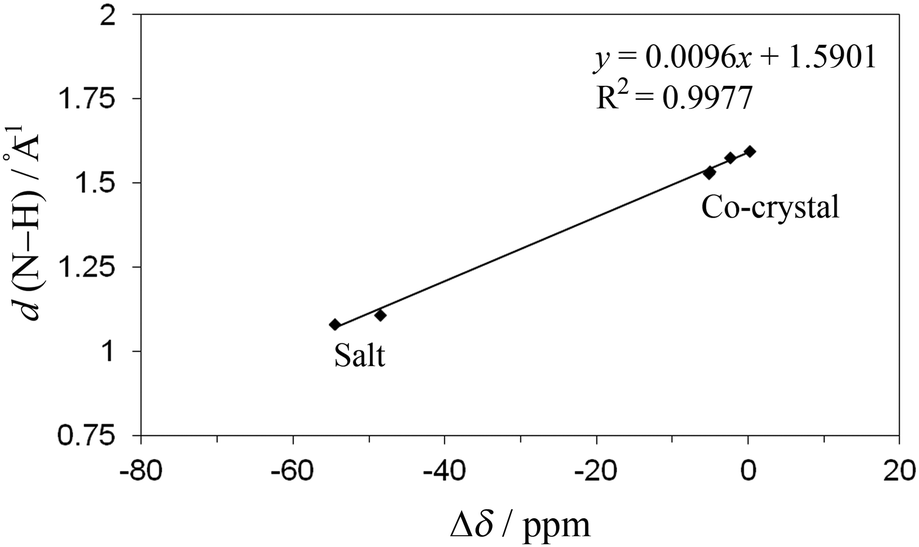 | ||
| Fig. 9 CASTEP DFT optimised H-bond lengths versus predicted chemical shift differences (Δδ). | ||
ΔpKa correlations
Plotting the nitrogen XPS and ssNMR values for the co-crystals and salts against ΔpKa (Fig. 10) shows the dependence of the extent of proton transfer on the strength of the acidic component. Of the 17 complexes, the co-crystals have ΔpKa ≤ 0.3, and the salts have ΔpKa ≥ 1.8, with ΔpKa ranging from −3.9 to 17.7, and four salts and two co-crystals have been assigned that are within the range for ΔpKa typically viewed as the transition region (0–3) for which formation of a salt vs. co-crystal is not predictable.12 Recently, pKa matching has been used as a tool for predicting H-bond strengths61 (including the proposed pKa slide rule62). A correlation between bond lengths in crystals and ΔpKa values was shown for N–H⋯O/O–H⋯N bonds.62 In line with this work, we find a clear separation between N 1s XPS chemical shifts of the co-crystals around 399.6 eV and salts around 401.7 eV (Fig. 10). Measurement of the N 1s binding energy unambiguously determines whether protonation has occurred, with a mean N 1s binding energy difference of +2.1 eV for N → NH+.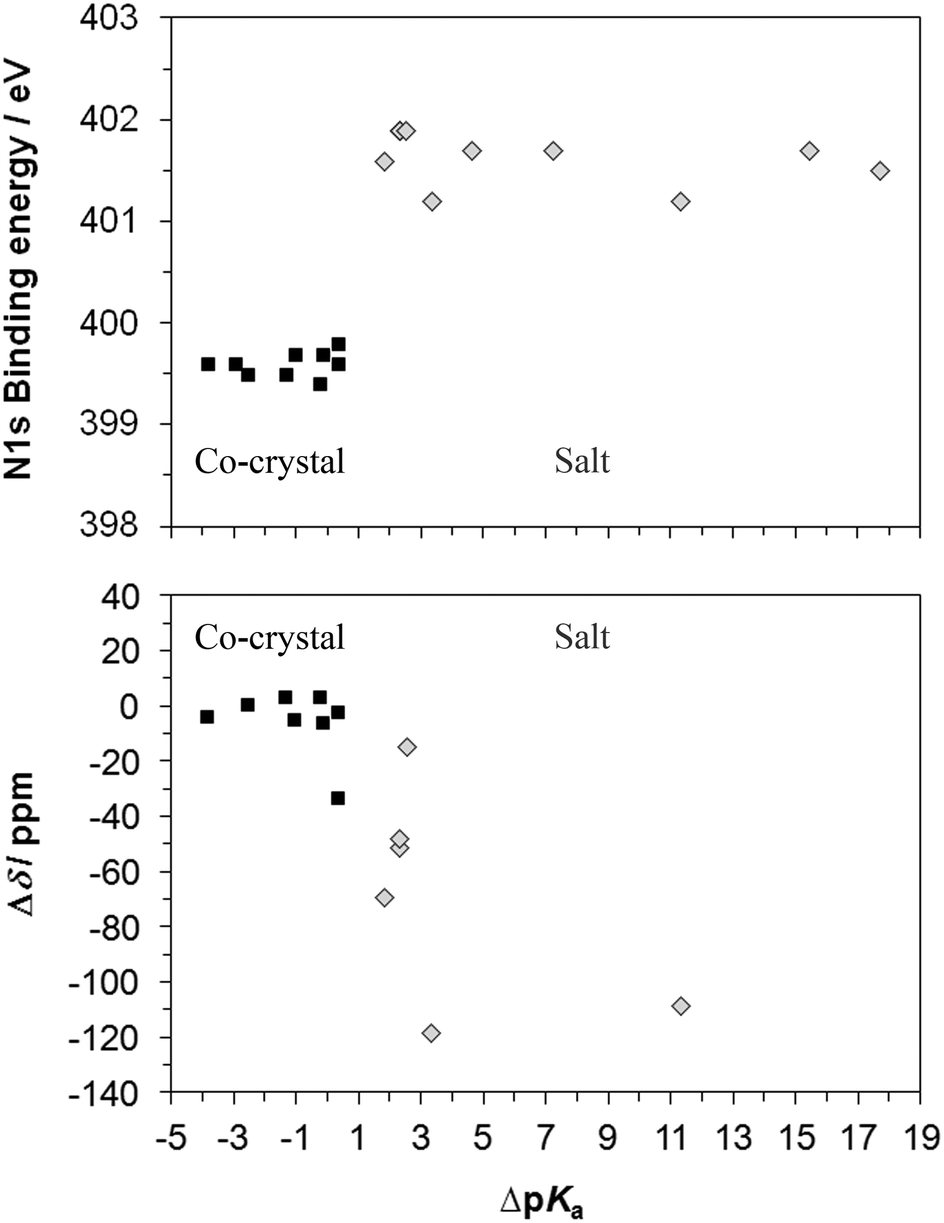 | ||
| Fig. 10 Correlations between N 1s XPS and 15N ssNMR chemical shifts with ΔpKa, illustrating the clear separation between protonated (salt) and unprotonated nitrogen (co-crystal) with XPS and the overlap for new H-bond formation in a co-crystal (1c) and protonation of aliphatic nitrogen in a salt (3,5-diamino/3,5-dinitro) with ssNMR. | ||
As the 15N ssNMR chemical shift δ varies significantly with each base moiety and thus the absolute value is not a unique identifier for protonation state, the chemical shift difference (Δδ, Fig. 10) is used to compare against the ΔpKa for the complexes (no ssNMR data for API 2, 3, and 4). There is a clustering of Δδ values for the co-crystals, although there is an overlap between co-crystals and salts. The smallest chemical shift observed for protonation of nitrogen (−14.11 ppm) and salt formation occurs for 3,5-diaminobenzoic acid/3,5-dinitrobenzoic acid and is a result of the reduced magnitude of the shift for aliphatic rather than heterocyclic aromatic nitrogen59 as mentioned previously, and the theophylline and isonicotinamide salts exhibit Δδ of −50 to 70 ppm while the di-salt API 1s shows larger magnitude shifts of around 110 ppm. The largest (most negative) shift for nitrogen within the co-crystals occurs for the development drug co-crystal 1c, causing it to be greater than that of the signal from the aliphatic salt – formation of a strong hydrogen bond that is not present in the free base form (API 1) is intimated for co-crystal 1c, influencing the chemical shift, as well as a possible contribution from conformational/crystallographic packing differences. Comparing shifts between compounds sharing a component (such as salts and co-crystals which share the same free base) rather than the whole series clearly identifies each form correctly (Fig. 7, Tables 3–5).
XPS core level chemical shifts reflect primarily the influence of the most immediate atomic neighbours on the electronic state of the photoexcited atom,33,48,50,63 with the effect of strong local intermolecular interactions such as ionic and H-bonding dominating over the comparatively weaker van-der-Waals and dipole interactions. It is for this reason that the core level binding energies reported in Table 2 and Fig. 10 are so universally sensitive to local structure and thus protonation state. In contrast, NMR is far more dependent on the extended environment and interactions, which leads to greater variance in Δδ as an indicator of protonation, although it does provide spatial resolution (allowing structurally inequivalent atoms of the same type of functional group to be distinguished). The combination of XPS and ssNMR to identify when hydrogen transfer has occurred from acid to base component is therefore especially strong, making assignment of salt or co-crystal unambiguous.
Conclusions
Nitrogen XPS and ssNMR chemical shifts clearly separate protonated from unprotonated nitrogen for a wide range of different solid-state donor–acceptor systems, including five pharmaceutical drug substances. The nature of the intermolecular interaction between the acid–base components was correctly identified for the eight salts and nine co-crystals. Brønsted proton transfer to nitrogen of the base component (salt formation) leads to chemical shifts towards higher core level binding energies in XPS and to lower frequency in ssNMR, while the hydrogen-bonded co-crystals exhibited only minor shifts compared to the free base form, with the exception of one of the pharmaceutical co-crystals (1c). The salt and co-crystal of the same API (1) allowed comparison between the magnitude of the shift for formation of a strong hydrogen bond (at a position where there was none in the free base) compared to that for complete hydrogen transfer (protonation) at the same position in the salt. DFT-calculated 15N ssNMR results show good correlation with experimental data, allowing prediction of chemical shifts and assignment of salt or co-crystal, and the strong influence of H-bond strength on the chemical shift.Our data demonstrate both the versatility and unambiguity of such a combined XPS/ssNMR approach, which has, to our knowledge, never been systematically pursued before across such a large body of samples. The results will inform a more critical assessment of the possibilities and limitations associated with XPS and ssNMR, and provide a reference database for future investigations of hydrogen-bonding and protonation in other systems, for example the study of solvation effects in the nucleation and growth of organic crystals.64
Acknowledgements
We thank EPSRC and Sanofi-Aventis for previous financial support for JSS through a DTA/CTA studentship, and the EPSRC for the PhD + fellowship for JSS. We gratefully acknowledge support for SLMS, JSS, GS, and RJD through an ESPRC Critical Mass Grant (EP/1013563/1).References
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 95, 499–516 Search PubMed.
- P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef CAS PubMed.
- C. B. Aakeroy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef PubMed.
- Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH, Weinheim, 2002 Search PubMed.
- Z. J. Li, Y. Abramov, J. Bordner, J. Leonard, A. Medek and A. V. Trask, J. Am. Chem. Soc., 2006, 128, 8199–8210 CrossRef CAS PubMed.
- D. P. McNamara, S. L. Childs, J. Giordano, A. Iarriccio, J. Cassidy, M. S. Shet, R. Mannion, E. O'Donnell and A. Park, Pharm. Res., 2006, 23, 1888–1897 CrossRef CAS PubMed.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114–123 CrossRef CAS PubMed.
- C. C. Sun and H. Hou, Cryst. Growth Des., 2008, 8, 1575–1579 CAS.
- M. Viertelhaus, R. Hilfiker, F. Blatter and M. Neuburger, Cryst. Growth Des., 2009, 9, 2220–2228 CAS.
- S. Chattoraj, L. Shi and C. C. Sun, CrystEngComm, 2010, 12, 2466–2472 RSC.
- C. M. Reddy, G. R. Krishna and S. Ghosh, CrystEngComm, 2010, 12, 2296–2314 RSC.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS PubMed.
- C. B. Aakeroy, A. Rajbanshi, Z. J. Li and J. Desper, CrystEngComm, 2010, 12, 4231–4239 RSC.
- M. Byres, P. J. Cox, G. Kay and E. Nixon, CrystEngComm, 2009, 11, 135–142 RSC.
- D. M. S. Martins, D. S. Middlemiss, C. R. Pulham, C. C. Wilson, M. T. Weller, P. F. Henry, N. Shankland, K. Shankland, W. G. Marshall, R. M. Ibberson, K. Knight, S. Moggach, M. Brunelli and C. A. Morrison, J. Am. Chem. Soc., 2009, 131, 3884–3893 CrossRef CAS PubMed.
- S. Mohamed, D. A. Tocher, M. Vickers, P. G. Karamertzanis and S. L. Price, Cryst. Growth Des., 2009, 9, 2881–2889 CAS.
- A. Parkin, S. M. Harte, A. E. Goeta and C. C. Wilson, New J. Chem., 2004, 28, 718–721 RSC.
- C. Wilson, Crystallogr. Rev., 2007, 13, 143–198 CrossRef CAS.
- R. Taylor and O. Kennard, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 133–138 CrossRef.
- A. V. Trask and W. Jones, in Organic Solid State Reactions, ed. F. Toda, Springer-Verlag, New York, 2005, pp. 41–70 Search PubMed.
- J. S. Stevens, S. J. Byard and S. L. M. Schroeder, Cryst. Growth Des., 2010, 10, 1435–1442 CAS.
- C. C. Wilson, K. Shankland and N. Shankland, Z. Kristallogr., 2001, 216, 303–306 CrossRef CAS.
- C. Wilson, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 435–439 CAS.
- J. A. Cowan, J. A. K. Howard, G. J. McIntyre, S. M. F. Lo and I. D. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 794–801 Search PubMed.
- T. Steiner, I. Majerz and C. C. Wilson, Angew. Chem., Int. Ed., 2001, 40, 2651–2654 CrossRef CAS.
- C. C. Wilson, N. Shankland and A. J. Florence, Chem. Phys. Lett., 1996, 253, 103–107 CrossRef CAS.
- C. C. Wilson, N. Shankland and A. J. Florence, J. Chem. Soc., Faraday Trans., 1996, 92, 5051–5057 RSC.
- J. S. Stevens, S. J. Byard, C. A. Muryn and S. L. M. Schroeder, J. Phys. Chem. B, 2010, 114, 13961–13969 CrossRef CAS PubMed.
- R. Gobetto, C. Nervi, E. Valfre, M. R. Chierotti, D. Braga, L. Maini, F. Grepioni, R. K. Harris and P. Y. Ghi, Chem. Mater., 2005, 17, 1457–1466 CrossRef CAS.
- J. S. Stevens, S. J. Byard and S. L. M. Schroeder, J. Pharm. Sci., 2010, 99, 4453–4457 CrossRef CAS PubMed.
- T. Friščić, L. Fábián, J. C. Burley, D. G. Reid, M. J. Duer and W. Jones, Chem. Commun., 2008, 1644–1646 RSC.
- R. Gobetto, C. Nervi, M. R. Chierotti, D. Braga, L. Maini, F. Grepioni, R. K. Harris and P. Hodgkinson, Chem.–Eur. J., 2005, 11, 7461–7471 CrossRef CAS PubMed.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Angew. Chem., Int. Ed., 2011, 50, 9916–9918 CrossRef CAS PubMed.
- P. A. Williams, C. E. Hughes, G. K. Lim, B. M. Kariuki and K. D. M. Harris, Cryst. Growth Des., 2012, 12, 3104–3113 CAS.
- F. Bisti, A. Stroppa, S. Picozzi, M. Donarelli, S. Picozzi and M. Coreno, J. Chem. Phys., 2013, 138, 014308 CrossRef CAS PubMed.
- J. S. Stevens and S. L. M. Schroeder, Surf. Interface Anal., 2009, 41, 453–462 CrossRef CAS.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond, Oxford University Press, Oxford, 2009 Search PubMed.
- Experimental pKa values were obtained using the ACD/I-Lab Web service (ACD/pKa DB 12.0), Advanced Chemistry Development, Inc. (ACD/Labs), Toronto, 2010 Search PubMed.
- Predicted pKa values were obtained using ACD/PhysChem Suite v12.0, Advanced Chemistry Development, Inc. (ACD/Labs), Toronto, 2010 Search PubMed.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Angew. Chem., 2011, 123, 10090–10092 CrossRef.
- J. Madarász, P. Bombicz, K. Jármi, M. Bán, G. Pokol and S. Gál, J. Therm. Anal. Calorim., 2002, 69, 281–290 CrossRef.
- C. C. Seaton, K. Chadwick, G. Sadiq, K. Guo and R. J. Davey, Cryst. Growth Des., 2009, 10, 726–733 Search PubMed.
- K. Chadwick, G. Sadiq, R. J. Davey, C. C. Seaton, R. G. Pritchard and A. Parkin, Cryst. Growth Des., 2009, 9, 1278–1279 CAS.
- Avantium Crystal16® parallel crystallizer, Avantium, Amsterdam Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- N. Fairley and A. Carrick, The Casa Cookbook – Part 1: Recipes for XPS Data Processing, Acolyte Science, Knutsford, Cheshire, 2005 Search PubMed.
- D. Briggs, M. P. Seah and P. M. A. Sherwood, in Practical Surface Analysis, Volume 1: Auger and X-ray Photoelectron Spectroscopy, ed. D. Briggs and M. P. Seah, John Wiley & Sons, Chichester, 2nd edn, 1990 Search PubMed.
- D. Briggs and G. Beamson, Anal. Chem., 2002, 64, 1729–1736 CrossRef.
- The XPS of Polymers Database, Surface Spectra Ltd., Manchester, 2000 Search PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef CAS.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- M. D. Segall, P. L. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2743 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, J. R. Yates and V. Zorin, Magn. Reson. Chem., 2007, 45, S174–S186 CrossRef CAS PubMed.
- Materials Studio version 4.4, Accelrys, Inc., San Diego, 2010 Search PubMed.
- J. S. Stevens, S. J. Byard, E. Zlotnikov and S. L. M. Schroeder, J. Pharm. Sci., 2010, 100, 942–948 CrossRef PubMed.
- C. G. Levy and R. L. Litcher, Nitrogen-15 Nuclear Magnetic Resonance Spectroscopy, Wiley, New York, 1979 Search PubMed.
- NMR Crsytallography, John Wiley & Sons Ltd, Chichester, 2009 Search PubMed.
- P. Gilli, L. Pretto and G. Gilli, J. Mol. Struct., 2007, 844–845, 328–339 CrossRef CAS PubMed.
- P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33–44 CrossRef CAS PubMed.
- K. Siegbahn, ESCA: Atomic, Molecular, and Solid State Structure Studied by Means of Electron Spectroscopy, Almqvist & Wiksells, Uppsala, 1967 Search PubMed.
- R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS PubMed.
Footnote |
| † CCDC 960081 and 960082. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cp53907e |
| This journal is © the Owner Societies 2014 |