 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sorption enhanced CO2 methanation
Andreas Borgschulte*a, Noris Gallandata, Benjamin Probsta, Riccardo Sutera, Elsa Callinia, Davide Ferrib, Yadira Arroyoc, Rolf Ernic, Hans Geerlingsd and Andreas Züttela
aLaboratory for Hydrogen & Energy, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland. E-mail: Andreas.borgschulte@empa.ch
bLaboratory for Solid State Chemistry and Catalysis, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
cElectron Microscopy Center, Empa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
dChemical Engineering, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands
First published on 15th April 2013
Abstract
The transformation from the fatuous consumption of fossil energy towards a sustainable energy circle is most easily marketable by not changing the underlying energy carrier but generating it from renewable energy. Hydrocarbons can be principally produced from renewable hydrogen and carbon dioxide collected by biomass. However, research is needed to increase the energetic and economic efficiency of the process. We demonstrate the enhancement of CO2 methanation by sorption enhanced catalysis. The preparation and catalytic activity of sorption catalysts based on Ni particles in zeolites is reported. The functioning of the sorption catalysis is discussed together with the determination of the reaction mechanism, providing implications for new ways in catalysis.
Introduction
The negative consequences of our actual energy economy (scarcity of fuels, climate change etc.) originate from an open cycle of materials carrying the energy, e.g. mining of crude oil and release of CO2 into the atmosphere. It is, however, possible to close the cycle of materials for energy harvesting, transport, storage and utilization by the production of renewable chemical energy carriers.1 The natural cycles in the atmosphere are able to transport only a few compounds, namely oxygen, nitrogen, water and carbon dioxide. Therefore, the combustion product of any kind of renewable fuel may only be water, carbon dioxide or nitrogen. Mimicking the natural carbon cycle has the potential of closing the energy circle by producing synthetic hydrocarbons. Their combustion products are water and CO2, which hitherto are starting compounds for the renewable synthesis of the desired high energy density fuels. The required energy might be directly introduced during synthesis, or originate from hydrogen, which can be produced from renewable energy via water electrolysis or directly from photoelectrolysis.2 Possible synthetic fuels are methane, methanol, dimethyl ether, and liquid hydrocarbons (CnH2n+2 with n > 6).3 The main advantage of methanol from CO2 and H2 is the rather simple and nearly energy-neutral synthesis process. However, its gravimetric energy density is half of that of methane and octane. For transportation, octane is the ultimate fuel – being liquid and comprising an energy density as high as 44 MJ kg−1 (13 kWh kg−1). Its disadvantage is the intricate synthesis process – usually via reversed water gas shift reaction combined with Fischer–Tropsch synthesis.4 Thus, from a practical point of view, methane is the easiest to synthesize chemical energy carrier with the potential of using the fully developed natural gas infrastructure but replacing the natural gas by environmentally friendly, renewable methane.Recently, this concept has been realized by replacing fossil natural gas with biogas,5i.e. methane produced from biological feedstock, using natural photosynthesis. Before feeding the natural gas grid, the raw biogas has to be purified.6 The CO2 separation process is particularly energy consuming (up to 10% of the heating value of the biogas).7 A further obstacle for biogas as a renewable energy carrier is the limited biologic feedstock.8 An improvement of the process has been proposed: the biogas upgrade9 converts the CO2 in the raw biogas to methane using hydrogen from renewable energy sources. The fundamental of the biogas upgrade process is the Sabatier reaction:
| 4H2 + CO2 → H2O + CO + 3H2 → 2 H2O + CH4. ΔH = −165 kJ mol−1 | (1) |
The heat release of approximately 17% of the heating value has to be considered the main efficiency loss of the Sabatier process. The strongly exothermic heat of formation allows theoretically a thermodynamically high reaction yield, in particular at low temperatures. However, this is not found practically, because the Sabatier reaction is a complex surface reaction, and is kinetically limited. This kinetic barrier can be overcome using catalysts. Commercial catalysts reach a conversion of more than 90%, however only above 250 °C.10 Besides a high activity, the catalyst should exhibit high selectivity for methane. However, toxic carbon monoxide is also formed at high temperatures. A further improvement of the catalytic activity is thus difficult and requires new concepts.
In this paper we demonstrate the utilization of the Le Chatelier principle to improve the reaction kinetics and yield by local absorption of water at the reaction centres.11,12 The so-called sorption enhanced catalysis implies that the reaction conditions can be controlled to modify directly the concentration of reactants and products at the reaction centres and thereby increase the reaction yield. The idea is depicted in Fig. 1: the kinetics of a normal catalyst depend on the adsorption and desorption of reactants and products, respectively. This is mainly determined by gas–solid thermodynamics, i.e. the binding strengths of the species to the surface.13 Using a catalyst that has a sorption function, we are able to tailor coverage of the reactions sites by removing water through a sorbent. The sorbent has a high affinity to water (see energy diagram in Fig. 1), and thereby actively removes the product from the reaction centres. The idea is experimentally realized by Ni particles on zeolites. We demonstrate the proof-of-principle of the water sorption enhanced CO2 methanation reaction, and shed light on the reaction mechanism by kinetic modelling and in situ spectroscopy.
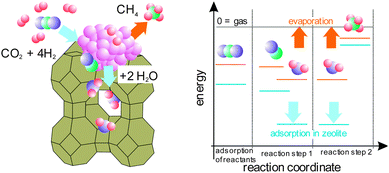 | ||
| Fig. 1 Left: structure model of a sorption catalyst based on Ni particles on zeolites; right: sketch of the free energy of reactants and products of the Sabatier reaction on the conventional metal catalyst (orange) and a sorption catalyst (blue), respectively, depicting the water-absorption-enhanced methanation of CO2 due to an additional driving force (exothermic water adsorption) both for reaction step 1 and 2. | ||
Materials and methods
Nickel nitrate (Ni(NO3)2·6H2O) (Sigma Aldrich) and molecular sieves (zeolite 5A) with pore sizes of 5 Å (pellets, 1.6 mm, Sigma-Aldrich) with linear formulae CanNa12−2n[(AlO2)12(SiO2)12]·xH2O, were used in the preparation of nickel sorption catalysts. The alkaline/earth alkaline zeolite was exchanged using nickel nitrate aqueous solution with different concentrations at room temperature for 24 h. Then, the zeolite was washed with deionized water and dried under an air stream at 100 °C for two days. Subsequently, the sample was reduced in a hydrogen flow at 650 °C for two hours. For comparison, a commercial Ni catalyst (Ni on silica/alumina, Sigma Aldrich) has been investigated.The measurement of the catalytic activity was performed using a stainless steel tubular flow reactor with a length of 450 mm and a diameter of 18 mm. The typical catalyst mass was 13 g (total) with a volume of 25 ml. The gas flows were controlled by thermal mass flow meters from MKS Instruments connected to a Labview interface. Typical flow rates were 50 ml min−1 CO2, and 400 ml min−1 H2 corresponding to a space velocity of 1000 h−1. The pressure was 1.2 bar. The exhaust gases were analysed by a Fourier transform infrared (FT-IR) spectrometer (Bruker Alpha equipped with an 8 cm gas cell). The exhaust gas was diluted with N2 to avoid saturation of the infrared spectrum and the condensation of water. Two measurement procedures were applied: for a general characterization of catalyst the temperature dependence of the conversion at constant reactants’ flows was monitored. For transient measurements at isothermal conditions, the catalyst was dried in hydrogen at high temperature. After equilibration to the desired measurement temperature, the CO2 flow was switched on.
The catalysts were characterized for their specific surface area (BET method) and Ni content, metal area and crystallite size. The BET surface area of the sample was extracted from nitrogen adsorption isotherms at 77 K measured with a BELSORPmax (BEL, Japan).
For microstructural characterization at the μm-scale, secondary electron SEM imaging was carried out with an FEI ESEM XL30 at an acceleration voltage of 20 kV. The SEM is equipped with an energy-dispersive X-ray spectroscopy (EDX) for elemental analysis. The structure at the nano-scale was examined by scanning transmission electron microscopy (STEM) with a JEOL 2200FS TEM.
Diffusive reflectance IR Fourier transform spectroscopy (DRIFTS) combined with mass spectrometry has been used to probe the surface species (reactants, intermediates and products) during reaction. The DRIFT spectra were collected using a Vertex 70 infrared spectrometer (Bruker Optics) equipped with a DRIFT unit (Praying Mantis, Harrick) and liquid nitrogen cooled MCT detector. The commercial Harrick cell (HVC-DRP-3) was attached to a gas manifold system. Gas exiting the cell was analysed online using a mass spectrometer (Pfeiffer, Omnistar).
Results
Catalyst characterization
There is a maximum load of Ni in zeolites, if prepared by ion exchange from 5 M Ni nitrate hexahydrate, corresponding to approximately 2.5 at% (6 wt%). Such catalysts show the best performance. Mapping the Ni-content over a cut through a catalyst pellet indicates a very homogenous distribution of Ni within the pellet. The Ni-ions partly exchange Ca/Na ions in the zeolite. With reduction and formation of Ni particles, the corresponding sites are re-occupied by hydrogen. These acidic sites may have an influence on the catalytic properties of the Ni–zeolite catalyst.14 Indeed, repeating the infiltration procedure twice leads to catalysts with diminished performance – stressing the importance of the surface structure of the zeolite. Further investigations are needed to clarify this effect. In this paper; we restrict the discussion to the system exhibiting the best catalytic performance. The surface area of the zeolite 5A is 440 m2 g−1 and the pore volume is 0.22 cm3 g−1. After ion exchange and calcination, the BET-surface and pore volume are only slightly reduced to 360 m2 g−1 and 0.21 cm3 g−1, respectively. This indicates that the Ni particles do not block the pores, and the overall structure of the zeolite remains unchanged.The SEM pictures display 5–10 μm large zeolite crystals, but do not resolve the fine structure of the catalysts (see Fig. 2). TEM shows Ni particles of the size of about 20 nm with a broad size distribution.
 | ||
| Fig. 2 Scanning electron microscopy image (left) and transmission electron microscopy image (right) on Ni–zeolite 5A. | ||
Performance of catalyst
The standard analysis to reveal catalytic performance is the measurement of the temperature dependence of the methanation rate at a given space velocity. To minimize the influence of systematic errors, such measurements were performed on the sorption catalyst and a commercial Ni catalyst. The latter has similar physical and chemical properties except that it is able to absorb water in large quantities. The measurements as shown in Fig. 3 were conducted after long term equilibration, thereby eliminating the sorption function of the zeolite. Indeed the catalytic properties of the sorption catalyst are only slightly improved, i.e. reaching maximum conversion at slightly lower temperatures and lower CO release than that of the commercial one.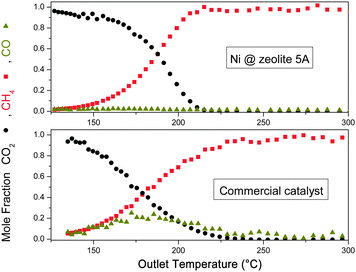 | ||
| Fig. 3 Temperature dependence of the exhaust gas content of Ni@zeolite 5A compared to that of a commercial catalyst Ni@silica/alumina. The amount of Ni is identical in both cases. The space velocity is around 1000 h−1. | ||
The strength of the sorption effect is demonstrated by the transient kinetics starting with a dry sorption catalyst. Fig. 4 shows the time evolution of the gas composition at the reactor outlet. Initially the gas contains 100% methane with only impurities of water and CO2. However, the produced water is adsorbed in the zeolite pores with limited sorption capacity. When the total amount of water exceeds this capacity, water also leaves the reactor. Simultaneously, the concentration and thus the reaction yield of methane decreases, demonstrating the interconnection of reaction yield and water concentration. With decreasing reaction yield, the reactant CO2 and intermediate product CO are released. Thus, despite the need for regeneration, the reactor is a chemical converter with 100% selectivity for a limited time. The difference in reaction yield between the dry and wet state of the sorption catalyst is well explained by the additional driving force originating from the exothermic adsorption of water (compare Fig. 1).
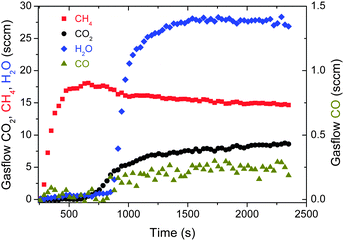 | ||
| Fig. 4 Isothermal measurements of the transient kinetics, T = 170 °C, 20 sccm CO2, 13 g Ni sorption catalyst. CO2 flux starts at t = 200 s. During 500 s (“sorption enhanced reaction”), water and CO are emitted below the detection limit. | ||
Fig. 5 shows the temperature dependence of this effect – the conversion with and without maximum sorption enhancement. The effect is most pronounced at intermediate conversion. At high conversion, the reaction rate is limited by the low CO2-concentration remaining in the process gas, while at low temperatures the intrinsic catalytic activity of the Ni is too low. We substantiate this qualitative explanation using a kinetic model.
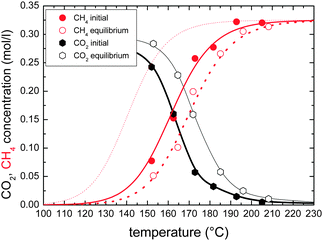 | ||
| Fig. 5 Temperature dependence of the outlet concentrations of CO2 and CH4 – with (“initial”) and without (“equilibrium”) maximum sorption enhancement. 50 sccm CO2, 11 g Ni@zeolite5A. The dots are measurement data, black lines are a guide to the eye. The red lines are fits to the measured CH4 yields using eqn (6), the fine dotted curve is a simulation with B′ = 0. | ||
The methanation kinetics on supported Ni particles is usually described by an empirical Langmuir-type rate equation:15
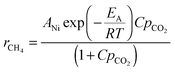 | (2) |
ANi is the constant and EA is the activation energy of the reaction, respectively, while C is the adsorption constant of CO2, and pCO2 the partial pressure of CO2.
The empirical finding is based on a reaction mechanism, which depends only on the number of reaction sites occupied by CO2:
| rCH4 ∝ θCO2 | (3) |
If we include competitive adsorption of two water molecules and CO2:
| rCH4 ∝ θCO2(1 − θH2O) | (4) |
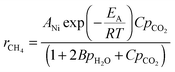 | (5) |
For fitting data, we have to consider that the CO2 partial pressure in the reactor depends on the reaction rate, particularly as the conversion yield reaches nearly 100%.
The corresponding implicit function is estimated by
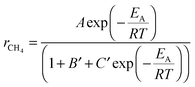 | (6) |
The data in Fig. 5 is fitted by this eqn (6). The important parameter, which is extracted from the fit, is B′, which describes the effect of occupied sites on the reaction yield. All other parameters (activation energy EA and A, C) are kept constant when fitting the CH4 reaction yield with and without maximum sorption enhancement. The good fit indicates that the temperature behaviour is solely explained by the number of sites occupied by water or CO. Furthermore, a simulation of the function with B′ = 0 (i.e. all water respective to CO is instantaneously removed from the reaction centre, Fig. 5) gives the maximum enhancement theoretically possible by the water sorption effect. The measured sorption enhancement does not reach the ideal curve; because the kinetics of mass transport from the reaction centres to the zeolite are limited (see discussion later).
Investigation of the reaction mechanism
The modelling of the performance data by a quantitative model gives clear indication for a mechanism of the sorption based on the removal of adsorbates from the reaction centres. Further details are found by probing the reaction using in situ spectroscopy, more specifically diffusive reflectance IR transform spectroscopy (DRIFTS) combined with mass spectrometry. By utilizing this technique it is possible to identify active adsorbates and follow the surface site occupancy the during reaction. In a typical experiment, after equilibration of the catalyst in an Ar–H2 mixture a DRIFT spectrum is taken, and the reactants are introduced. The first spectrum is considered to be the reference spectrum and is subtracted from all following spectra, hence only showing changes due to evolution of adsorbed species on the surface of the catalyst. The thus measured DRIFT spectrum of Ni on zeolite 5A after long-term exposure to a H2–CO2–Ar-mixture at 693 K (“kinetic equilibrium”) is shown in Fig. 6.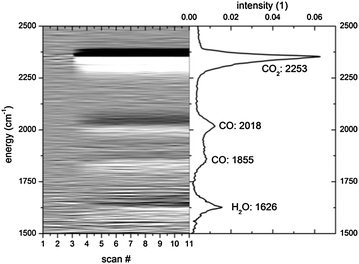 | ||
| Fig. 6 Transient kinetics of methanation on sorption catalysts measured by time-resolved DRIFTS measurements. The right panel shows a DRIFTS spectrum after reaching kinetic equilibrium. The 1st differentials of the measurements are assembled as a grey-scale plot to highlight the changes taking place after exposing the sample to hydrogen and CO2 (after scan #2). | ||
The spectrum shows four main peaks between 1500 and 2500 cm−1. Additional structures are visible at higher and lower energies, which can be attributed to the same species as indicated in Fig. 6: 2353 cm−1 corresponds to the ν3 stretching vibrations of physisorbed CO2 on zeolites;16 2018 cm−1 and 1855 cm−1 (and 1918 cm−1) are vibrations of strongly adsorbed CO on Ni, and the region around 1626 cm−1 is water adsorbed on the zeolite. Surnev et al. studied17 the CO adsorption on Ni(111) by the use of infrared reflection absorption spectroscopy. By a comparison with their study we assign the peak at 2018 cm−1 to linear CO species and the ones at 1855 cm−1 and 1918 cm−1 to two-fold bridged CO species. Additional intermediates/side products have been proposed for the CO2 methanation:18,19 carbonate species (strong band at 1420–1470 cm−1)20 and formate species (giving bands at 2996, 2901, 1592 and 1395 cm−1).21 As none of the bands occurs, these species are either not formed or their concentration is below the detection limit. The observed shift of the ν3 stretching vibrations of physisorbed CO2 corroborates that adsorption of CO2 on zeolites is enhanced when compared to CO2 on Ni as sketched in Fig. 1. The greyscale plot shows the time evolution of the spectra. No new peaks develop, but strong intensity changes are evident.
The intensity is a measure of the coverage of the active sites by the corresponding adsorbents during sorption reaction. The normalized intensities are compared to the normalized CH4 concentration of the product gas in Fig. 7 (“1” corresponds to the equilibrium value). At first glance, the graph already confirms the enhancement of the CO2 methanation by water adsorption: the higher the amount of water adsorbed on the catalysts the lower is the reaction yield. For quantitative analysis, we fit the decay/growths of the data by an exponential growth function of the first order giving a time constant for the change of kinetics. The reciprocal time constant is taken as a measure of the rate constant. With τH2O = −23.4 s and τCH4 = 11.6 s for the methane yield and amount of adsorbed water, respectively, the methanation rate is proportional to (1 − θH2O),2 as assumed in eqn (4).
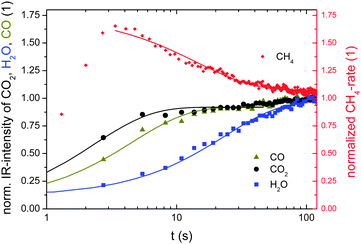 | ||
| Fig. 7 Normalized CH4 rate from mass spectrometry and IR intensities as a measure of the amount of water and CO absorbed in the zeolite and on the Ni particles during transient kinetics. Lines are fit to the data by an exponential function giving characteristic time constants. | ||
If CO produced by the first step (reversed water–gas shift) is not reacting fast enough to eventually form CH4 during the subsequent reaction step(s), CO will poison the reaction centres (⇒θCO ∼ 1); for low coverage (⇒θCO ∼ 0), the situation is reversed:22
| rCH4 ∝ θCO (1 − θCO) ≈ −θCO ∀ θCO → 1, and ∝ θCO ∀ θCO → 0 | (7) |
If τCH4 ∼ −τCO (see Fig. 7), the methanation rate would be proportional to −θCO as proposed for θCO ∼ 1. This is not the case; the CO-coverage on the Ni-catalysts quickly reaches a steady state below full coverage. Still, the coverage increases slowly, i.e. the steady state coverage of CO depends on the methanation yield, which decreases slowly. This means that the removal of water from the reaction centres increases the reaction yield of CH4 and decreases the coverage of CO and thereby minimizes the release of this unwanted side-product.
Apart from their ratio, the absolute number of the time constants is an important piece of information. The enhancement of the reaction vanishes after approximately 40 s. Experiments with different flows show that the time constant scales with the absolute reaction yield (e.g. increasing the reactant flow by a factor of two reduces the time constant by a factor of two as well). This means that the transport of species from the reaction centres to the “storage sites” on the zeolites is fast enough to allow for a reduction of the blocking of the reaction centres. A “steady-state” of both CO and CO2 is reached within seconds, which requires fast diffusion within a zeolite crystallite. Diffusion parameters of the order of 10−11 m2 s−1 and 10−9 m2 s−1 for CO and water23 have been found in zeolites. For diffusion times of one second, diffusion lengths of several micrometers are obtained: the size of the zeolite crystallites (compare Fig. 2), which is also the penetration depth of the IR probe. Thus, the above interpretation of the inverse time constant as a measure of the coverage of the active sites (both reactions centres and storage sites) is well founded. On the other hand, a physical mixture of a commercial Ni catalyst with zeolites does not show a sorption effect. That means that the coupling of reaction and sorption is suboptimal when proceeding via the gas phase. Probably the additional desorption and adsorption steps slow down the effective transport of species. The effect emphasises the need for a nanostructured sorption catalyst.
Conclusions and outlook
We have investigated the sorption effect on the reaction yield of CO2 methanation on Ni particles. The obtained sorption catalyst is readily applicable to be used in biogas upgrade reactors with excellent properties and advantages compared to commercial catalysts:• The catalytic activity for methanation of CO2 using sorption catalysts exceeds that of commercial catalysts.
• The sorption of the products increases the reaction yield up to 100%.
• With proper engineering, a chemical reactor producing pure CH4 from CO2 and hydrogen can be designed.
Water and carbon monoxide are emitted below the detection limit. While the removal of water (process gas drying) is relatively simple after reaction, the separation of CO from methane is technically demanding. In addition, the study reveals interesting findings from a scientific point of view: the spectroscopic evidence of the sorption enhancement confirms the long-postulated reaction mechanism of CO2 methanation on Ni (-particles). Several studies show the existence of CO as a species occurring during the reaction. However, various competitive reaction paths such as the formation of formate species were proposed.24 As it was shown that an increasing amount of water reduces the reaction rate while the CO coverage grows simultaneously, CO is the important intermediate.
Zeolites are chemically active nanostructures,25 which can be tailored to exhibit defined chemical properties such as surface acidity, pores size, etc. There is thus space for improvement for enhancing the sorption properties of the presented sorption catalysts. Furthermore, we did not change the catalytic metal (i.e. Ni). Also, space for improvement exists. Finally, the application of the sorption effect to other chemical reactions is possible.
We demonstrated the design of nano-structured catalysts by growing nano-particles inside a pre-structured support. These catalysts have their analogues in biologic enzymes, which have channels pre-selecting the process molecules (for example the hydrogenase enzyme26). We want to further develop this line of thought: such enzymes are usually implemented in membranes, which separate reactants and products on macroscopic scale. Such a setup may be realized by designing nano-structured membranes made of zeolite membranes.27
Acknowledgements
This work was financially supported by CCEM and Swisselectric research through the HyTech project. The authors are grateful to M. Gorbar for technical support.Notes and references
- A. Züttel, A. Remhof, A. Borgschulte and O. Friedrichs, Hydrogen: the future energy carrier, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 3329–3342 CrossRef
.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS
- G. Centi and S. Perathoner, Opportunities and prospects in the chemical recycling of carbon dioxide to fuels, Catal. Today, 2009, 148, 191–205 CrossRef CAS
- See, e.g., www.fischer-tropsch.org/.
- J. D. Murphy, E. McKeogh and G. Kiely, Technical/economic/environmental analysis of biogas utilization, Appl. Energy, 2004, 77, 407–427 CrossRef CAS
- B. K. Richards, F. G. Herndon, W. J. Jewell, R. J. Cummings and T. E. White, In situ methane enrichment in methanogenic energy crop digesters, Biomass Bioenergy, 1994, 6, 275–282 CrossRef CAS
- M. Persson, Evaluation of upgrading techniques for biogas (http://www.sgc.se/dokument/Evaluation.pdf), 2003; the electric energy consumption is 3–6% of the heating value, in addition there is a methane loss up to 10% Search PubMed.
- A. D. Cuellar and M. E. Webber, Cow power: the energy and emissions benefits of converting manure to biogas, Environ. Res. Lett., 2008, 3, 034002–034010 CrossRef
- M. Specht, et al., Storage of renewable energy in the natural gas grid, Erdoel, Erdgas, Kohle, 2010, 126, 342–346 CAS
- S. K. Hoekman, A. Broch, C. Robbins and R. Purcell, CO2 Recycling by reaction with renewably-generated hydrogen, Int. J. Greenhouse Gas Control, 2010, 4, 44–50 CrossRef CAS
- B. T. Carvill, J. R. Hufton, M. Anand and S. Sircar, Sorption-enhanced reaction process, AIChE J., 1996, 42, 2765–2772 CrossRef CAS
- H. Habazaki, M. Yamasaki, P.-B. Zhang, A. Kawashima, S. Kohno, T. Takai and K. Hashimoto, Co-methanation of carbon monoxide and carbon dioxide on supported nickel and cobalt catalysts prepared from amorphous alloys, Appl. Catal., A, 1998, 172, 131–140 CrossRef CAS
- J. K Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1, 37–46 CrossRef
- C. V. McDaniel and P. K. Maher, in Zeolite Chemistry and Catalysis, ed. J. A. Rabo, ACS Monogr., 1976, p. 171 Search PubMed
- T. van Herwijnen, H. van Doesburg and W. A. de Jong, Kinetics of the methanation of CO and CO2 on a nickel catalyst, J. Catal., 1973, 28, 391–402 CrossRef CAS
- V. B. Kazansky, V. Y. Borovkov, A. I. Serykh and M. Bulow, First observation of the broad-range DRIFT spectra of carbon dioxide adsorbed on NaX zeolite, Phys. Chem. Chem. Phys., 1999, 1, 3701–3702 RSC
- L. Surnev, Z. Xu and J. T. Yates, Jr., IRAS Study of the adsorption of CO on Ni(111) – interrelation between various bonding modes of chemisorbed CO, Surf. Sci., 1988, 201, 1–13 CrossRef CAS
- S. I. Fujita, M. Nakamura, T. Doi and N. Takezawa, Mechanisms of methanation of carbon dioxide and carbon monoxide over nickel/alumina catalysts, Appl. Catal., A, 1993, 104, 87–100 CrossRef CAS
- D. J. Darensbourg, C. G. Bauch and C. Ovalles, Mechanistic aspects of catalytic carbon dioxide methanation, Rev. Inorg. Chem., 1985, 7, 315–339 CrossRef CAS
- L. H. Little, Infrared Spectra of Adsorbed Species, Academic Press, New York, 1966 Search PubMed
- Y. Amenomiya, Active-sites of solid acidic catalysts. 3. Infrared study of the water gas conversion reaction on alumina, J. Catal., 1979, 57, 64–71 CrossRef CAS
- M. Agnelli, H. M. Swaan, C. Marquez-Alvarez, G. A. Martin and C. Mirodatos, CO hydrogenation on a nickel catalyst, J. Catal., 1998, 175, 117–128 CrossRef CAS
- C. Parravano, J. D. Baldeschwieler and M. Boudart, Diffusion of water in zeolites, Science, 1967, 155, 1535–1536 CAS
- E. Vesselli, et al., Hydrogen-assisted transformation of CO2 on nickel: The role of formate and carbon monoxide, J. Phys. Chem. Lett., 2010, 1, 402–406 CrossRef CAS
- B. Smit and T. L. M. Maesen, Towards a molecular understanding of shape selectivity, Nature, 2008, 451, 671–678 CrossRef CAS
- W. Lubitz, E. J. Reijerse and J. Messinger, Solar water-splitting into H2 and O2: design principles of photosystem II and hydrogenases, Energy Environ. Sci., 2008, 1, 15–31 CAS
- F. Gallucci, A. Basile and F. I. Hai, Introduction – A Review of Membrane Reactors, in Membranes for Membrane Reactors: Preparation, Optimization and Selection, ed. A. Basile and F. Gallucci, John Wiley & Sons, 2011 Search PubMed
| This journal is © the Owner Societies 2013 |