 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarrier dynamics in quantum-dot sensitized solar cells measured by transient grating and transient absorption methods†
Naotaka
Maeda
ab,
Hiroaki
Hata
ab,
Naoya
Osada
ab,
Qing
Shen
*bc,
Taro
Toyoda
bc,
Shota
Kuwahara
a and
Kenji
Katayama
*a
aDepartment of Applied Chemistry, Chuo University, 1-13-27, Kasuga, Bunkyo, Tokyo 112-8551, Japan. E-mail: kkata@kc.chuo-u.ac.jp; Fax: +81-3-3817-1913; Tel: +81-3-3817-1913
bDepartment of Engineering Science, The University of Electro-Communications, 1-4-1 Chofugaoka, Chofu, Tokyo 182-8585, Japan. E-mail: shen@pc.uec.ac.jp
cCREST, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 7th May 2013
Abstract
Carrier dynamics in quantum-dot sensitized solar cells (QDSSCs) was clarified by combining the information obtained by the heterodyne transient grating (HD-TG), transient absorption (TA) and transient photocurrent (TP) measurements under the short circuit conditions in the time range from microseconds to seconds. The HD-TG signal is sensitive to the ionic species at the electrode/electrolyte interface, and the electrons in the titanium oxide layer injected from quantum dots (QDs) were monitored by the TA signal, and the photocurrent as a final output was monitored by the TP signal. By using the compensating information, the whole picture of the charge dynamics was obtained in the time region after the initial electron injection from QDs into the titanium oxide layer. In the former part of this paper, the assignment of the responses for each measurement was clarified based on the previous paper on dye sensitized solar cells (S. Kuwahara, et al. Phys. Chem. Chem. Phys., 2013, 15(16), 5975–5981). In the latter part, the effect of the device parameters for actual QDSSCs, such as electrolyte concentrations, and coating times of surface passivation of QDs were investigated.
Introduction
As promising devices for generating electricity from solar energy, dye-sensitized solar cells (DSSCs) have been intensively developed1–6 and a number of them have been commercialized. In the last ten years, a new type of solar cells where quantum dots composed of inorganic materials (QDSSCs) were used as sensitizers, have been studied. Since QDs have some unique properties due to the quantum confinement effect, such as a tunable optical absorption spectrum, high absorption coefficient and high efficiency multiple exciton generation (MEG),7–9 the theoretical maximum efficiency for QDSSCs (44%) is expected to be larger than that of DSSCs (30%).10 However, QDSSCs present lower energy conversion efficiency at the moment. Typically InAs, CdS, CdSe,11,12 and PbS13 were used as QDs, but the efficiency for most QDSSCs was around 1–4%.12,14,15 As for the other advantages of QDSSCs over DSSCs, the lower production cost and higher resistance to degradation are expected for QDSSCs because they are composed of inorganic materials.To improve the conversion efficiency, many research studies have focused on the search for optimal conditions for QDs on the size,16,17 binding on the substrate,18and for structure and materials of a semiconductor layer14,19–21 or their optimal combinations,11 but the understanding of the working mechanism is also necessary. The DSSCs and QDSSCs are very complicated electrochemical systems; typically a transparent electrode, a nanoporous semiconductor layer for electron transport such as TiO2, sensitizers, an electrolyte and redox solution, and a counter electrode. There are many electron or hole transfer processes involved in the photoelectrochemical process, and the time scale for these processes ranges from femtoseconds to seconds.22 Furthermore, not only an electron transfer in the expected direction, but also the back electron transfer or recombination processes are known to lower the cell efficiency.18,23,24
To investigate such processes of photogenerated carriers (electrons and holes), time-resolved measurement methods such as transient absorption (TA),22–24 transient photocurrent and transient photovoltage measurements have been generally used,25–28 and provided much information on the carrier dynamics of DSSCs and QDSSCs. However, each time-resolved method has some limitations to study the dynamics; for the TA method, optical absorption of the target molecules must be within the detection spectral range; intermediate states are not observed by current or voltage because they are the final output of a photovoltaic cell; and then it is difficult to clarify the whole process of carrier dynamics. As similar techniques in the frequency domain, intensity-modulated photovoltage and photocurrent spectroscopies,29,30 and impedance spectroscopy31 also have given fruitful information on the interfacial charge transfer, but they also do not give the whole information.
Although there are a number of studies of the carrier dynamics in DSSCs, there are much less data available for QDSSCs, and only a few groups lead this field. Hodes32 reviewed the difference of the carrier dynamics between DSSCs and QDSSCs, and it is explained that the main differences are caused by 1: the trap states at the QD/semiconductor interface, 2: different applicable electrolytes, and 3: multiple layers of QDs. The initial electron injection occurs on the picosecond order, where an electron–hole recombination process competes. There is another recombination process, where electrons injected into the semiconductor layer back to QDs via surface states. The holes generated in QDs are likely to be trapped on the picosecond time order, and they are transferred to redox species, whose charge transfer time ranges from picoseconds to milliseconds, depending on the redox species.33 Similarly to DSSCs, there is back electron transfer from the semiconductor to the electrolyte, competing with the electron diffusion in the semiconductor on the order of milliseconds. Different from DSSCs, the charge transfer time for each process has a large variation for QDSSCs probably due to the various QD states such as size, aggregation, etc. Such dynamic studies for QDSSCs are not so many, compared with the enormous number of studies for DSSCs. There are several important reports on carrier dynamics. Kamat, et al.7,17,34 studied the initial electron injection dynamics depending on the size of QDs, and clarified that the electron injection rate was beautifully explained by the energy difference in the conduction band between QDs and semiconductors. Shen and Gomez et al.27,28,35 measured electron and hole dynamics, and clarified that holes are trapped by surface states of QDs and/or scavenged by electrolytes within several picoseconds, and electrons are trapped at the interface and then injected into the semiconductor over several hundreds of picoseconds besides some direct injection of electrons within a few picoseconds. Although there have been studies on the carrier dynamics in QDSSCs on the picosecond time order, few data are available on the microsecond order, when back electron transfer, electrolyte diffusion, etc should be involved.
In the previous study,36,37 the carrier dynamics of DSSCs was investigated using the heterodyne transient grating (HD-TG) method in addition to TA and transient photocurrent (TP) methods on a nanoseconds to seconds time scale. Since the change in the refractive index is monitored using the HD-TG technique, various carrier dynamics can be monitored even if optical absorption is negligible, such as carrier transfer between TiO2, dyes and electrolyte species, carrier transport in a TiO2 layer or an electrolyte solution, etc. This advantage is suitable for DSSCs or QDSSCs, whose component materials or intermediate species do not necessarily have optical absorption, and such optically silent carrier dynamics is expected to be clarified using the TA method. In fact we could successfully observe the carrier dynamics at the electrode/electrolyte interface, especially on the electrolyte side; rearrangement or diffusion motion of charged electrolyte species occurred when the injected electrons were trapped at the TiO2 interface and when the reverse electron transfer occurred, which were not detected using the TA method.22 By combination of HD-TG, TA and TP measurements, we can expect to understand the whole dynamics in the time region from nanoseconds to seconds.
Similarly, in this study, we studied the carrier dynamics in QDSSCs using HD-TG, TA and TP methods, and clarified the dynamics involved in the working processes of QDSSCs in the temporal range from nanoseconds to seconds, from the investigations on the dependence on the applied voltage, concentration of electrolyte or redox species and the coating times of surface passivation of QDs.
Experimental method
The principle and the setup of the HD-TG method were reported in detail in the previous papers,36,38,39 and in ESI.† In this study, the pump light source was an OPO (Surelite II – 10FP) output excited by using a Nd:YAG laser (Panther, Continuum, Electro-Optics Inc.), and the pulse light had a wavelength of 500 or 570 nm, a pulse width of 5 ns, and a repetition rate of 0.5 Hz. The repetition rate was decided as it is slow enough to recover the original baseline for the HD-TG and TA measurements. The pump intensity was 0.65 mJ pulse−1 unless otherwise specified. The probe light was a fiber coupled CW semiconductor laser with a wavelength of 658 nm for all the HD-TG (10 mW) and the TA measurements (4 mW). The pump and probe lights were incident from the FTO substrate side to avoid the absorption of the pump light by a polysulfide redox solution. Transient photocurrent (TP) was measured using a potentiostat (Nikko Keisoku, time resolution: 10 μs), and it was recorded on an oscilloscope at the same time as the detection of the HD-TG response. TA responses were measured at the same probe wavelength just by removing a glass plate of a transmission grating used for the HD-TG measurement.The method for the preparation of the working electrodes was reported in a previous paper.40 AanataseTiO2 paste was prepared with TiO2 nanoparticles with an average diameter of 15 nm and polyethylene glycol with a molecular weight of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The resulting paste was cast onto a glass substrate coated with fluorine doped tin oxide (FTO) with a Scotch tape as a frame and spacer, raking off the excess solution with a glass rod (squeegee technique). The TiO2 electrodes were dried in air at room temperature for 10 min, and annealed at 450 °C for 30 min in a furnace, and then cooled down to room temperature. The film thickness was around 7 ± 1 μm. In the next step, CdSe was deposited on the electrode. The CdSe QDs were prepared using a chemical bath method, which was explained in the paper by Gorer and Hodes.41 Firstly, as the Se source, an 27 mM sodium selenosulphate (Na2SeSO3) solution was prepared by dissolving elemental Se powder in a 67 mM Na2SO3 solution. Secondly, 27 mM CdSO4 and 40 mM trisodium salt of nitrilotriacetic acid (N(CH2CooNa)3) were mixed in a volume ratio of 1:1. Finally, both solutions were mixed in a volume ratio of 1:2. The TiO2 electrodes were placed in darkness in a glass container filled with the final solution for 24 h. The samples were coated with ZnS for surface passivation of the QDs by using successive ionic layer adsorption and reaction (SILAR) in 100 mM Zn(CH2COO) and 100 mM Na2S aqueous solution for 1 min for each dip.27 The coating procedure was typically repeated twice unless otherwise specified. We confirmed the surface topography using SEM measurements before and after the growth of CdSe quantum dots (Fig. S2 in ESI†). The pore size of the electrode was on the order of tens of nanometers. The chemical composition of quantum dots was confirmed by XRD.
000. The resulting paste was cast onto a glass substrate coated with fluorine doped tin oxide (FTO) with a Scotch tape as a frame and spacer, raking off the excess solution with a glass rod (squeegee technique). The TiO2 electrodes were dried in air at room temperature for 10 min, and annealed at 450 °C for 30 min in a furnace, and then cooled down to room temperature. The film thickness was around 7 ± 1 μm. In the next step, CdSe was deposited on the electrode. The CdSe QDs were prepared using a chemical bath method, which was explained in the paper by Gorer and Hodes.41 Firstly, as the Se source, an 27 mM sodium selenosulphate (Na2SeSO3) solution was prepared by dissolving elemental Se powder in a 67 mM Na2SO3 solution. Secondly, 27 mM CdSO4 and 40 mM trisodium salt of nitrilotriacetic acid (N(CH2CooNa)3) were mixed in a volume ratio of 1:1. Finally, both solutions were mixed in a volume ratio of 1:2. The TiO2 electrodes were placed in darkness in a glass container filled with the final solution for 24 h. The samples were coated with ZnS for surface passivation of the QDs by using successive ionic layer adsorption and reaction (SILAR) in 100 mM Zn(CH2COO) and 100 mM Na2S aqueous solution for 1 min for each dip.27 The coating procedure was typically repeated twice unless otherwise specified. We confirmed the surface topography using SEM measurements before and after the growth of CdSe quantum dots (Fig. S2 in ESI†). The pore size of the electrode was on the order of tens of nanometers. The chemical composition of quantum dots was confirmed by XRD.
The electrolyte solution was a polysulfide aqueous solution (1 M) consisting of 1 M Na2S and 1 M S, unless otherwise specified. The sulfur ion species worked as both redox and electrolyte species. The counter electrode was copper sulfide (Cu2S). The working electrode and the counter electrode were connected with a copper wire to a potentiostat, which was used for an ammeter, and no bias voltage was applied unless otherwise specified. The counter electrode was used as a reference electrode, too.
Results and discussion
Assignment of the HD-TG, TA and TP responses
Firstly, we investigated optimal pump irradiation intensity for dynamics measurements. The pump intensity dependencies of the HD-TG, TA and TP measurements were studied for the pump intensity range of 0.35–0.85 mJ pulse−1 as shown in Fig. 1. Basically the signal intensities for all the responses for the HD-TG, TA and TP measurements increased as the pump intensity, and the responses seemed to be saturated at higher intensities.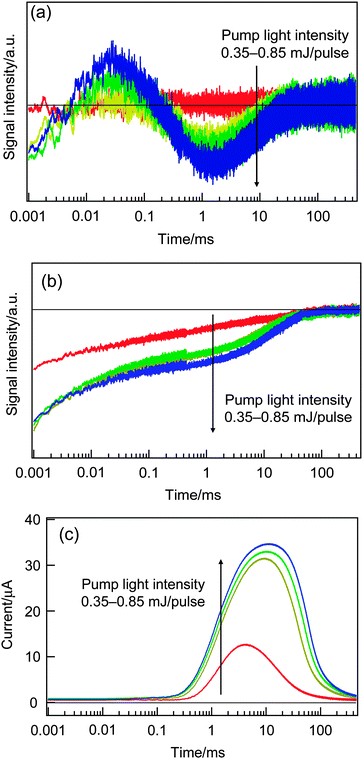 | ||
| Fig. 1 The dependencies of the HD-TG (a), TA (b) and TP (c) responses on the pump intensities in the range from 0.35 to 0.85 mJ pulse−1. | ||
In all the measurements, the response waveforms showed discontinuous changes between 0.35 and 0.5 mJ pulse−1. In the HD-TG responses, all the components in the response increased similarly; in the TA responses, a new component arose on the order of 10 ms; TP increased suddenly and looked saturated for a pump intensity of over 0.5 mJ pulse−1. It is supposed that there would be some threshold pump intensity for this QDSSC system.
As we explain later, the lifetime of electrons under this short circuit condition is on the order of several tens of milliseconds. When a single optical pulse with an intensity of 1 mJ pulse−1 is irradiated during the lifetime, the corresponding irradiated photon number is roughly on the same order as that for 1 sun. (When an optical pulse with 1 mJ pulse−1 is irradiated over an area of 1 cm2, the photon energy is equal to 10 J m−2, while 1 sun is equal to 1000 W m−2 and the irradiated photon number during 10 ms is 10 J m−2.) Considering the saturation of the photocurrent for the pump intensity >0.5 mJ per pulse, the number of the injected electrons was enough to saturate the number of trapped electrons in TiO2, which build up the photovoltage to flow current. This condition is considered to correspond to a typical working condition of this QDSSC. Since the temporal responses were the same for different intensities of the pump intensities >0.5 mJ pulse−1, the following experiments were performed for 0.65 mJ pulse−1 throughout the experiments.
Next, the dependence on the materials of the counter electrodes was studied to check the effect of the reaction rate of redox species in solutions. At the counter electrodes, oxidized redox species at the working electrode are reduced to an original species, and if the reaction rate is not fast enough, the oxidized redox species are accumulated in the electrolyte solution and it may affect the HD-TG, TA and TP responses in the slower time region for repeated experiments. The influence was observed in the TP responses for a Pt counter electrode in polysulfide solution by repeating the pulse irradiation, as shown in Fig. S3 (ESI†). By repeating the pulse irradiation, the photocurrent was gradually reduced. It is known that a Cu2S counter electrode is more active for the redox reaction of polysulfide, compared with the Pt counter electrode,42 and the counter electrode was replaced with a Cu2S counter electrode, which avoided the photocurrent reduction. On the other hand, the dependence on the counter electrode was not observed for the HD-TG responses for Pt and Cu2S counter electrodes as shown in Fig. S4 (ESI†). Even after 100 measurements, the difference was smaller than the error range and the effect of the counter electrode was negligible for the HD-TG responses.
The origin of the TA components was considered, and the components after microseconds were not caused by the excited holes left in QDs by initial electron injection into TiO2, which occurs within several picoseconds,27,28 and also were not caused by the excited state of QDs, which decays on the order of nanoseconds.7 The TA component in 1–100 ms was investigated. This component was observed for DSSCs in the previous paper,36 and was assigned to the decay of the trapped electrons in TiO2. To confirm the assignment, the applied voltage dependence was investigated. The dependencies of anodic and cathodic applied potentials are shown in Fig. 2(a) and (b), respectively. (It is noted that the solar cell typically works under cathodic applied potential in the range from 0 – Voc.) When the absolute value of the applied potentials, |V|, was less than 0.5 V, the component in 1–100 ms was observed, but the component was lost for |V| > 0.5 V. For the anodic applied potential, by decreasing the conduction band of TiO2, the trapped electrons were taken away from the circuit via the FTO electrode. For the cathodic applied potential, by increasing the conduction band of TiO2, the trapped electrons were taken by the redox species in the electrolyte solution. A weak response observed in the wide time range even for |V| > 0.5 V was assumed due to inhomogeneously distributed free electrons in TiO2. This cell structure has a Voc value of 0.52 V, corresponding to the cathodic potential, and the electrons should be still trapped for a cathodic potential of 0.5 V, but the electrons are taken away probably due to the non-perfect cell system designed for the HD-TG and TA measurements.
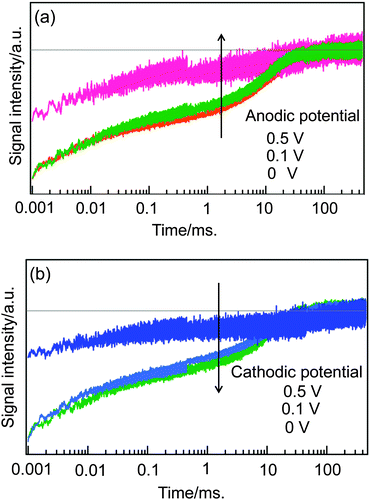 | ||
| Fig. 2 Dependence of TA responses on the applied bias voltage is shown for anodic (a) and cathodic (b) applied potentials, respectively. | ||
Next, the responses of the HD-TG and TA were compared in the presence and the absence of the working electrode, namely only the electrolyte solution and the same solution including the working electrode. In the absence of the working electrode, the pump wavelength was set at 500 nm, and it was set at 570 nm in the presence of the working electrode. Since the absorbance of an electrolyte solution was 3.0 for 500 nm, and 0.01 for 570 nm, respectively, only the polysulfide was excited in the absence of the working electrode, and only the QDs were excited in the presence of the working electrode (the IPCE of this type of cell is shown in Fig. S5 in ESI†).
By comparing the TA responses in the presence or absence of the electrode (Fig. 3), the first component (i) in 1–100 μs was observed for both the cases, and the second component (ii) in 1–100 ms was observed only in the presence of the electrode. It is supposed that the (i) component corresponds to the decay of S−˙, which is generated via the oxidization of S2− in the earlier time scale, because this component originates from the species in the electrolyte solution.35,43 In the case of the direct excitation of S2−, there are no reports on the formation of S−˙, but the similarity of the (i) component in both cases indicated that (i) has the same origin, S−˙. With regard to the (ii) component, as we have already explained, it is due to the trapped electrons in TiO2, and the fact that this component was observed only in the presence of the electrode supports the assignment.
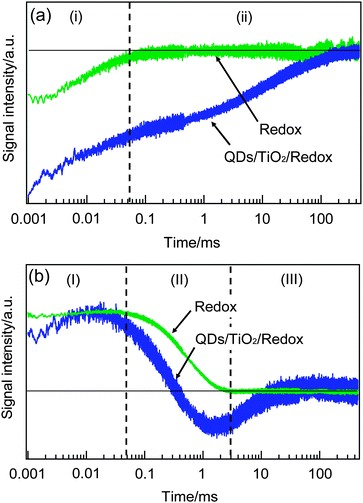 | ||
| Fig. 3 The TA (a) and HD-TG (b) responses only for the redox solution and the QDSSC system including the working electrode. Time region was separated for each observed component for easy comparison. | ||
By comparing the HD-TG responses in the presence or absence of the electrode, the first and third components, (I) and (III) were observed only in the presence of the electrode, and the second component (II) was observed in both cases. The (I) component had a time constant different from that of the (i) component in TA, and corresponds to the process after the electron injection from QDs and the following hole compensation from the electrolyte species, and also this occurred before the photocurrent increase in the circuit shown in Fig. 1(c). We assume that this process corresponds to the reorganization of electrolyte species at the electrode/electrolyte interface in order to compensate for the accumulated electron charge at the TiO2 surface. This is supported by the fact that similar dynamics was observed for DSSCs in the similar time region, reported recently.36 The (II) component corresponds to the thermal diffusion process in the electrode–electrolyte sample or the electrolyte solution, which is a typical response observed in the HD-TG signals, originating from the refractive index change caused by the temperature change. The (III) component is the process only observed in the presence of the electrode, and the assignment was performed based on the following data.
To confirm the assignment, we compared the responses of HD-TG and TA when the electrolyte solution included electrolytes or not, as shown in Fig. 4. In the HD-TG responses, (I) and (III) components were observed only when the solution included electrolytes, while the (II) component was observed for both the cases. Since the (II) component was assigned to thermal diffusion processes in the electrode/solution sample, it is reasonable that the process was observed for both the cases. The electrolytes are necessary to observe (I) and (III) components, and it is reasonable that the (I) component corresponds to the reorganization process of redox species at the electrode/electrolyte interface. With regard to the (III) component, considering the time region on the order of 1–100 ms, which was on the same order as the photocurrent decay in the TP response and the decay of the trapped electrons of TiO2 observed in the TA response, it is related to the electron decay processes involving both the TiO2 layer and the redox species, thus it should be reverse electron transfer from TiO2 to redox species. The signal intensities for (I) and (III) were decreased when the solution was water, which can be understood that the trapped electrons in TiO2 were reduced due to the lack in counter cations and that caused to reduce the process of the reorganization of electrolyte species and the reverse electron transfer processes. The signal intensity of (II) reflects the difference in the refractive index change due to thermal property of water and an electrolyte solution.44
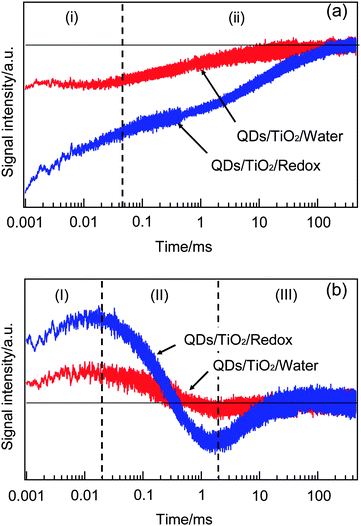 | ||
| Fig. 4 The TA (a) and HD-TG (b) responses when the solution included electrolyte species or not are shown. | ||
As for the TA responses, the (i) component was lost and the (ii) component was reduced considerably in the absence of electrolyte species. Since the (i) component corresponds to the decay of S−˙ species, the disappearance of the (i) component in the absence of redox species agrees with the assignment. The decrease in the signal intensity of (ii) is understood that a considerable number of electrons cannot be trapped stably in the absence of the counter cations.
Dependence on various electrode and electrolyte conditions
At first, we investigated how the electrolyte concentration affects the carrier dynamics. The HD-TG, TA, and TP results are shown in Fig. 5. In the HD-TG responses, with increasing electrolyte concentration the (I) process was accelerated; the signal intensity of (II) increased; the (III) signal intensity decreased and accelerated. The change in the (I) process indicated that the reorganization of electrolyte species at the interface became faster, and it is reasonable because the concentration of ionic species was increased. The increase in (II) intensity is just due to the optical property change due to the electrolyte concentration variation; the refractive index with respect to the temperature is typically positive for various electrolyte solutions,44 although there are no data available for polysulfide solutions. As for the (III) component, the acceleration of the process can be explained in the same manner as for the (I) component. The signal intensity decrease indicated the reduction of the electron acceptors (oxidized species), which are generated via the hole transfer from QDs to the redox species. It is supposed that the oxidized species at the interface disappeared more quickly by hole transfer due to higher redox concentration.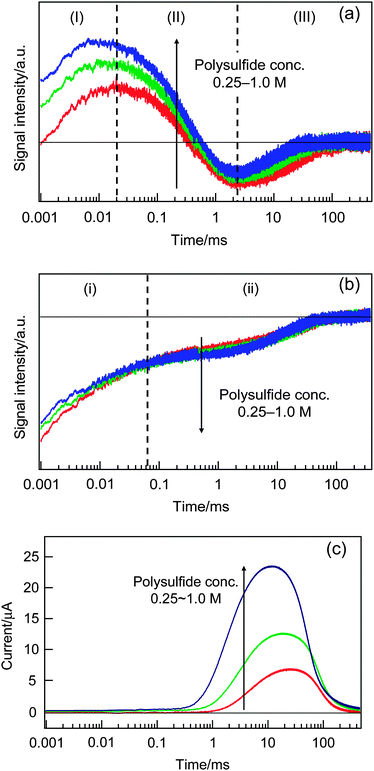 | ||
| Fig. 5 The dependencies of the HD-TG (a), TA (b) and TP (c) responses on the polysulfide concentration. | ||
Looking at the TA responses, the (i) component decreased, while the (ii) component increased slightly as the concentration increased. Since the (i) component corresponds to the decay of S−˙, the signal intensity increase by increasing the electrolyte concentration is reasonable. The increase in the (ii) intensity indicated that the trapped electrons in TiO2 increased, which agreed with the HD-TG result that indicated that the reverse electron transfer processes were reduced, because more electrons can be retained in TiO2. The photocurrent increased and the rising time was accelerated with the concentration as shown in Fig. 5(c), indicating smoother hole acceptance by the redox species and faster ambipolar diffusion of electrons in the TiO2 layer as the electrolyte concentration increases.
Next, the effect of the ratio between Na2S and S as an electrolyte solution was investigated (Fig. 6). The ratio was 1:0, 1:0.25, 1:0.5 and 1:1. By increasing the ratio of S, a multi-atomic state, Sn2− was increased and S2− was decreased, and the former and the latter species work as an oxidized and reduced redox species, while the cation concentration was maintained to be 1 M. As the ratio of sulfur was increased, only the signal intensity of (II) was increased and the other components did not show any dependence on the HD-TG responses, and no dependence for all the components was observed on the TA responses, and a gradual increase in photocurrent was observed for the TP responses. The only change observed in the HD-TG response is probably due to the similar reason for the electrolyte concentration dependence; namely the temperature coefficient of the refractive index was different depending on the ingredients in the electrolyte solution, though there are no literature data to explain it. The photocurrent increase indicated the efficient hole acceptance by increasing the sulfur ratio. It was concluded that this ratio did not affect the carrier dynamics.
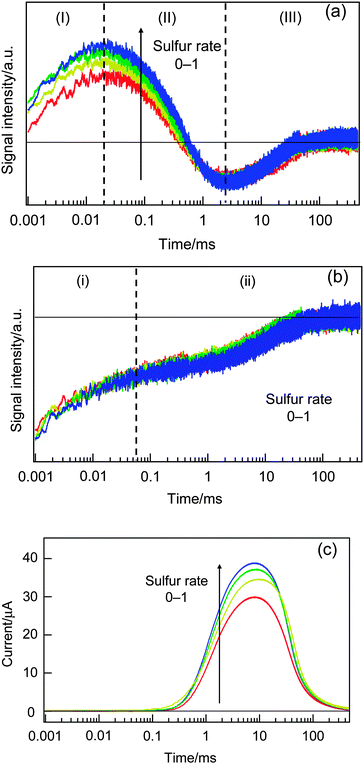 | ||
| Fig. 6 The dependencies of the HD-TG (a), TA (b) and TP (c) responses on the ratio between Na2S and S as an electrolyte solution. | ||
Finally, the effect of zinc sulfur coating was investigated. This coating is necessary for this kind of QDSSCs not only for preventing the corrosion of CdSe QDs, but also for stopping the reverse electron transfer from TiO2 to electrolytes,18 and actually the efficiency was doubled by this coating. Based on the SILAR method,45,46 the electrode was coated by ZnS on the order of a few nanometers in thickness, but the deposition times of ZnS is empirically decided and it is explained that the coating works for the blockage of the TiO2 surface and passivation of QDs, and also it promotes electron transfer.47 But the effect on the carrier dynamics is still not clear. We compared the HD-TG, TA and TP responses for different coating times, as shown in Fig. 7. By increasing the coating times, the signal intensity was gradually decreased, the (I) component was delayed and the intensity of the (III) component was much reduced in the HD-TG responses; the TA signal intensity was reduced, and the TP signal was reduced over 3 times coating.
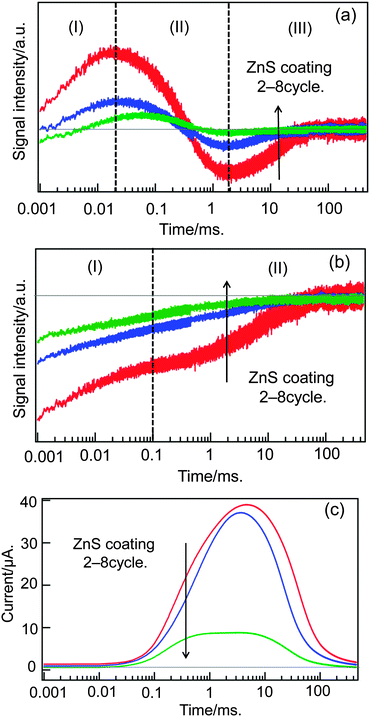 | ||
| Fig. 7 The dependencies of the HD-TG (a), TA (b) and TP (c) responses on the times of ZnS coating. | ||
The delay of the (I) component of the HD-TG signal can be understood that the coating of ZnS screened the charge of the trapped electrons in TiO2 from the electrolyte solution and that the effect of the Coulomb charge was weakened to de-accelerate the reorganization process of electrolyte species at the interface. The reduction of the (III) component indicated that the reverse electron transfer was blocked by the ZnS layer. The decrease in the TA and HD-TG signal intensity indicated that the trapped electrons were reduced by ZnS coating, and it is understood that the counter cations for stabilizing the trapped electrons in TiO2 cannot migrate into the TiO2 layer and a smaller portion of electrons can stay in the TiO2 layer. The photocurrent was reduced due to the difficulty in charge transfer between the QDs and the redox species. Thus, it is concluded that the ZnS coating has a favorable effect on the reverse electron transfer, but has an unfavorable effect on the stabilization of trapped electrons and hole compensation of QDs, and the balance is important for optimization of the photovoltaic performance of QDSSCs.
Conclusion
We investigated the carrier dynamics in QDSSCs by combination of the HD-TG, TA, and TP methods. Since each technique gives different information, we could get the whole picture of the carrier dynamics in QDSSCs in the time range from microseconds to seconds. HD-TG gives information on the motion of ionic species at the interface; TA gives the electron lifetime in TiO2; TP gives the final output of current. It was clarified that this approach was effective not only for understanding the mechanism of the QDSSCs, but also for optimizing various device parameters. We expect that this approach will boost the development of new QDSSCs, and enhance their efficiency.Acknowledgements
This research was supported by PRESTO and CREST programs of Japan Science and Technology Agency (JST).Notes and references
- A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS
.
- T. W. Hamann, R. A. Jensen, A. B. F. Martinson, H. Van Ryswyk and J. T. Hupp, Energy Environ. Sci., 2008, 1, 66 CAS
- J. K. Lee and M. J. Yang, Mater. Sci. Eng., B, 2011, 176, 1142 CrossRef CAS
- B. C. O'Regan and J. R. Durrant, Acc. Chem. Res., 2009, 42, 1799 CrossRef CAS
- D. Wei, Int. J. Mol. Sci., 2010, 11, 1103 CrossRef CAS
- Q. F. Zhang and G. Z. Cao, Nano Today, 2011, 6, 91 CrossRef CAS
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737 CAS
- S. Ruhle, M. Shalom and A. Zaban, Chemphyschem, 2010, 11, 2290 CrossRef
- Q. Shen, K. Katayama, T. Sawada, S. Hachiya and T. Toyoda, Chem. Phys. Lett., 2012, 542, 89 CrossRef CAS
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef
- S. Gimenez, I. Mora-Sero, L. Macor, N. Guijarro, T. Lana-Villarreal, R. Gomez, L. J. Diguna, Q. Shen, T. Toyoda and J. Bisquert, Nanotechnology, 2009, 20 Search PubMed
- Q. X. Zhang, X. Z. Guo, X. M. Huang, S. Q. Huang, D. M. Li, Y. H. Luo, Q. Shen, T. Toyoda and Q. B. Meng, Phys. Chem. Chem. Phys., 2011, 13, 4659 RSC
- S. Hachiya, Q. Shen and T. Toyoda, J. Appl. Phys., 2012, 111 Search PubMed
- S. Q. Huang, Q. X. Zhang, X. M. Huang, X. Z. Guo, M. H. Deng, D. M. Li, Y. H. Luo, Q. Shen, T. Toyoda and Q. B. Meng, Nanotechnology, 2010, 21 Search PubMed
- Q. Shen, A. Yamada, S. Tamura and T. Toyoda, Appl. Phys. Lett., 2010, 97 Search PubMed
- Q. Shen, J. Kobayashi, L. J. Diguna and T. Toyoda, J. Appl. Phys., 2008, 103 Search PubMed
- I. Robel, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2007, 129, 4136 CrossRef CAS
- I. Mora-Sero, S. Gimenez, F. Fabregat-Santiago, R. Gomez, Q. Shen, T. Toyoda and J. Bisquert, Acc. Chem. Res., 2009, 42, 1848 CrossRef CAS
- L. J. Diguna, Q. Shen, J. Kobayashi and T. Toyoda, Appl. Phys. Lett., 2007, 91 Search PubMed
- T. Toyoda and Q. Shen, J. Phys. Chem. Lett., 2012, 3, 1885 CrossRef CAS
- M. Samadpour, S. Gimenez, P. P. Boix, Q. Shen, M. E. Calvo, N. Taghavinia, A. I. Zad, T. Toyoda, H. Miguez and I. Mora-Sero, Electrochim. Acta, 2012, 75, 139 CrossRef CAS
- A. Y. Anderson, P. R. F. Barnes, J. R. Durrant and B. C. O'Regan, J. Phys. Chem. C, 2010, 114, 1953 CAS
- S. Ardo, Y. Sun, F. N. Castellano and G. J. Meyer, J. Phys. Chem. B, 2010, 114, 14596 CrossRef CAS
- J.
G. Rowley and G. J. Meyer, J. Phys. Chem. C, 2011, 115, 6156 CAS
- P. R. F. Barnes, A. Y. Anderson, J. R. Durrant and B. C. O'Regan, Phys. Chem. Chem. Phys., 2011, 13, 5798 RSC
- H. K. Dunn and L. M. Peter, J. Phys. Chem. C, 2009, 113, 4726 CAS
- N. Guijarro, T. Lana-Villarreal, Q. Shen, T. Toyoda and R. Gomez, J. Phys. Chem. C, 2010, 114, 21928 CAS
- N. Guijarro, Q. Shen, S. Gimenez, I. Mora-Sero, J. Bisquert, T. Lana-Villarreal, T. Toyoda and R. Gomez, J. Phys. Chem. C, 2010, 114, 22352 CAS
- G. Schlichthörl, S. Y. Huang, J. Sprague and A. J. Frank, J. Phys. Chem. B, 1997, 101, 8141 CrossRef
- L. Dloczik, O. Ileperuma, I. Lauermann, L. M. Peter, E. A. Ponomarev, G. Redmond, N. J. Shaw and I. Uhlendorf, J. Phys. Chem. B, 1997, 101, 10281 CrossRef CAS
- J. van de Lagemaat, N. G. Park and A. J. Frank, J. Phys. Chem. B, 2000, 104, 2044 CrossRef CAS
- G. Hodes, J. Phys. Chem. C, 2008, 112, 17778 CAS
- N. S. Lewis, Annu. Rev. Phys. Chem., 1991, 42, 543 CrossRef CAS
- A. Kongkanand, K. Tvrdy, K. Takechi, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2008, 130, 4007 CrossRef CAS
- Q. Shen, Y. Ayuzawa, K. Katayama, T. Sawada and T. Toyoda, Appl. Phys. Lett., 2010, 97, 263113 CrossRef
- S. Kuwahara, H. Hata, S. Taya, N. Maeda, Q. Shen, T. Toyoda and K. Katayama, Phys. Chem. Chem. Phys., 2013, 15, 5975 RSC
- Q. Shen, Y. Ogomi, B. W. Park, T. Inoue, S. S. Pandey, A. Miyamoto, S. Fujita, K. Katayama, T. Toyoda and S. Hayase, Phys. Chem. Chem. Phys., 2012, 14, 4605 RSC
- K. Katayama, M. Yamaguchi and T. Sawada, Appl. Phys. Lett., 2003, 82, 2775 CrossRef CAS
- M. Yamaguchi, K. Katayama and T. Sawada, Chem. Phys. Lett., 2003, 377, 589 CrossRef CAS
- Q. Shen and T. Toyoda, Thin Solid Films, 2003, 438–439, 167 CrossRef CAS
- S. Gorer and G. Hodes, J. Phys. Chem., 1994, 98, 5338 CrossRef CAS
- G. Hodes, J. Manassen and D. Cahen, J. Electrochem. Soc., 1980, 127, 544 CrossRef CAS
- L. Min and R. J. D. Miller, Appl. Phys. Lett., 1990, 56, 524 CrossRef CAS
- K. Katayama, T. Sawada, I. Tsuyumoto and A. Harata, Bull. Chem. Soc. Jpn., 1999, 72, 2383 CrossRef CAS
- A. Braga, S. Giménez, I. Concina, A. Vomiero and I. n. Mora-Seró, J. Phys. Chem. Lett., 2011, 2, 454 CrossRef CAS
- H. M. Pathan and C. D. Lokhande, Bull. Mater. Sci., 2004, 27, 85 CrossRef CAS
- N. Guijarro, J. M. Campina, Q. Shen, T. Toyoda, T. Lana-Villarreal and R. Gomez, Phys. Chem. Chem. Phys., 2011, 13, 12024 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp51542g |
| This journal is © the Owner Societies 2013 |