 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural specificity of alkoxy radical formation in crystalline carbohydrates†
Siv G.
Aalbergsjø
*a,
Ewald
Pauwels
bcd,
Hendrik
De Cooman
bc,
Eli O.
Hole
a and
Einar
Sagstuen
a
aDepartment of Physics, University of Oslo, P. O. Box 1048 Blindern, N-0316, Oslo, Norway. E-mail: s.g.aalbergsjo@fys.uio.no
bCenter for Molecular Modeling, Ghent University, Technologiepark 903, B-9052 Zwijnaarde, Belgium
cQCMM – alliance Ghent-Brussels, Belgium
dUGent HPC, Ghent University, Krijgslaan 281 S9, B-9000 Gent, Belgium
First published on 22nd April 2013
Abstract
A DFT study of radiation induced alkoxy radical formation in crystalline α-L-rhamnose has been performed to better understand the processes leading to selective radical formation in carbohydrates upon exposure to ionizing radiation at low temperatures. The apparent specificity of radiation damage to carbohydrates is of great interest for understanding radiation damage processes in the ribose backbone of the DNA molecule. Alkoxy radicals are formed by deprotonation from hydroxyl groups in oxidized sugar molecules. In α-L-rhamnose only one alkoxy radical is observed experimentally even though there are four possible sites for alkoxy radical formation. In the present work, the origin of this apparently specific action of radiation damage is investigated by computationally examining all four possible deprotonation reactions from oxygen in the oxidized molecule. All calculations are performed in a periodic approach and include estimates of the energy barriers for the deprotonation reactions using the Nudged Elastic Band (NEB) method. One of the four possible radical sites is ruled out due to the lack of a suitable proton acceptor. For the other three possible sites, the reaction paths and energy profiles from primary cationic radicals to stable, neutral alkoxy radicals are compared. It is found that deprotonation from one site (corresponding to the experimentally observed radical) differs from the others in that the reaction path is less energy demanding. Hence, it is suggested that the alkoxy radical formation is not necessarily site specific, but that the observed radical is formed in much greater abundance than the others due to the different energetics of the processes and reaction products.
1. Introduction
Crystalline carbohydrates are relevant and convenient model systems to examine radical formation induced by ionizing radiation.1–8 Also, as ribose is an integral part of all nucleotides, sugars are frequently used as models for studying the reactions and molecular structures relevant to radiation-induced DNA damage. These processes have attracted great interest, and insight into them is essential to understand radiation damage to DNA and related consequences.9–13 One intriguing aspect is the apparent specificity of radiation damage to the ribose sugar. Although it is commonly considered that each component of DNA is randomly ionized, remarkable selectivities have been observed for primary ribose radical formation. In particular, one of the carbohydrate carbons, C1′, seems to be predominantly targeted, illustrating that the effect of radiation damage is structurally selective.14,15 Understanding which factors direct this specificity is important, and some studies have therefore focused on model nucleoside systems.16 In some crystalline carbohydrates, however, radiation-induced processes take place that display an analogous specificity. A prime example is the α-L-rhamnose single crystal, in which experimental data show that alkoxy radical formation at low temperatures is limited to one site, even though three other similar sites are available.17,18 This ‘simple’ and structurally well-characterized system can therefore yield unique insight into radiation specificity from a more general point of view.The structure of free radicals induced by ionizing radiation may be investigated experimentally using electron paramagnetic resonance (EPR) spectroscopy and related techniques.19 By irradiation and measurement at very low temperatures, primary radicals may be stabilized, thus enabling their characterization. Throughout the last decade, these experiments are increasingly being complemented by molecular modeling. The computational methods have proven to be an important, often essential, tool for understanding and predicting structural and mechanistic aspects of radical reactions.2,4,5,20–22
In irradiated single crystals of α-L-rhamnose (see Fig. 1), several different types of radicals have previously been identified using EPR techniques17,18,23 and molecular modeling studies.22 One of these is an O4-centered alkoxy radical17,18 (see Fig. 1 for the atom numbering scheme), which is a primary radiation product and has only been observed to be stable at temperatures below 77 K. Previous computational investigations24 have provided a likely explanation for how this radical is formed. Following a one-electron oxidation event, a primary cation radical is generated, in which a proton subsequently detaches from the O4 hydroxyl group. This proton travels along an infinite hydrogen bond chain throughout the crystal via succeeding proton shuffles towards a suitable acceptor, leaving behind a stable, neutral O4-centered alkoxy radical. However, as there are four hydroxyl groups in the rhamnose molecule, in principle deprotonation from each of these four sites might occur, giving rise to O1-, O2-, O3- as well as O4-centered radical species. Yet, only the last alkoxy radical has been experimentally observed and characterized. The origin of this apparently specific radiation damage is so far unknown. In the present work, this question is addressed in detail by computationally examining all four hydroxyl deprotonation reactions of the α-L-rhamnose cation radical.
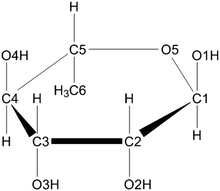 | ||
| Fig. 1 The chemical structure of α-L-rhamnose. | ||
2. Model system and computational details
2.1 The rhamnose crystal structure
α-L-Rhamnose in the solid state is a hexose carbohydrate and its chemical structure is shown in Fig. 1. The crystallographic unit cell is monoclinic with cell parameters a = 7.901 Å, b = 7.922 Å, c = 6.670 Å and β = 95.52°.25 The unit cell contains two rhamnose molecules and two waters of crystallization. Of particular interest in this work is the hydrogen-bonding scheme in the crystal: two infinite hydrogen bond chains extend throughout the crystal in the b- and c-directions. The chain along the b-direction is composed of alternating rhamnose and crystal water hydroxyl groups, in an infinite [O4–H → Ow–H →]∞ pattern. The one along the c-direction is a bit more intricate and alternates between the hydroxyls of two rhamnose molecules and crystal water [O1–H → O2–H → Ow–H →]∞. Every water molecule in the crystal is at the intersection between these two infinite hydrogen bond chains: one proton is involved in the chain along the b-direction, the other water proton in the chain along the c-direction. These chains are illustrated in Fig. 2.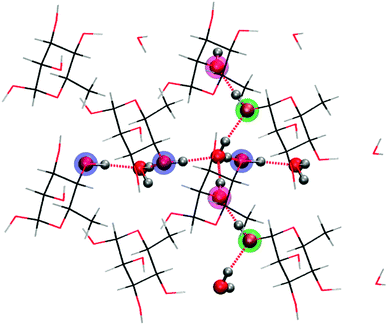 | ||
| Fig. 2 Four co-planar unit cells from the crystal lattice for rhamnose depicting the two infinite hydrogen bond chains along the crystal axes b (horizontal) and c (vertical). The highlighted atoms are oxygen atoms on rhamnose taking part in the hydrogen bond chains. | ||
Each water molecule is involved in four hydrogen bonds. Along the c-direction, the donating H-bond distance Ow–H → O1 is 1.98 Å and the accepting distance HO2 → Ow is 1.81 Å. The atoms in the proton donating reaction Ow–H⋯O1 are far from collinear but make an angle of 160.3°. Along the b-direction, on the other hand, the donating hydrogen bond distance is much smaller and the proton donation angle is more linear. The Ow–H → O4 distance is only 1.77 Å, and the accepting HO4 → Ow distance is 1.82 Å. The Ow–H⋯O4 atoms make an angle of 175.2°. The angles of the accepting reactions are 171.8° and 173.2° in the b- and c-directions respectively.
2.2 Computational procedures
All calculations were performed in a periodic approach using the CP2K software package26 on a singly oxidized 〈2a2b2c〉 supercell (net charge +1e), obtained by doubling the original rhamnose unit cell in all directions. This ensured that the radical was well separated from its periodic images, and that a sufficient amount of molecules involved in the hydrogen bond chains were included. Structural models for the four possible alkoxy radicals were then obtained by restrained geometry optimization, enforcing a proton transfer from the hydroxyl oxygen to its hydrogen bound partner. This was followed by unrestrained optimization. Each time, the Gaussian and Plane Waves (GPW) dual basis set method27 was used, employing a TZVP Gaussian basis set28 and plane waves (280 Ry density cut-off) with GTH pseudo potentials.29–31 The BLYP functional32,33 was chosen.Estimates of the energy barriers for the deprotonation reactions were obtained using the Nudged Elastic Band (NEB) method.34–36 In this approach, the minimal energy path between two stable molecular conformations is sought by optimizing the energy of a set of intermediate replica that represent the gradual transition between both end points and that are successively connected by harmonic springs. Here, we employed 8 replicas, with the reference optimized supercell structure and the results of the unrestrained optimizations as starting and end points, respectively. The same level of theory was adopted as in the geometry optimizations except that a DZVP-GTH basis set was used to ease the computational cost.
EPR properties were calculated for all optimized alkoxy radical geometries using the BLYP functional. The same 〈2a2b2c〉 supercell was employed under periodic boundary conditions, but the all-electron Gaussian and Augmented Plane Wave (GAPW) dual basis set scheme variant37,38 was selected, in order to explicitly describe core electrons. All atoms were treated in this GAPW scheme to calculate the hyperfine coupling tensors. A TZVP all-electron basis set39 and 250 Ry plane-wave cutoff were chosen. For the g-tensor calculations, a hybrid GAPW/GPW approach was used to lift the computational burden as much as possible: only the atoms of the radical were considered at the all-electron level, whereas all other atoms in the supercell were treated using the GPW method. The plane wave cutoff for the electron density was set to 400 Ry and all atoms in the GPW scheme were described using a DZVP basis set and GTH pseudo potentials.29–31 The all-electron DZVP basis set39 was used to describe the atoms of the radical.
3. Results and discussion
3.1 Structure of the alkoxy radicals
Deprotonation of the hydroxyl groups in the ionized rhamnose molecule yields stable alkoxy radical structures, except for O3. The O3 hydroxyl group is hydrogen bound to the ring oxygen (O5) of a neighboring rhamnose molecule and proton transfer is not stable. The global conformations of the other alkoxy radicals differ only marginally from the pristine structure, apart from the hydrogen bonding pattern in the vicinities of the radical centers which has changed significantly. The formation of these three radicals proceeds each time via two subsequent proton transfers towards other rhamnose molecule(s) and/or crystal water. Fig. 3 illustrates the results of these three transfer routes.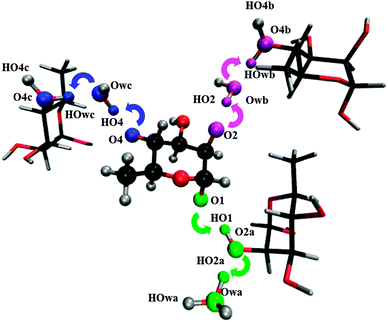 | ||
| Fig. 3 Illustration of the three possible deprotonation pathways in rhamnose single crystals. In all three cases, the (final) proton shuffled structures are shown. | ||
Deprotonation from O1 occurs via the infinite hydrogen bond chain along the c-direction. The HO1 hydroxyl proton is transferred to the O2 oxygen of a neighboring rhamnose molecule which, in turn, donates its excess HO2 proton to crystal water. There, the charge remains localized, in the H3O+ hydronium form.
Deprotonation from O2 initially also proceeds via the infinite hydrogen bond chain along the c-direction. The HO2 proton is transferred to crystal water, but then the proton transfer changes direction, which is possible because each water molecule is at the intersection between the b- and c-direction hydrogen bond chains (see also Fig. 2). The subsequent proton transfer proceeds towards the O4 oxygen of the next rhamnose molecule in the b-direction, generating an –OH2+ oxonium species.
Deprotonation from O4 proceeds entirely along the b-direction hydrogen bond chain. The HO4 hydroxyl proton is first transferred to crystal water, which then protonates the next rhamnose molecule at the O4 site. Again, charge is finally localized on an –OH2+ oxonium species.
EPR properties, g-tensors and hyper fine coupling tensors, were calculated for all three stable alkoxy radicals. These results correspond well with previous studies. In particular, only the g-tensor of the O4 centered radical coincides with the available experimental data, thus confirming that it is not the O1- or O2-centered species that are observed in experiment. Details and discussions of the calculated EPR properties are found in the ESI.†
3.2 Deprotonation energy paths
The energy profiles for O1, O2 and O4 deprotonation calculated using the NEB method are shown in Fig. 4. The variation in the distances between the oxygens and protons involved throughout the described minimum energy paths is given in Fig. 5; they effectively make up the reaction coordinate of the energy path.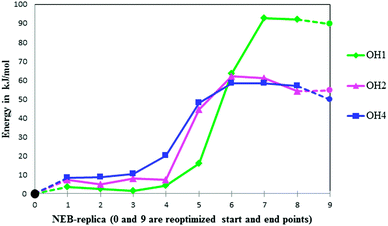 | ||
| Fig. 4 Energy profiles for deprotonation reactions from O1, O2 and O4. Optimized points are indicated by circles, NEB-points are indicated by diamonds, triangles and squares. Each NEB replica refers to a geometry along the reaction path (compare to Fig. 5). | ||
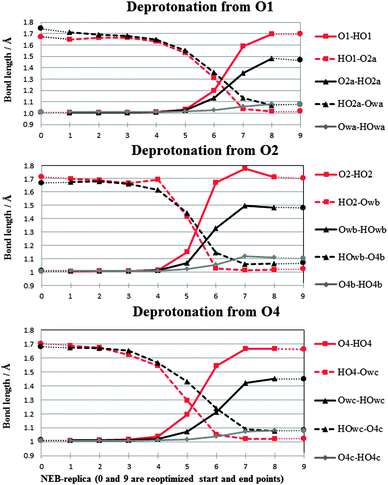 | ||
| Fig. 5 Decomposition of the NEB reaction coordinate in terms of O–H bond lengths. Optimized points are indicated by circles, NEB-points are indicated by diamonds, triangles and squares. Intersection of two lines associated with the same symbol indicates proton transfer between two oxygen atoms. | ||
The reaction energy profiles in Fig. 4 show that deprotonation reactions from all hydroxyl groups are endothermic, with energy barriers of 60–90 kJ mol−1 (approximated as the difference between the minimal and maximal energies encountered along the NEB profiles). This corresponds to less than 1 eV. In the experimental setup the alkoxy radical is observed after γ- and keV X-ray irradiation at low temperatures, which means that the energy available from the primary photon interactions is of the order of keV per photon. This energy is deposited in the crystal as kinetic energy by cascading events of ejecting electrons, or by electronically and/or vibrationally exciting molecules. Such events leave behind enough energy at the oxidation site for the deprotonation reactions to occur. Exactly how this energy is distributed and converted is however beyond the scope of this study.
The deprotonation energy barrier is substantially greater for the O1 route than for the other two processes. In turn, the energy barrier for the O4 route is slightly lower than that for O2. Reoptimization of the conformations at the end points of each NEB profile, followed by energy calculation, yields information on the thermodynamics: the alkoxy radical resulting from deprotonation at O4 has a lower absolute energy than the alkoxy radicals formed through the other routes.
Even though the energy profiles for deprotonation from O2 and O4 appear to be quite similar, simple Boltzmann statistics reveals that at the low temperatures of 4 and 77 K, the feasibility of radical formation is quite different. Even these small energy differences result in a population difference that will render the O2 product in such small amounts that it will be impossible to observe relative to the O4 product. This is found irrespective of whether kinetic control (regulated by the reaction barrier) or thermodynamic control (regulated by the energy of the end product) is assumed. This presents an explanation for the observed selective action of radiation damage to crystalline rhamnose.
A closer inspection of the reaction coordinate in Fig. 5 indicates that in each deprotonation process two proton shuffles occur, one swiftly following after the other. This suggests that the two proton transfers in each reaction occur concomitantly, and that the first transfer is largely determining each process. Previous investigations of proton transfer in rhamnose24 found that the number of proton shuffles along the infinite hydrogen bond chain in the b-direction occurring in a computational simulation is determined by the size of the periodic supercell used in the calculations. This led Pauwels et al.24 to suggest that in a physical system not being bound by computational (periodic) restraints, any number of proton shuffles along this infinite hydrogen bond chain may occur at a very low energetic cost. In this way, the positive charge can migrate away from the initial site of radiation damage, possibly until another irregularity (e.g. radiation damaged site) is encountered. Although we did not explicitly extend our supercell in a similar fashion in the present study due to computational restrictions, the results at hand indicate that only the infinite hydrogen bond chain along the b-direction, and not the one along the c-direction, acts as the proton conduit proposed by Pauwels et al.24 Both the O1 and O2 deprotonation processes initially follow the hydrogen bond chain in the c-direction of the crystal. In the case of O1, deprotonation ends with the excess proton stabilized on a water molecule at the intersection between the two infinite hydrogen bond chains in the crystal. In the case of O2, the proton transfer process changes direction upon charge migration to this water at the intersection and continues along the b-direction chain after this point.
A possible explanation for the inefficiency of the c-direction hydrogen bond chain as a proton conduit is found in the crystal structure itself, by considering the hydrogen bond distances with the crystal water molecule at the intersection between both chains (see also Section 2). Although the proton accepting distances for the water are similar in b- and c-directions, the hydrogen bond distance for the proton donation along the c-direction is 0.2 Å longer than in the b-direction. In addition the angle of the proton donation along the c-direction is 160.3° as opposed to 175.2° in the b-direction. The evidence then suggests that the c-direction hydrogen bond chain is less effective for proton transfer through the crystal than the b-direction chain.
4. Conclusions
The question of the selective formation of the O4-centered alkoxy radical in α-L-rhamnose over the other possible hydroxyl sites, O1, O2 or O3, has been examined by explicit calculation of the oxidative deprotonation energy profiles along each reaction path. Calculations of EPR parameters of the end products of these reactions and comparison with experimental data found in the literature confirm that it is the O4-centered alkoxy radical which is observed. The reaction paths and energy profiles of the different deprotonation routes are compared, and it is found that the deprotonation from O4 differs from the others in that it is less energy demanding and takes place along the infinite hydrogen bond chain in the b-direction of the crystal. It is suggested that the protons in a physical crystal will migrate via the b-direction hydrogen bond chain until some interruption in the crystal and/or chemical structure is met. This hydrogen bond chain appears to be a more efficient proton transfer mediator by way of shorter and more linear hydrogen bonds as compared to the c-direction chain. This renders the product of the O4 deprotonation process far more abundant than the other possible alkoxy radicals, and experimental observation of the other alkoxy radicals is not expected.Acknowledgements
This work is supported by the Fund for Scientific Research – Flanders (FWO) and the Research Board of the Ghent University. Computational resources (Stevin Supercomputer Infrastructure) and services were provided by Ghent University, the Hercules Foundation and the Flemish Government – department EWI. Part of the work was performed on the Titan cluster owned by the University of Oslo and the Norwegian metacenter for High Performance Computing (NOTUR). A grant for computing resources at NOTUR from the Research Council of Norway is acknowledged.References
- A. Adhikary, D. Becker, B. J. Palmer, A. N. Heizer and M. D. Sevilla, J. Phys. Chem. B, 2012, 116, 5900–5906 CrossRef CAS.
- H. De Cooman, E. Pauwels, H. Vrielinck, E. Sagstuen, S. Van Doorslaer, F. Callens and M. Waroquier, Phys. Chem. Chem. Phys., 2009, 11, 1105–1114 RSC.
- H. De Cooman, G. Vanhaelewyn, E. Pauwels, E. Sagstuen, M. Waroquier and F. Callens, J. Phys. Chem. B, 2008, 112, 15045–15053 CrossRef CAS.
- E. Pauwels, H. De Cooman, G. Vanhaelewyn, E. Sagstuen, F. Callens and M. Waroquier, J. Phys. Chem. B, 2008, 112, 15054–15063 CrossRef CAS.
- M. Tarpan, E. Sagstuen, E. Pauwels, H. Vrielinck, M. Waroquier and F. Callens, J. Phys. Chem. A, 2008, 112, 3898–3905 CrossRef CAS.
- M. A. Tarpan, H. De Cooman, E. Sagstuen, M. Waroquier and F. Callens, Phys. Chem. Chem. Phys., 2011, 13, 11294–11302 RSC.
- A. Adhikary, D. Becker, S. Collins, J. Koppen and M. D. Sevilla, Nucleic Acids Res., 2006, 34, 1501–1511 CrossRef CAS.
- D. M. Close, J. Phys. Chem. A, 2008, 112, 8411–8417 CrossRef CAS.
- D. M. Close, in Radiation Induced Molecular Phenomena in Nucleic Acids, ed. M. K. Shukla and J. Leszczynski, 2008, pp. 493–529 Search PubMed.
- D. Becker, A. Adhikary and M. D. Sevilla, in Charged particle and photon interactions with matter: recent advances, applications, and interfaces, ed. Y. Hatano, Y. Katsumura and A. Mozumder, CRC Press, Boca Raton, 2010, pp. 503–541 Search PubMed.
- D. Becker, A. Adhikary and M. D. Sevilla, in Charge Migration in DNA: Perspectives from Physics, Chemistry, and Biology, ed. T. Chakraborty, Springer-Verlag, Berlin, Heidelberg, 2007, pp. 139–175 Search PubMed.
- W. A. Bernhard, in Radical and Radical Ion Reactivity in Nucleic Acid Chemistry, ed. M. D. Greenberg, John Wiley & Sons, New Jersey, 2009, pp. 41–68 Search PubMed.
- W. A. Bernhard and D. M. Close, in Charged particle and photon interactions with matter chemical, physicochemical, and biological consequences with applications, ed. A. Mozumder and Y. Hatano, Marcel Dekker, New York, 2004, pp. 431–470 Search PubMed.
- M. Roginskaya, W. A. Bernhard, R. T. Marion and Y. Razskazovskiy, Radiat. Res., 2005, 163, 85–89 CrossRef CAS.
- M. Roginskaya, Y. Razskazovskiy and W. A. Bernhard, Angew. Chem., Int. Ed., 2005, 44, 6210–6213 CrossRef CAS.
- A. Kumar, V. Pottiboyina and M. D. Sevilla, J. Phys. Chem. B, 2012, 116, 9409–9416 CrossRef CAS.
- P. O. Samskog and A. Lund, Chem. Phys. Lett., 1980, 75, 525–527 CrossRef CAS.
- E. E. Budzinski and H. C. Box, J. Chem. Phys., 1985, 82, 3487–3490 CrossRef CAS.
- E. Sagstuen and E. O. Hole, in Electron Paramagnetic Resonance: A Practitioner's Toolkit, ed. M. Brustolon and G. Giamello, John Wiley & Sons, Inc, Hoboken, New Jersey, 2009 Search PubMed.
- V. G. Malkin, M. Kaupp and M. Bühl, Calculation of NMR and EPR parameters: theory and applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- E. Pauwels, H. De Cooman, M. Waroquier, E. O. Hole and E. Sagstuen, Phys. Chem. Chem. Phys., 2010, 12, 8733–8736 RSC.
- E. Pauwels, V. Van Speybroeck and M. Waroquier, J. Phys. Chem. A, 2006, 110, 6504–6513 CrossRef CAS.
- E. Sagstuen, M. Lindgren and A. Lund, Radiat. Res., 1991, 128, 235–242 CrossRef CAS.
- E. Pauwels, R. Declerck, V. Van Speybroeck and M. Waroquier, Radiat. Res., 2008, 169, 8–18 CrossRef CAS.
- S. Takagi and G. A. Jeffrey, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2551–2555 CrossRef.
- The CP2K developers group, CP2K version 2.3.43, 2011.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–487 CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Elber and M. Karplus, Chem. Phys. Lett., 1987, 139, 375–380 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- M. Krack and M. Parrinello, Phys. Chem. Chem. Phys., 2000, 2, 2105–2112 RSC.
- G. Lippert, J. Hutter and M. Parrinello, Theor. Chem. Acc., 1999, 103, 124–140 CrossRef CAS.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information about calculated EPR properties of the radical structures, along with a comparison of these data with experimental results. See DOI: 10.1039/c3cp50789k |
| This journal is © the Owner Societies 2013 |