 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Different formation kinetics and photoisomerization behavior of self-assembled monolayers of thiols and dithiolanes bearing azobenzene moieties
Chun L.
Yeung†
a,
Scott
Charlesworth†
ab,
Parvez
Iqbal
a,
James
Bowen
a,
Jon A.
Preece
b and
Paula M.
Mendes
*a
aSchool of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: p.m.mendes@bham.ac.uk; Tel: +44 (0)121-414-5343
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 9th May 2013
Abstract
Self-assembled monolayers (SAMs) containing azobenzene moieties are very attractive for a wide range of applications, including molecular electronics and photonics, bio-interface engineering and sensoring. However, very little is known about the aggregation and photoswitching behavior that azobenzene units undergo during the SAM formation process. Here, we demonstrate that the formation of thiol-based SAMs containing azobenzenes (denoted as AzoSH) on gold surfaces is characterised by a two-step adsorption kinetics, while a three-step assembly process has been identified for dithiolane-based SAMs containing azobenzenes (denoted AzoSS). The H-aggregation on the AzoSS SAMs was found to be remarkably dependent on the time of self-assembly, with less aggregation as a function of time. While photoisomerization of the AzoSH was suppressed for all different assembly times, the reversible trans–cis photoisomerization of AzoSS SAMs formed over 24 hours was clearly observed upon alternating UV and Vis light irradiation. We contend that detailed information on formation kinetics and related optical properties is of crucial importance for elucidating the photoswitching capabilities of azobenzene-based SAMs.
Introduction
Photo-switchable self-assembled monolayers (SAMs) have been receiving considerable attention, motivated by their potential applications in molecular electronics and photonics, bio-interface engineering, catalysis and sensoring.1–4 One of the most frequently studied classes of photo-switchable molecules is the azobenzene-containing molecules, since the azobenzene moiety is known to undergo cis–trans isomerism.5 Photoisomerization from the thermodynamically more stable trans isomer to the cis isomer and vice versa can be selectively induced by irradiation with either ultraviolet (UV) light or visible (Vis) light. The free volume required for the cis form is larger than for the trans form assuming either an inversion or a rotation mechanism of the azo bond.6–8 Thus, photoisomerization rates strongly depend on the local free volume available for the conformation change. In this regard, an azobenzene-terminated alkanethiol SAM hardly exhibits any photoisomerization owing to the existence of both H-aggregation and spatial constraints.9–11 Regarding the former, neighboring azobenzenes have a high tendency to form head-to-head H-aggregates in a SAM due to π–π stacking interactions, leading to a dense molecular packing, that suppresses azobenzene photoisomerization.10Efforts have been made to increase the spacing between the azobenzene moieties in order to promote the reversible trans-to-cis photoisomerization process in SAMs on gold.12–21 Photoswitching of azobenzene SAMs has been achieved by employing asymmetrical disulfides,12 asymmetrical thioethers,13 bulky alkyl groups into the benzene ring of the azobenzene unit,14–16 or by inclusion of a bulky carborane unit para to the diazo functionality in order to space out the backbones.17 Other strategies relied on azobenzene derivatives with bulkier terminal headgroups than thiols, including a tripodal adamantane-based thioacetate headgroup,18 an asparagusic acid-based 1,2-dithiolane headgroup19 and an α-lipoic acid-based 1,2-dithiolane headgroup.21 Regarding the latter, no photoisomerization was observed when the molecules were absorbed from the trans isomer, whereas molecules adsorbed in cis configuration were photoreactive. The inhibition of structural arrangement associated with trans–cis isomerization when the molecules were absorbed from the trans isomer is most probably due to hydrogen bonds between the amide groups by which the azobenzene moieties were attached to the dithiolane headgroup moiety.21 Although SAMs of an azobenzene terminated dithiolane analogue, in which the amide group in the spacer was replaced by an ester moiety, were prepared by the same authors,21 no photoisomerization studies were conducted. Thus, the photoisomerization capabilities of α-lipoic acid-based SAMs containing azobenzene moieties are not fully understood and remain to be further investigated. Furthermore, in this study and others,17–21 little attention has been paid to the aggregation and photoswitching behavior that azobenzene units undergo during the SAM formation process. Single monolayer formation times have been used for accessing the switching of SAMs containing azobenzene moieties and times of formation have been varied from 0.5 h to 24 h.17,18,20,21
Stimulated by the lack of information in certain aspects of azobenzene-based SAMs as discussed above, in this study we investigate in detail monolayers formed from an α-lipoic acid-based azobenzene (AzoSS, Fig. 1), in which an ester linkage is incorporated into the chain between the α-lipoic acid and the azobenzene, and a thiol-based azobenzene analogue (AzoSH, Fig. 1), in which the α-lipoic acid is replaced by an alkanethiol which is linked to the azobenzene using an ether bond. The goal of this work is to extend our knowledge on azobenzene-based SAMs, to answer two principal questions. First, how does H-aggregation and the optically induced switching depend on the time of SAM formation, and are these properties dependent on the headgroup? Second, are α-lipoic acid-based azobenzene SAMs capable of undergoing reversible photoisomerization in the absence of hydrogen bonding on the SAM structure? The adsorption kinetics of the AzoSS and AzoSH SAMs are studied by means of contact angle, ellipsometry, surface plasmon resonance (SPR) spectroscopy and X-ray photoelectron spectroscopy (XPS). The relation between time of SAM formation and its effect on H-aggregation and photoswitching capabilities for both SAMs, AzoSH and AzoSS SAMs, is also ascertained by UV/Vis spectroscopy using the spectroscopic characteristics of the azo chromophores in the cis and trans conformations.
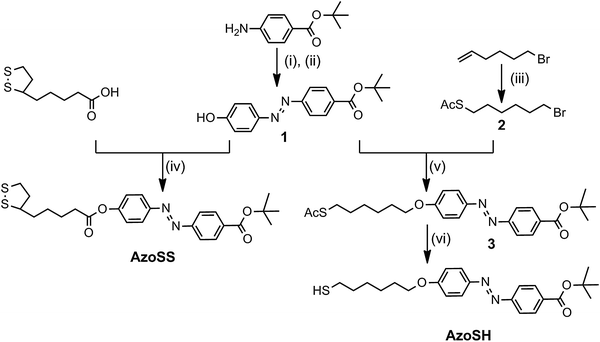 | ||
| Fig. 1 Synthesis of AzoSS and AzoSH: (i) NaNO2, 1.1 M HCl, MeOH, 0 °C; (ii) Phenol, KOH(aq), MeOH, rt, 4 h; (iii) CH3COSH, AIBN, PhMe, reflux, 2 h; (iv) DCC, DMAP (cat), DCM, rt, N2(g) atm, 16 h; (v) K2CO3, acetone, refluxed, 16 h; (vi) 0.1 M HCl, MeOH, reflux, N2(g) atm, 4 h. | ||
Results and discussion
Two new azobenzene derivatives, AzoSS and AzoSH were synthesized as outlined in Fig. 1. Synthesis of AzoSS was initiated through diazotisation of the 4-aminobenzoic acid tert-butyl ester, which was reacted with phenol under basic conditions to give the azobenzene 1. The azobenzene 1 was coupled with thioctic acid using N,N′-dicyclohexylcarbodiimide (DCC) in the presence of 4-dimethylaminopyridine (DMAP) to afford the AzoSS. The synthesis of AzoSH involved initial formation of the thioacetate 2via thioacetylation of 6-bromo-1-hexene in the presence of CH3COSH and the radical initiator, 2,2′-azobisisobutyronitrile (AIBN), followed by alkylation with the phenoxide moiety in 1, to form the azobenzene thioacetate 3. Subsequently, the thioacetate 3 was hydrolysed under acidic conditions to obtain the desired AzoSH. These hydrolysis conditions did not hydrolyse the tert-butyl ester group.Fig. 2 shows the UV absorption spectra of AzoSS and AzoSH in ethanol (0.0625 mM) before and after irradiation with UV (365 nm) and subsequent visible light irradiation (436 nm). Typical of azobenzene derivatives, the trans form is more stable and is the dominant isomer before UV irradiation. The trans form of AzoSS exhibits a strong π–π* absorption peak with a maximum wavelength (λmax) at 329 nm and a weak n–π* band at around 445 nm. The corresponding bands for the trans form of AzoSH are found at 360 nm and 445 nm. Thus, the λmax of the π–π* absorption band is influenced by the linking functionality (i.e. ether or ester group) between the azobenzene and the headgroup, with λmax of the AzoSH being bathochromically shifted by 25 nm with respect to that of AzoSS. For both compounds, AzoSS and AzoSH, upon UV irradiation for 1 min, a drastic reduction in the π–π* band is observed, whereas the n–π* becomes more intense. These results suggest that the azobenzene molecules are converted into cis isomers in both solutions. Subsequent visible light irradiation for 1 min gives rise to cis to trans isomerization, maximizing the π–π* absorption band. Although the cis to trans isomerization is fully reversible for the AzoSS molecule, the AzoSH molecule displays slightly less reversible behaviour. This photoequilibrium composition remained unchanged even after the irradiation was prolonged to 5 min.
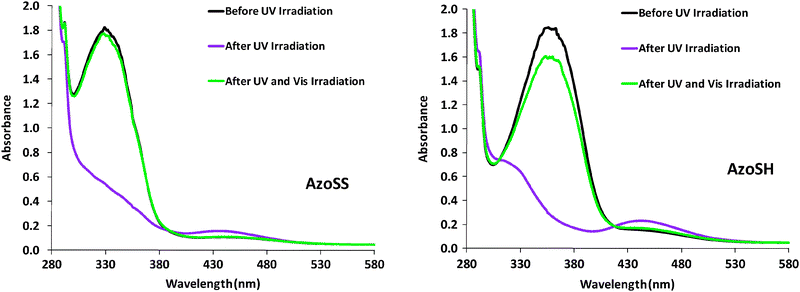 | ||
| Fig. 2 UV/Vis absorption spectra of AzoSS and AzoSH in ethanol before and after UV and Vis irradiation. UV irradiation at 365 nm led to trans → cis isomerization, whereas Vis irradiation at 436 nm drove the back reaction (cis → trans). | ||
SAM formation was evaluated using ellipsometry, contact angle and XPS (Fig. 3–5) after 24 h immersion time of a cleaned gold substrate in a 0.0625 mM ethanolic solution of AzoSS or AZoSH. The ellipsometric thicknesses of the fully formed SAMs are 1.8 nm (AzoSS) and 1.6 nm (AzoSH), and both are less than the theoretical molecular length of the molecules (both 2.5 nm). This discrepancy, between molecular length and SAM thickness, is expected and is in agreement with the literature, being ascribed to both the tilt angle and density of the SAM surfactants.22,23 The advancing (θAdv) and receding (θRec) contact angles for AzoSS (≈90° and ≈75°, respectively) and AzoSH (≈90° and ≈80°, respectively) are in good agreement with the literature24,25 for tert-butyl ester SAMs, noting that the final hysteresis values (θAdv–θAdv) of AzoSS (∼15°) are slightly larger than AzoSH (10°) indicating a less dense packed SAM for AzoSS. XPS analysis confirms the presence of the elemental species N, C, O and S on the AzoSS SAMs (Fig. 5). The single peak at 400.5 eV in the N (1s) spectrum is assignable to the nitrogen of the azo group.16 The C (1s) spectrum can be deconvoluted into three peaks, which are attributed to five different binding environments (Fig. 5). The main, predominant peak (284.7 eV) is attributed to C–C bonds26 within both the alkyl chains and phenyl rings. The first of the two smaller peaks (286.4 eV) is attributed to C (1s) of the three binding environments of C–S, C–N and C–O–C.26 The third and final peak (288.7 eV) is attributed to the C (1s) photoelectron of the carbonyl moiety, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.26 The O (1s) spectra are de-convoluted into two different peaks, corresponding to two different binding environments, arising from the ester moieties, C–O–C (533.9 eV) and the carbonyl oxygen CO (532.3 eV).26 The S (2p) spectrum displays a doublet structure at 163.6 eV (S (2p1/2)) and 162.4 eV (S (2p3/2)), which is assignable to the thiolate-type sulfur bound to the gold surface as previously reported for SAMs using a 1,2-dithiolane headgroup.21 The AzoSH SAMs exhibit similar N (1s) and S (2p) spectra (Fig. 5). The C (1s) and O (1s) are also similar, but with lower intensities for the C (1s) peak at 288.7 eV and O (1s) at 532.3 eV due to the absence of the second carbonyl moiety on the AzoSH molecule.
O.26 The O (1s) spectra are de-convoluted into two different peaks, corresponding to two different binding environments, arising from the ester moieties, C–O–C (533.9 eV) and the carbonyl oxygen CO (532.3 eV).26 The S (2p) spectrum displays a doublet structure at 163.6 eV (S (2p1/2)) and 162.4 eV (S (2p3/2)), which is assignable to the thiolate-type sulfur bound to the gold surface as previously reported for SAMs using a 1,2-dithiolane headgroup.21 The AzoSH SAMs exhibit similar N (1s) and S (2p) spectra (Fig. 5). The C (1s) and O (1s) are also similar, but with lower intensities for the C (1s) peak at 288.7 eV and O (1s) at 532.3 eV due to the absence of the second carbonyl moiety on the AzoSH molecule.
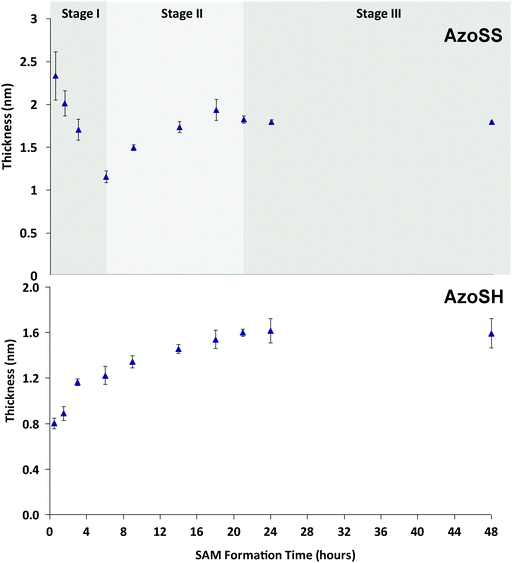 | ||
| Fig. 3 Ellipsometry kinetic study, thickness versus the formation time for AzoSS and AzoSH SAMs prepared by immersion of a clean gold substrate into the sample solution for 0.5 h, 1.5 h, 3 h, 6 h, 9 h, 18 h, 21 h, 24 h and 48 h. | ||
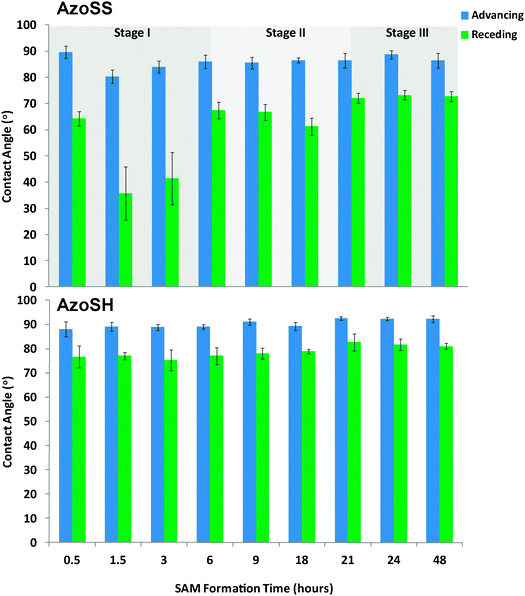 | ||
| Fig. 4 Water contact angle kinetics of AzoSS and AzoSH SAMs. | ||
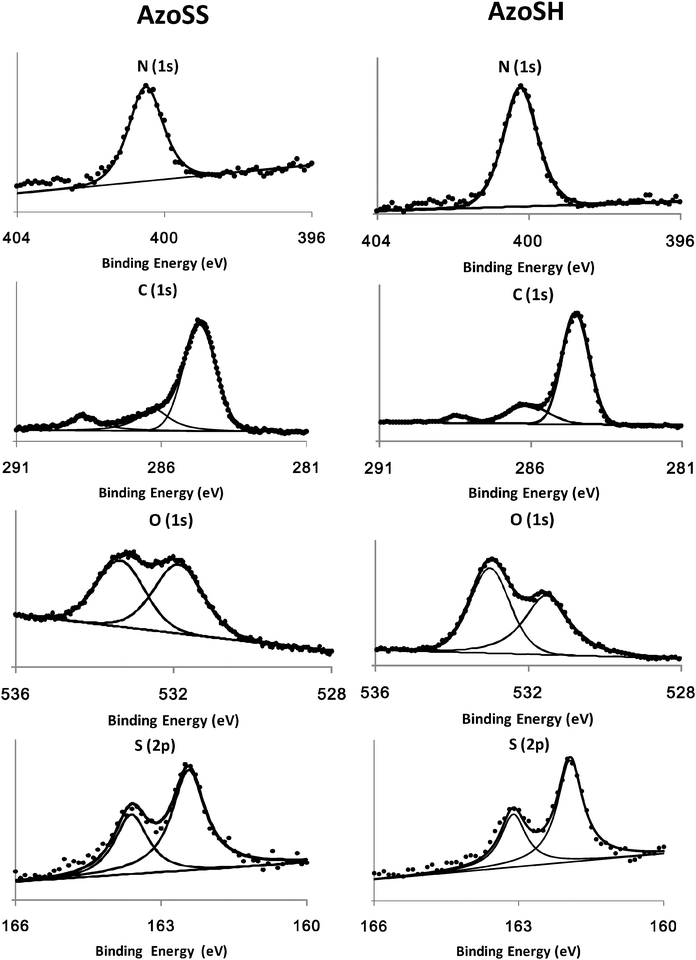 | ||
| Fig. 5 N (1s), C (1s), O (1s) and S (2p) XPS spectra of AzoSS and AzoSH SAMs, together with the corresponding fits. | ||
The kinetics of formation of the AzoSS and AzoSH SAMs was investigated by ellipsometry and contact angle. SAMs were formed by immersion of freshly cleaned Au substrates in 0.0625 mM ethanolic solutions of either AzoSS or AzoSH for 0.5 h, 1.5 h, 3 h, 6 h, 9 h, 18 h, 21 h, 24 h and 48 h, followed by rinsing with ethanol. Ellipsometric thickness and contact angles (θAdv and θRec) were recorded for each time interval (Fig. 3 and 4). In the case of AzoSS monolayers, three distinct SAM formation stages can be discerned in both Fig. 3 and 4 when considering the ellipsometric thickness and the θRec angle, which is significantly different to a simple alkyl thiol and indeed the AzoSH growth kinetics (see later). These stages occur between 0 and 6 h (Stage I), 6 and 21 h (Stage II) and after 21 h (Stage III). Considering the very initial stage (Stage I), the thickness rises to ≈2.4 nm within 0.5 h and subsequently drops to 1.2 nm over the first 6 h. Over the next 15 h, the AzoSS SAM undergoes a growth in thickness, as might be expected for SAM formation (Stage II). Finally, after 21 h the thickness plateaus at ≈1.8 nm, indicating that the SAM is fully formed (Stage III).
In contrast, for AzoSH there is no Stage I as for AzoSS, and the SAM thickness rises to ≈0.80 nm within 0.5 h and then to ≈1.2 nm by 6 h and reaches a plateau of ≈1.6 nm by 24 h, following standard alkyl thiol growth kinetics.27 This two-stage adsorption behavior of AzoSH is also in agreement with adsorption kinetic studies conducted by Tamada et al.11 on azobenzene-containing alkanethiol SAMs on gold. As a control, and to rule out the initial increase in thickness of the AzoSS surface we considered that a bare Au surface is a relatively high energy surface and is prone to the physisorption of airborne contaminants.27 Thus, we carried experiments without the AzoSS in the solution and measured the ellipsometric thickness after several time intervals over 6 h, which showed the ellipsometric thickness of the contaminant layer to be ≈1.24 nm, irrespective of immersion time. This thickness is lower than the values observed (2.4 nm) within the first 4 h of SAM formation with AzoSS, and hence Stage I in Fig. 3 cannot be attributed to the non-specific physisorption of airborne contaminants.
The θRec angles of the AzoSS reveal similar three stage behavior to the ellipsometric data. The θRec reaches ≈65° within 0.5 h, and then decreases to ≈35° over 1.5 h (Stage I), then increases to ≈70° over the following 4.5 h (Stage II), and remains constant for the next 12 h (Stage III), suggesting the presence of a more sparsely packed monolayer at 24 h, as indicated by a large hysteresis of ∼15° (θAdv–θRec) relative to AzoSH. In contrast the AzoSHθRec angle increases consistently over the SAM formation time, starting at ∼75° and reaching a plateau after 21 h at ∼80°, giving a final smaller hysteresis of 10° (θAdv–θRec), indicating a more ordered and packed monolayer structure than the AzoSS.
Label-free sensing technologies such as SPR and quartz crystal microbalance (QCM) allow for real-time monitoring of adsorption processes and kinetics.28–30 Thus, in addition to the ellipsometry and contact angle measurements, we have carried out SPR analysis in order to monitor the adsorption kinetics of the AzoSS and AzoSH onto the gold surfaces (Fig. 6). The SPR baseline for the clean gold chips was established using ethanol, following which the AzoSS or AzoSH in ethanol (0.0625 mM) was introduced into the SPR flow cell at the rate of 10 μL min−1 (Fig. 6). Data were collected for 21 h, followed by washing with ethanol. SPR reveals clear differences in the kinetics of monolayer formation for AzoSS and AzoSH SAMs. The AzoSS SAM formation proceeds through three stages. In Stage I (0–2.3 h), an initial adsorption of molecules on the surface that peaks at 1950 response units is followed by a 15% decrease in the SPR signal after 30 minutes. This decrease suggests a significant level of desorption of molecules from the gold surface. After Stage I, the formation kinetics proceeds into two more stages – Stage II and Stage III. Stage II occurs between 2.3 h and 6.6 hours and Stage III from 6.6 hours to the time the rinsing is initiated. Both stages are characterised by an initial increase in the amount of AzoSS on the surface, evidenced by the increase in the SPR response units, that plateaus for Stage II and Stage III after 1.5 h and 3 h, respectively. When ethanol replaces the AzoSS solution, there is a significant drop in the SPR signal to 1980 response units, indicating the removal of a significant amount of non-chemisorbed AzoSS molecules from the surface.
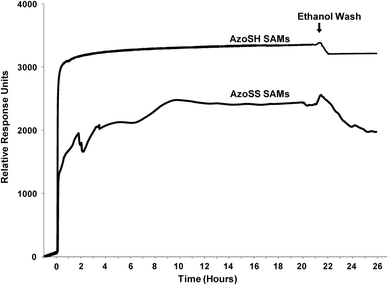 | ||
| Fig. 6 SPR sensorgram traces showing the formation of the AzoSS and AZoSH SAMs in real time for 21 h. Following SAM formation, the surfaces were washed with ethanol. | ||
The AzoSH SAM formation proceeds through a two stage process, involving a rapid initial adsorption of AzoSH on the surface followed by a much slower adsorption that plateaus at ∼3330 response units. After contact with the AzoSH thiol solution is finished, the signal decreases slightly with only a small amount of AzoSH being removed from the surface.
These distinct stages between the kinetics of SAM formation of the AzoSS and AzoSH, as shown in the ellipsometric (Fig. 3), contact angle (Fig. 4) and SPR (Fig. 6) analysis, suggest that the interplay between the dithiolane and the azobenzene moiety interacting with the Au substrate plays an important role in defining the SAM adsorption process for AzoSS, and an inherently different interplay to the thiol and azobenzene in AzoSH, which follows classical alkyl thiol absorption kinetics.27
Thus, it is clear that the adsorption kinetics of the AzoSS and AzoSH SAM formation are significantly different, which may be accounted for by the competing interactions of the azobenzene moiety and either the thiol or the dithiolane for the gold surface. If we consider that the RS–Au bond forms more rapidly from a thiol than it does from a disulfide or dithiolane as previously suggested by Whitesides and co-workers,31 the initial physisorption of the azobenzenes in a lying-down configuration on the gold surface may persist for longer with the AzoSS than the AzoSH. This behaviour allows a multilayer of AzoSS molecules to form by virtue of van der Waals and π–π interactions between surface physisorbed AzoSS and those approaching the interface. Indeed, the high thickness of AzoSS SAMs at lower formation times (Stage I) supports such a hypothesis when coupled to the rapid decline in thickness, such that at low formation times for AzoSS (Stage I) a transient multilayer adsorption process occurs with the azobenzenes lying-down on the gold surface before the S–S bond can cleave and form the Au–S bond. This explanation is consistent with the literature32–34 on the growth of SAMs in which the SAM formation phases (initial physisorbed, lying-down phase, followed by a chemisorbed, standing-up phase) are determined, among several others parameters, by the structure of the SAM molecule, i.e. headgroup, backbone and endgroup.
A striking difference between the ellipsometry and SPR results is that in the former characterisation, the initial adsorption of AzoSS molecules on the gold surface is much more pronounced and the desorption occurs more slowly. Ellipsometry results demonstrate that the desorption occurs for a period of 5.5 h while SPR desorption takes place for 30 min. The differences obtained by ellipsometry and SPR are believed to be related to the flow-induced shear stress applied during the SPR analysis which is not present during the preparation of the SAM surfaces used for ellipsometry measurements. These findings suggest that flow-induced shear stress may prevent to a certain extent lateral stacking of the AzoSS molecules on the surface. Despite the use of different SAM preparation conditions for both analyses, both ellipsometry and SPR clearly illustrate the differences in kinetics of AzoSS and AzoSH SAM formation.
The UV/Vis spectra of both AzoSS and AzoSH SAMs (Fig. 7) were investigated as a function of time. Post SAM formation samples were washed and dried, then immediately analysed using UV/Vis absorption spectroscopy. The UV/Vis spectra of AzoSS SAMs show the three formation stages (I–III), in accordance with those seen by ellipsometry and contact angle. At low formation times (0.5–6 h, Stage I), λmax (Table 1) is hypsochromically shifted with respect to λmax = 329 nm in an ethanolic solution, which can be interpreted in terms of strong intermolecular interactions in the film leading to formation of H-aggregates. In Stage II (6–12 h of SAM formation), λmax is still hypsochromically shifted with respect to the solution value (329 nm), but the hypsochromic shifts are generally not as large as in Stage I, suggesting that the degree of aggregation is reduced with increasing formation time. In Stage III (24 h of SAM formation), the spectrum revealed an intense π–π* absorption band at ≈329 nm, which is in agreement with the solution value, therefore indicating that azobenzene is not aggregated.
AzoSH UV/Vis spectra are characterized by two absorption bands (Fig. 7), with the absorption at lower wavelengths (273–279 nm, λmax 1 in Table 1) representing H-aggregates that are strongly packed on the surface since they are significantly hypsochromically shifted from the solution value of 360 nm. For lower formation times (0.5–3 h), the absorption at higher wavelengths (λmax 2 in Table 1) is similar to the solution value, indicating that some of the azobenzene molecules are adsorbed on the surface in a non-aggregated state. By increasing the time of SAM formation, λmax 2 increasingly shifted to lower wavelengths, representing the formation of H-aggregates with increased order of the monolayer.35,36
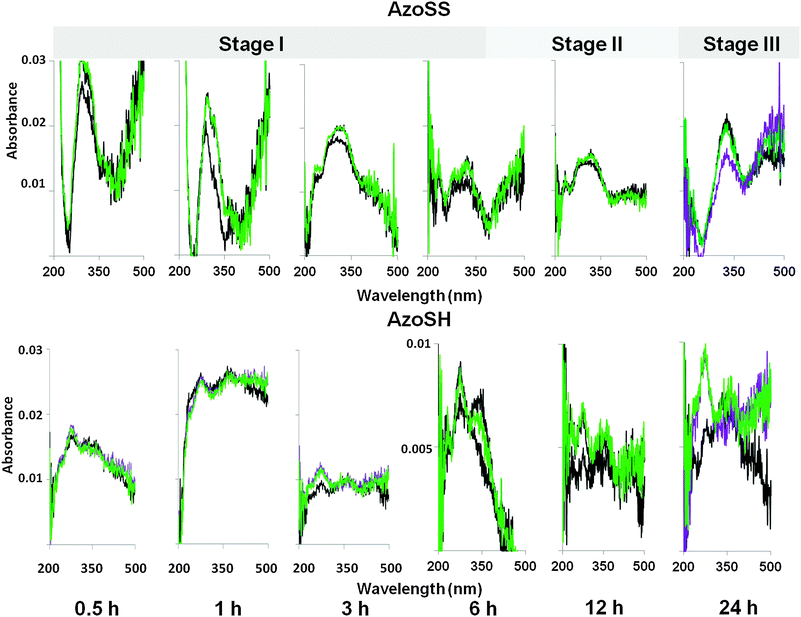 | ||
Fig. 7 Typical UV/Vis spectra of AzoSS and AzoSH SAMs at different formation times (i.e. 0.5 h, 1 h, 3 h, 6 h, 12 h and 24 h) on transparent gold substrates, (——) before and ( ) after irradiation at 360 nm for 3 min to switch the molecules in their cis isomer and ( ) after irradiation at 360 nm for 3 min to switch the molecules in their cis isomer and ( ) subsequent irradiation at 480 nm for 3 min to switch in the trans isomer. ) subsequent irradiation at 480 nm for 3 min to switch in the trans isomer. | ||
| Time (h) | AzoSS | AzoSH | |
|---|---|---|---|
| λ max (nm) | λ max 1 (nm) | λ max 2 (nm) | |
| 0.5 | 294 | 275 | — |
| 1 | 287 | 278 | 361 |
| 3 | 318 | 273 | 360 |
| 6 | 322 | 277 | 349 |
| 12 | 323 | 274 | 346 |
| 24 | 329 | 279 | 344 |
The photoisomerization of both AzoSS and AzoSH SAMs was also investigated as a function of SAM formation time, using the samples discussed above. In order to examine reversible changes in the molecular conformation and the resulting spectroscopic features of the AzoSS and AzoSH SAMs under UV and Vis light irradiation, SAMs were subjected to alternating irradiation for 3 min with UV and Vis light and the UV/Vis absorption spectroscopy employed to follow such a process (Fig. 7). It should be mentioned that for the samples where a photoreaction was achieved (i.e.AzoSS SAMs formed over 24 h), the photostationary equilibrium by UV and Vis irradiation was reached after 2 min.
For AzoSS SAMs, at low formation times (0.5 h and 1 h), the samples show optical activity atypical of azobenzenes. Irradiation with UV light increases absorption, while exposure to Vis light had little further effect, suggesting that irradiation induces aggregation. The samples having formation times between 3 h and 12 h exhibit a small degree of isomerization. In contrast, at 24 h AzoSS SAMs, upon UV light irradiation, a significant reduction in the π–π* absorption band at 329 nm was observed, indicating that azobenzene molecules undergo a conformation change from the trans to the cis form. Subsequent visible light irradiation (436 nm) gave rise to cis-to-trans back isomerization, maximizing the π–π* absorption band at ≈329 nm. Generally, for AzoSH SAMs, exposure to UV results in an increase in the absorption of λmax 1 corresponding to a higher amount of H-aggregates that are strongly packed on the surface. The absorption of λmax 1 remains unchanged upon Vis irradiation. UV and Vis irradiation has a small effect on the absorption of λmax 2 for times of SAM formation between 6 and 12 h. However, by 24 h of SAM formation the degree of isomerization on the AzoSH SAMs is completely suppressed.
Conclusions
We have compared the formation kinetics and photoisomerization properties of azobenzene SAMs comprising either a thiol or a dithiolane headgroup. The azobenzene dithiolane SAMs were found to follow a three-stage assembly process, with ellipsometry, contact angle, SPR and UV/Vis spectroscopy data suggesting that the AzoSS moieties initially adsorb on the surface as multilayer aggregates. The results highlight the critical role of time of SAM formation in dictating H-aggregation and photoisomerization on AzoSS SAMs. AzoSS SAMs are aggregated at low formation times and the degree of aggregation decreases with increasing formation time, until non-aggregated SAMs are formed after 24 h. Azobenzene, within AzoSH SAMs are generally H-aggregated, irrespective of formation time. While SAMs of AzoSH do not exhibit photoisomerization, clear and highly reversible photoinduced effects are observed for AzoSS SAMs.Experimental
Chemicals and materials
Commercially available chemicals and solvents were purchased from Aldrich Chemicals and Fisher Chemicals and were used as received. Thin-layer chromatography (TLC) was carried out on aluminium plates coated with silica gel 60 F254 (Merck 5554). The TLC plates were either air-dried and analysed under a short wave UV lamp (254 nm) or developed in either permanganate solution and heat-dried. Column chromatographic separations were performed using silica gel 120 (ICN Chrom 32–63, 60 Å).Synthesis of AzoSH and AzoSS
Compound characterisation
SAM characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30% H2O2) at room temperature for 10 min, rinsing with Ultra High Pure (UHP) H2O and then HPLC grade EtOH thoroughly for 1 min. (Caution: piranha solution reacts violently with all organic compounds and should be handled with care.) Immediately after cleaning, the substrates were immersed in freshly prepared ethanolic solutions of AzoSS (0.0625 mM) and AzoSH (0.0625 mM) in the trans form.
:30% H2O2) at room temperature for 10 min, rinsing with Ultra High Pure (UHP) H2O and then HPLC grade EtOH thoroughly for 1 min. (Caution: piranha solution reacts violently with all organic compounds and should be handled with care.) Immediately after cleaning, the substrates were immersed in freshly prepared ethanolic solutions of AzoSS (0.0625 mM) and AzoSH (0.0625 mM) in the trans form.
Post-immersion in the SAM forming solution, the substrates were rinsed with HPLC EtOH and dried under a stream of N2.
Acknowledgements
We acknowledge the Leverhulme Trust (F/00094/AW) for financial support. We would like to thank the University of Leeds EPSRC Nanoscience and Nanotechnology Facility (LENNF) for access to the XPS. This research was in part supported through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), supported by Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).References
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512 RSC.
- S. Diegoli, C. A. E. Hamlett, S. J. Leigh, P. M. Mendes and J. A. Preece, Proc. Inst. Mech. Eng., Part G, 2007, 221, 589 CAS.
- C. L. Yeung, P. Iqbal, M. Allan, M. Lashkor, J. A. Preece and P. M. Mendes, Adv. Funct. Mater., 2010, 20, 2657 CrossRef CAS.
- B. H. Northrop, A. B. Braunschweig, P. M. Mendes, W. R. Dichtel and J. F. Stoddart, in Handbook of Nanoscience, Engineering, and Technology, ed. W. A. Goddard, III, D. W. Brenner, S. E. Lyshevski and G. J. Iafrate, CRC Press, Boca Raton, 2nd edn, 2007, vol. 1 Search PubMed.
- F. Ercole, T. P. Davis and R. A. Evans, Polym. Chem., 2010, 1, 37 RSC.
- Y. S. Dou, Y. Hu, S. A. Yuan, W. F. Wu and H. Tang, Mol. Phys., 2009, 107, 181 CrossRef CAS.
- C. M. Stuart, R. R. Frontiera and R. A. Mathies, J. Phys. Chem. A, 2007, 111, 12072 CrossRef CAS.
- A. Shaabani and M. Zahedi, J. Mol. Struct., 2000, 506, 257 CrossRef CAS.
- S. D. Evans, S. R. Johnson, H. Ringsdorf, L. M. Williams and H. Wolf, Langmuir, 1998, 14, 6436 CrossRef CAS.
- W. Freyer, D. Brete, R. Schmidt, C. Gahl, R. Carley and M. Weinelt, J. Photochem. Photobiol., A, 2009, 204, 102 CrossRef CAS.
- K. Tamada, J. Nagasawa, F. Nakanishi, K. Abe, T. Ishida, M. Hara and W. Knoll, Langmuir, 1998, 14, 3264 CrossRef CAS.
- K. Tamada, H. Akiyama and T. X. Wei, Langmuir, 2002, 18, 5239 CrossRef CAS.
- S. Sortino, S. Petralia, S. Conoci and S. Di Bella, J. Mater. Chem., 2004, 14, 811 RSC.
- K. Tamada, H. Akiyama, T. X. Wei and S. A. Kim, Langmuir, 2003, 19, 2306 CrossRef CAS.
- M. N. Han, T. Honda, D. Ishikawa, E. Ito, M. Hara and Y. Norikane, J. Mater. Chem., 2011, 21, 4696 RSC.
- M. Han, D. Ishikawa, T. Honda, E. Ito and M. Hara, Chem. Commun., 2010, 46, 3598 RSC.
- M. Ito, T. X. Wei, P. L. Chen, H. Akiyama, M. Matsumoto, K. Tamada and Y. Yamamoto, J. Mater. Chem., 2005, 15, 478 RSC.
- S. Wagner, F. Leyssner, C. Kordel, S. Zarwell, R. Schmidt, M. Weinelt, K. Ruck-Braun, M. Wolf and P. Tegeder, Phys. Chem. Chem. Phys., 2009, 11, 6242 RSC.
- U. Siemeling, C. Bruhn, F. Bretthauer, M. Borg, F. Trager, F. Vogel, W. Azzam, M. Badin, T. Strunskus and C. Woll, Dalton Trans., 2009, 8593 RSC.
- U. Jung, O. Filinova, S. Kuhn, D. Zargarani, C. Bornholdt, R. Herges and O. Magnussen, Langmuir, 2010, 26, 13913 CrossRef CAS.
- T. Weidner, F. Bretthauer, N. Ballav, H. Motschmann, H. Orendi, C. Bruhn, U. Siemeling and M. Zharnikov, Langmuir, 2008, 24, 11691 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533 CrossRef CAS.
- H. L. Zhang, J. Zhang, H. Y. Li, Z. F. Liu and H. L. Li, Mater. Sci. Eng., C: Biomimetic Supramol. Syst., 1999, 8–9, 179 CrossRef.
- K. J. Lee, F. Pan, G. T. Carroll, N. J. Turro and J. T. Koberstein, Langmuir, 2004, 20, 1812 CrossRef CAS.
- P. Iqbal, K. Critchley, J. Bowen, D. Attwood, D. Tunnicliffe, S. D. Evans and J. A. Preece, J. Mater. Chem., 2007, 17, 5097 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp, Eden Prairie, MN, USA, 1992 Search PubMed.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321 CrossRef CAS.
- A. Pranzetti, S. Mieszkin, P. Iqbal, F. J. Rawson, M. E. Callow, J. A. Callow, P. Koelsch, J. A. Preece and P. M. Mendes, Adv. Mater., 2013, 25, 2181 CrossRef CAS.
- A. Pranzetti, S. Salauen, S. Mieszkin, M. E. Callow, J. A. Callow, J. A. Preece and P. M. Mendes, Adv. Funct. Mater., 2012, 22, 3672 CrossRef CAS.
- N. J. Cho, C. W. Frank, B. Kasemo and F. Hook, Nat. Protocols, 2010, 5, 1096 CAS.
- C. D. Bain, H. A. Biebuyck and G. M. Whitesides, Langmuir, 1989, 5, 723 CrossRef CAS.
- Y. Ahn, J. K. Saha, G. C. Schatz and J. Jang, J. Phys. Chem. C, 2011, 115, 10668 CAS.
- F. Schreiber, J. Phys.: Condens. Matter, 2004, 16, R881 CrossRef CAS.
- H. Kondoh, C. Kodama, H. Sumida and H. Nozoye, J. Chem. Phys., 1999, 111, 1175 CrossRef CAS.
- J. M. Kuiper and J. Engberts, Langmuir, 2004, 20, 1152 CrossRef CAS.
- T. Sato, Y. Ozaki and K. Iriyama, Langmuir, 1994, 10, 2363 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Owner Societies 2013 |