Halogen bonding and other σ-hole interactions: a perspective
Peter
Politzer
*ab,
Jane S.
Murray
*ab and
Timothy
Clark
cd
aDepartment of Chemistry, University of New Orleans, New Orleans, LA 70148, USA. E-mail: ppolitze@uno.edu; jsmurray@uno.edu
bCleveTheoComp, 1951 W. 26th Street, Suite 409, Cleveland, OH 44113, USA
cComputer-Chemie-Centrum and Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universität, Erlangen-Nürnberg, Nägelbachstrasse 25, 91052 Erlangen, Germany
dCentre for Molecular Design, University of Portsmouth, King Henry Building, King Henry I Street, Portsmouth PO1 2DY, UK
First published on 19th February 2013
Abstract
A σ-hole bond is a noncovalent interaction between a covalently-bonded atom of Groups IV–VII and a negative site, e.g. a lone pair of a Lewis base or an anion. It involves a region of positive electrostatic potential, labeled a σ-hole, on the extension of one of the covalent bonds to the atom. The σ-hole is due to the anisotropy of the atom's charge distribution. Halogen bonding is a subset of σ-hole interactions. Their features and properties can be fully explained in terms of electrostatics and polarization plus dispersion. The strengths of the interactions generally correlate well with the magnitudes of the positive and negative electrostatic potentials of the σ-hole and the negative site. In certain instances, however, polarizabilities must be taken into account explicitly, as the polarization of the negative site reaches a level that can be viewed as a degree of dative sharing (coordinate covalence). In the gas phase, σ-hole interactions with neutral bases are often thermodynamically unfavorable due to the relatively large entropy loss upon complex formation.
Noncovalent interactions: the σ-hole
It seems appropriate to begin with a recent quote: “with courageous simplification, one might assert that the chemistry of the last century was largely the chemistry of covalent bonding, whereas that of the present century is more likely to be the chemistry of noncovalent binding”.1 For example, the number of papers mentioning “halogen bonding”, the noncovalent interaction R–X⋯B, where X is a halogen and B a negative site, rose from 10 in 2002 to 141 in 2011!2It should not be inferred that halogen bonding was essentially unknown prior to this century. On the contrary, complexes of Cl2, Br2 and I2 with amines were observed already in the 19th century,3,4 and a number of others involving organic halides and oxygen/nitrogen Lewis bases were characterized crystallographically in the mid-20th century.5,6 Some years later, solution studies demonstrated that halogen bonding can be competitive with hydrogen bonding.7,8 Particularly important were the crystallographic surveys by Murray-Rust et al.,9–11 which revealed that covalently-bonded halogen atoms can interact attractively and highly directionally with both nucleophiles and electrophiles (structures 1 and 2).

It is the interaction with a nucleophile that we now call halogen bonding. To our knowledge, this specific term was not used until the work of Dumas et al.,12,13 although Hassel and Rømming referred to “halogen molecule bridges” in crystallographically-observed chains of alternating halogen and 1,4-dioxane molecules,5
Two developments in the 1990's can be viewed as important factors in the subsequent burst of activity relating to halogen bonding:
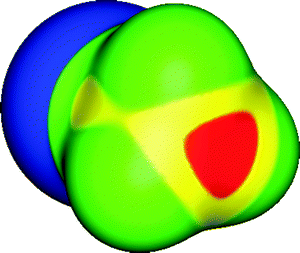
(1) The nature of this interaction had appeared to be somewhat enigmatic: covalently-bonded halogens are viewed as usually being negative in character; why should they interact attractively with negative sites? This was resolved through a surprising finding by Brinck et al.:14,15 Many covalently-bonded halogens have unexpected polar regions of positive electrostatic potential along the extensions of the covalent bonds; the potentials on the equatorial sides of these halogens are usually negative. These features can be seen in Fig. 1 on an iodine in 1,2-diiodoperfluoroethane. (Note that the surfaces of the fluorines are entirely negative.) Thus, as was pointed out,14–16 halogen bonding can be understood as the attraction between the positive outer region and the negative site. The observations of Murray-Rust et al.9–11 (linear interactions with nucleophiles, 1, and lateral interactions with electrophiles, 2) can also be explained. Analogous interpretations of halogen bonding, based upon computed electrostatic potentials, were later reached by Auffinger et al.17 and by Awwadi et al.18 The regions of positive potential are called “σ-holes”;19 they will be discussed in a later section. Since the positive σ-holes are usually accompanied by negative equatorial sides, it is possible to have “like attracting like”, a halogen atom in one molecule interacting through its σ-hole with the negative side of the same halogen in another, perhaps identical molecule.20–22
 | ||
| Fig. 1 Molecular surface electrostatic potential of 1,2-diiodoperfluoroethane (ICF2CF2I), computed on the 0.001 au contour of the electronic density. Color ranges, in kcal mol−1, are: red, greater than 25; yellow, between 15 and 25; green, between 0 and 15; blue, less than 0 (negative). One of the iodines is facing the viewer. The iodine σ-hole is shown in red and is along the extension of a C–I bond; its VS,max is 31 kcal mol−1. The fluorines are entirely negative. Computational level: M06-2X/6-311G(d). | ||
(2) At about the same time as the electrostatic aspect of halogen bonding was being revealed, significant applications were being demonstrated. Imakubo et al.23 and Amico et al.24 showed that halogen bonding can be an effective driving force for the self-assembly of molecules in predictable patterns to produce new materials with desirable properties, i.e. crystal engineering. This early work has evolved into a rapidly growing field, with relevance to magnetics, optics and electronics.25–28
What has also spurred interest in halogen bonding, although somewhat later than the two developments that have been mentioned, was the recognition of its widespread occurrence in biological systems. This was demonstrated by the survey of crystal structures in the Protein Data Bank that was carried out by Auffinger et al.17 The role of halogen bonding in a variety of biological and medicinal areas is now being investigated; among these are protein–ligand interactions, conformational stabilization, drug design, docking processes, etc.17,25,29,30
Accompanying the activity related to halogen bonding during the first years of this century has been the “discovery”, or more accurately rediscovery, that covalently-bonded atoms of Groups IV–VI can also interact noncovalently with negative sites. For Groups V and VI, this is sometimes labeled “pnicogen” and “chalcogen” bonding, respectively. These are on occasion described as “new” interactions, recent discoveries (two of the present authors have also been guilty of this31), overlooking the fact that such noncovalent interactions involving Group IV–VI atoms have been known for decades. For instance:
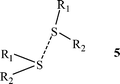
(1) Surveys of close contacts in divalent sulfide crystals revealed patterns analogous to those found for halides; nucleophiles interact along the extension of one of the covalent bonds, and electrophiles above and below the molecular plane:32,33

“Like attracting like” was also observed:33
(2) PF3 was listed as a Lewis acid in a 1991 tabulation,34 indicating the capacity to interact with negative sites B, i.e. F3P⋯B.
(3) S⋯O close contacts were found crystallographically35 and by neutron diffraction.36
(4) P⋯P interactions have been observed through structural analyses,37 as have P⋯N.38
(5) Si⋯N and Ge⋯N attractions have been shown to govern anomalous Si–O–N and Ge–O–N angles.39,40
Many more early examples, dating back as far as 40 years, have been cited by Iwaoka et al.,41 Cozzolino et al.42 and Bleiholder et al.43
Noncovalent interactions by Group IV–VI atoms are accordingly not a recent discovery. What is new, however, is the recognition that – like halogen bonding – they can, to a large extent, be explained in terms of positive σ-holes, i.e. regions of positive electrostatic potential along the extensions of one or more of the covalent bonds to the atom. The presence of positive σ-holes on many Group IV–VI atoms was initially demonstrated by Murray et al.,31,44,45 and was further discussed in several reviews.29,30,46,47Fig. 2–4 illustrate σ-holes on some molecules containing Group IV–VI atoms; these atoms can have as many σ-holes as covalent bonds. It is now evident that halogen bonding is simply a subset of the more general category of σ-hole bonding.
 | ||
| Fig. 2 Molecular surface electrostatic potential of GeH3Br, computed on the 0.001 au contour of the electronic density. The germanium and the hydrogens are in the foreground; the bromine in the rear. Color ranges, in kcal mol−1, are: red, greater than 25; yellow, between 15 and 25; green, between 0 and 15; blue, less than 0 (negative). The σ-hole along the extension of the Br–Ge bond is shown in red; that along the extension of an H–Ge is in yellow. Their VS,max are 39 and 20 kcal mol−1, respectively. Computational level: M06-2X/6-311G(d). | ||
 | ||
| Fig. 3 Molecular surface electrostatic potential of PH2Cl, computed on the 0.001 au contour of the electronic density. The phosphorus and hydrogens are in the foreground; the chlorine is in the rear. Color ranges, in kcal mol−1, are: red, greater than 25; yellow, between 15 and 25; green, between 0 and 15; blue, less than 0 (negative). The σ-hole along the extension of the Cl–P bond is shown in red; its VS,max is 39 kcal mol−1. The σ-hole along the extension of one of the H–P bonds is within the yellow region at left; its VS,max is 20 kcal mol−1. Note the negative region (blue) associated with the lone pair of the phosphorus, toward the top. Computational level: M06-2X/6-311G(d). | ||
 | ||
| Fig. 4 Molecular surface electrostatic potential of SeFCl, computed on the 0.001 au contour of the electronic density. The selenium is in the foreground, the chlorine toward the left rear and the fluorine toward the right rear. Color ranges, in kcal mol−1, are: red, greater than 34; yellow, between 17 and 34; green, between 0 and 17; blue, less than 0 (negative). The two selenium σ-holes are shown in red, and are located along the extensions of the F–Se and Cl–Se bonds; their VS,max are 46 and 40 kcal mol−1, respectively. Note the negative region (blue) associated with one of the lone pairs of the selenium, toward the top. Computational level: M06-2X/6-311G(d). | ||
The positive σ-holes on Groups V and VI atoms are generally found in conjunction with regions of negative electrostatic potential on the same atoms; see Fig. 3 and 4. “Like attracting like” is then possible, just as for halogens; an example is shown in Fig. 5, in which a positive σ-hole on the phosphorus in each PH2Cl molecule interacts with a negative region on the phosphorus in a second PH2Cl. Other Group V and VI like–like complexes have been displayed earlier: H2FAs⋯AsFH2 and Cl2S⋯SCl2,22 Cl2Se⋯SeCl247 and H2FP⋯PFH2.48 With regard to covalently-bonded tetravalent Group IV atoms, our experience is that they do not have negative regions; the σ-holes are simply more positive than their surroundings (Fig. 2). Accordingly, while these atoms do interact attractively with negative sites through their σ-holes,45,46 they do not form like–like complexes.
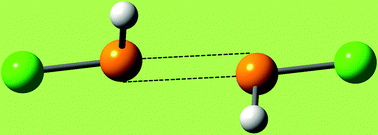 | ||
| Fig. 5 Ball-and-stick model of ClH2P⋯PH2Cl interaction. Phosphorus atoms are displayed in gold, chlorines in green and hydrogens in white. The P⋯P separation is 2.87 Å, the Cl–P⋯P angles are 168°, and the interaction energy ΔE is −4.1 kcal mol−1 (M06-2X/aug-cc-pVTZ). This complex is an example of a double σ-hole interaction, with the σ-hole on the extension of each Cl–P bond interacting with the negative region of the other phosphorus (see Fig. 3). The presence of two simultaneous interactions is the reason for the Cl–P⋯P angles deviating from 180°. | ||
Since 2008, the σ-hole concept is increasingly being invoked to interpret noncovalent interactions of Group IV–VI atoms.49–59 Particularly notable is the study by Mohajeri et al.,53 who characterized 30 complexes between molecules containing Group V and VI atoms and the Lewis bases NH3 and H2O. The importance of S⋯O and Se⋯O interactions in biological systems is well known. The Protein Data Bank shows a large number of S⋯O close contacts that can be understood in terms of sulfur σ-holes.55 An interesting case is that of thiazole and selenazole nucleoside drugs having antitumor activity that depends upon particular conformations being stabilized by intramolecular S⋯O and Se⋯O interactions, which were concluded (in 1992) to be electrostatic in nature60 and in 2008 shown to involve sulfur and selenium σ-holes.61 In the realm of crystal engineering, solid Se(CN)2 has been found to have a layered structure with Se⋯N σ-hole interactions,50 analogous to what had been predicted computationally for S(CN)2.44
Origins and properties of σ-holes
As was pointed out in the last section, many of the noncovalent interactions of Group IV–VII atoms can be explained in terms of electrostatic attractions between positive σ-holes on the Group IV–VII atoms and negative sites. (Polarization and dispersion effects also have significant roles, as will be discussed later.) What is the origin of these σ-holes?A free, ground-state atom has, on the average, a spherically-symmetrical electronic charge distribution.62 The electrostatic potential created around the atom by its nucleus and electrons is positive everywhere,63 the contribution of the nucleus dominating that of the dispersed electrons. When the atom forms a covalent bond, some of its electronic charge is polarized toward the bond region, leading to the atom's electronic density being diminished in its outer region (along the extension of the bond) but increased on its equatorial sides. This anisotropy, which is well known,18,64–69 can be seen in figures presented by Tsirelson et al.67 and by Bilewicz et al.69 The usual result is that a negative electrostatic potential is developed around the sides of the atom, while its outer portion becomes more positive – the σ-hole. (There are some exceptions to this pattern that will be discussed later in this section.)
A convenient way of showing these features is by plotting the molecule's electrostatic potential on its surface, which is commonly taken to be the 0.001 au (electrons per bohr3) contour of its electronic density ρ(r), as suggested by Bader et al.70Fig. 1–4 are examples, showing the positive σ-holes on the extensions of the covalent bonds to the Group IV–VII atoms, most of them with adjacent negative regions.
The electrostatic potential in the space around a molecule can be evaluated rigorously using the formula,
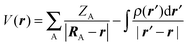 | (1) |
When plotted on a molecular surface, V(r) is designated VS(r) and its local most positive and most negative values (of which there may be several) are identified as the VS,max and VS,min. Thus a σ-hole typically has a VS,max, while the negative regions of the same atom may have VS,min. We normally obtain VS(r) and the VS,max and VS,min with the WFA-Surface Analysis Suite.73
Since σ-holes are due to the polarization of an atom's electronic charge toward the covalent bonds that it forms, any factor that enhances this polarization will strengthen the σ-hole. Accordingly, a σ-hole VS,max tends to be more positive the greater is the atom's polarizability and the less is its electronegativity relative to the remainder of the molecule. These trends can be seen from the representative examples in Table 1. For example, the VS,max of the halogens in the methyl halides increase in the order F < Cl < Br < I, as they become more polarizable and less electronegative. However the greater electron-withdrawing power of the F3C group compared to the H3C makes the VS,max of the halogen X more positive in F3C–X than in H3C–X. When an atom has two or more σ-holes, the one on the extension of the bond to the more electron-attracting atom or group will be the more positive. Thus in Se(CH3)CN, the NC–Se bond produces a σ-hole VS,max of 35 kcal mol−1 on the selenium, compared to 17 kcal mol−1 for the H3C–Se.
| Molecule | Atom | Bond producing VS,max | V S,max | V S,min | Ref. |
|---|---|---|---|---|---|
| a Present work. | |||||
| H3C–F | F | C–F | — | −25 | 74 |
| H3C–Cl | Cl | C–Cl | — | −16 | 74 |
| H3C–Br | Br | C–Br | 6 | −15 | 74 |
| H3C–I | I | C–I | 13 | −13 | 74 |
| F3C–F | F | C–F | — | −3 | 74 |
| F3C–Cl | Cl | C–Cl | 20 | −1 | 74 |
| F3C–Br | Br | C–Br | 25 | −2 | 74 |
| F3C–I | I | C–I | 32 | −2 | 74 |
| F3Si–Cl | Cl | Si–Cl | 12 | — | 74 |
| F3Si–Br | Br | Si–Br | 18 | — | 74 |
| F2 | F | F–F | 14 | −3 | 46 |
| Br2 | Br | Br–Br | 29 | −5 | 46 |
| F–CN | F | C–F | 16 | — | 46 |
| Cl–CN | Cl | C–Cl | 35 | — | 46 |
| C6H5Br | Br | C–Br | 12 | −14 | 75 |
| 1,3,5-C6H3F2Br | Br | C–Br | 18 | −8 | 75 |
| SCl2 | S | Cl–S | 25(2) | −6(2) | 22 |
| Cl | S–Cl | 13 | −6 | 22 | |
| S(CN)2 | S | C–S | 43(2) | — | 44 |
| Se(CN)2 | Se | C–Se | 47(2) | — | 44 |
| Se(CH3)CN | Se | NC–Se | 35 | −7(2) | 76 |
| Se | H3C–Se | 17 | −7(2) | 76 | |
| (H3C)3N | N | C–N | — | −34 | 31 |
| PF3 | P | F–P | 28(3) | — | 31 |
| PH2Cl | P | Cl–P | 37 | −10 | |
| P | H–P | 20 | −10 | ||
| Cl | P–Cl | — | −15 | ||
| (H3C)3As | As | C–As | 8(3) | −25 | 31 |
| AsF3 | As | F–As | 35(3) | — | 31 |
| As(CN)3 | As | C–As | 51(3) | — | 31 |
| SiH4 | Si | H–Si | 13(4) | — | 45 |
| SiCl4 | Si | Cl–Si | 20(4) | — | 45 |
| Cl | Si–Cl | 9 | −2 | 45 | |
| GeCl4 | Ge | Cl–Ge | 21(4) | — | 45 |
| Cl | Ge–Cl | 10 | −2 | 45 | |
As mentioned earlier, tetravalent Group IV atoms always have entirely positive surfaces. This can happen for Groups V–VII as well if the remainder of the molecule is highly electron-withdrawing. Examples are Cl–CN, Se(CN)2 and As(CN)3, in which the chlorine, sulfur and arsenic have no VS,min (Table 1). Nevertheless the σ-holes are still the most positive portions of the surfaces.
The opposite situation arises when a Group V–VII atom is much more electronegative than a bonding partner. Then the covalent bond may be polarized toward the Group V–VII atom sufficiently to more than neutralize the σ-hole; it will now be negative (although still less negative than its surroundings). This is why the fluorine in H3C–F and the nitrogen in (H3C)3N have no VS,max given in Table 1; their VS,max are negative. (See also the fluorines in Fig. 1.) Accordingly they are not expected to participate in σ-hole interactions. For some time it was in fact believed that fluorine does not halogen bond at all. However it is now well established that it can have a positive σ-hole and form halogen bonds when it is linked to a very electron-withdrawing group;77–79 note F2 and F–CN in Table 1.
The problem of atomic charges
It is well known that there is no rigorous basis for assigning charges to atoms in molecules,80 and that the many procedures that have been proposed for doing so are arbitrary and can lead to dramatically varying results. For instance, seven different methods predict the carbon in H3C–NO2 to have charges ranging from −0.478 to +0.564 electron units!81An intrinsic problem with the concept of atomic charge (which is neither a physical observable nor uniquely defined) is that attributing a single positive or negative value to each atom in a molecule implicitly assumes that their charge distributions are spherically-symmetrical. This was pointed out by Price,68 who referred to it as “a travesty of bonding theory”, since it ignores the anisotropies of the electronic densities around atoms in molecules.18,64–69
The fallacy of assigning a single charge to an atom in a molecule is clearly brought out by the numerous demonstrations, cited earlier, that many atoms in molecules have regions of both positive and negative electrostatic potential (e.g.Fig. 1, 3 and 4). Atomic charges cannot account for halogen bonding (since covalently-bonded halogens are usually given negative charges) nor for many other σ-hole interactions, and they cannot explain the close contacts with both electrophiles and nucleophiles found in the crystallographic surveys of Murray-Rust et al.9–11 and Parthasarathy et al.32,33
It was recognized by Auffinger et al.,17 and discussed in detail by Politzer et al.,22 that the force fields used in molecular mechanics and molecular dynamics often involve atomic charges and thus are likely to miss many noncovalent interactions. (For an example, see Dobeš et al.82) Accordingly there have been a number of efforts to design more realistic force fields.83–86
σ-Hole bonding
In Table 2 are listed a selection of noncovalent complexes between molecules containing Group IV–VII atoms and the nitrogen lone pairs of the Lewis bases NH3 and HCN. In each case are presented the σ-hole potential of the Group IV–VII atom, the VS,max, and the computed interaction energy ΔE. The latter is defined as,| ΔE = E(complex) − [E(Group IV–VII molecule) + E(NH3 or HCN)] | (2) |
| Complex | V S,max (kcal mol−1) | ΔE (kcal mol−1) |
|---|---|---|
| a Computational levels: VS,max: M06-2X/6-311G(d); ΔE: M06-2X/aug-cc-pVTZ. b The VS,max are for the Group IV–VII atoms prior to any interaction. | ||
| FF⋯NH3 | 11 | −1.5 |
| F3CCl⋯NH3 | 20 | −2.5 |
| F3CBr⋯NH3 | 25 | −3.7 |
BrC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CBr⋯NH3 CBr⋯NH3 |
30 | −4.2 |
| H2FP⋯NH3 | 39 | −7.2 |
| H2FAs⋯NH3 | 44 | −8.7 |
| FCl⋯NH3 | 45 | −10.3 |
| HFS⋯NH3 | 46 | −8.4 |
| ClF3Si⋯NH3 | 48 | −10.5 |
| HFSe⋯NH3 | 51 | −11.3 |
| F3CCl⋯NCH | 20 | −1.6 |
Br2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CBr2⋯NCH CBr2⋯NCH |
24 | −2.1 |
| BrCCBr⋯NCH |
30 | −2.7 |
| H3FSi⋯NCH | 35 | −4.2 |
| Cl2Se⋯NCH | 36 | −4.0 |
| H2FP⋯NCH | 39 | −4.7 |
| F2S⋯NCH | 40 | −4.5 |
| H3FGe⋯NCH | 43 | −4.9 |
| H2FAs⋯NCH | 44 | −5.7 |
| FBr⋯NCH | 53 | −7.1 |
It has been demonstrated several times, for complexes of halides with a given negative site, that ΔE correlates well with the VS,max of the halogen σ-hole;30,74,75,88 ΔE becomes more negative as VS,max is more positive. In Fig. 6 and 7, the data in Table 2 are used to show (for the first time, as far as we know) that Groups IV–VII can all be included in the same correlation, with NH3 as the negative site in Fig. 6 and HCN in Fig. 7. The squared correlation coefficients R2 are 0.95 and 0.98, respectively. (Note that the VS,max pertain to the Group IV–VII atoms prior to formation of the complex.)
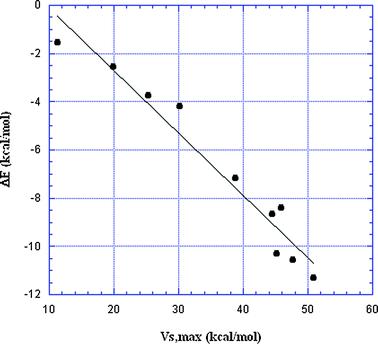 | ||
| Fig. 6 Plot of interaction energies vs. σ-hole VS,max for 10 σ-hole-bonded complexes with NH3 (Table 2). The VS,max are on the Group IV–VII atoms prior to interaction. R2 = 0.95. | ||
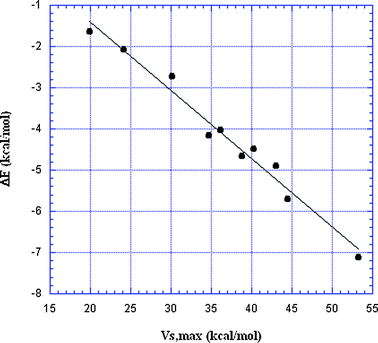 | ||
| Fig. 7 Plot of interaction energies vs. σ-hole VS,max for 10 σ-hole-bonded complexes with HCN (Table 2). The VS,max are on the Group IV–VII atoms prior to interaction. R2 = 0.98. | ||
Can the results for the two different negative sites be combined into one relationship? This would need to explicitly take into account the respective negative potentials VS,min of the two bases, −47 kcal mol−1 for NH3 and −33 kcal mol−1 for HCN, since these clearly help to determine the strengths of the interactions. One option for representing ΔE as a function of both the VS,max of the σ-holes and the VS,min of the Lewis bases is,
| ΔE = α1VS,max + β1VS,min + γ1 | (3) |
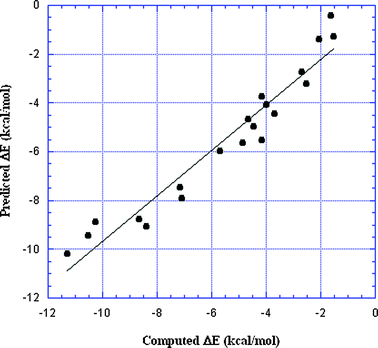 | ||
| Fig. 8 Plot of ΔE predicted by double regression analysis vs. ΔE computed with eqn (2), for the 20 σ-hole-bonded complexes listed in Table 2. R2 = 0.94. | ||
Another possibility, suggested by a reviewer, is eqn (4),
| ΔE = α2[VS,max × VS,min] + β2 | (4) |
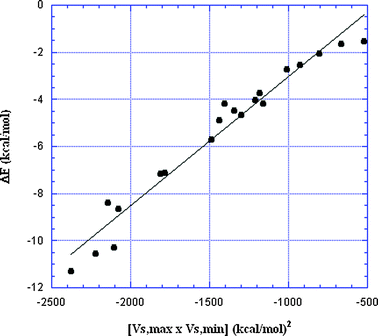 | ||
| Fig. 9 Plot of interaction energies vs. the product of the σ-hole VS,max and the Lewis base VS,min for the 20 σ-hole-bonded complexes listed in Table 2. The VS,max and VS,min are prior to interaction. R2 = 0.96. | ||
Fig. 6–9 are strong indicators of the electrostatically-driven natures of σ-hole interactions and of their fundamental similarity whether the atom having the σ-hole is in Group IV, V, VI or VII. It is all simply σ-hole bonding! It can readily be understood in terms of the electrostatic attraction between the positive σ-hole and a negative site – which might be a lone pair of a Lewis base, π electrons of unsaturated molecules, an anion or a hydride, the negative regions of another covalently-bonded atom, etc. Again, these are all σ-hole bonds.
An intrinsic component of an electrostatic interaction (unless it involves only point charges) is polarization. In σ-hole interactions, the electric fields of the positive σ-hole and the negative site induce some mutual rearrangements of electronic densities.29,89,90 This can be seen graphically in electronic density difference plots, which show that electronic charge on the negative site is polarized toward the σ-hole, while on the σ-hole atom it is polarized away from the negative site.47,91,92 This is exactly what would be expected from the respective electric fields. Dispersion can also have an important role, as was demonstrated by Riley et al.88,93,94 There is further a repulsive component to the interaction, which rapidly increases at short separations. For more extensive discussions of these factors in σ-hole bonding, see Politzer et al.30,47
The electrostatics/polarization plus dispersion interpretation of σ-hole interactions is in the tradition of earlier treatments of noncovalent bonding.68,95,96 For instance, Legon and Millen97 and subsequently Buckingham and Fowler98,99 showed that the angular geometries of hydrogen-bonded complexes can be predicted on the basis of electrostatics, and this was extended to halogen-bonded ones as well.99,100 In particular, electrostatics/polarization explains the linearity that is characteristic of σ-hole bonding: since the σ-hole lies along the extension of the covalent bond to the atom, it follows that the σ-hole interaction will generally be essentially co-linear with that covalent bond. In the gas phase, the angle between them is usually at least 175°.53 Deviations from this can usually be attributed to other simultaneous interactions30,75 (examples of which will be given later); these are particularly prevalent in the solid phase. Electrostatics/polarization also accounts very satisfactorily for other features of the interactions being discussed, such as the close contacts with both nucleophiles and electrophiles that have been observed in crystalline halides9–11 and sulfides.32,33
The “weakness” of the electrostatics/polarization plus dispersion interpretation is that it is distressingly straightforward. There is consequently a tendency among some researchers to attribute major or even dominant roles to additional factors, most notably charge transfer from the negative site to an antibonding molecular orbital involving the atom with the σ-hole. However there is no real physical distinction between charge transfer and polarization. This has been pointed out by a number of different persons.96,101–103 The difference is more semantics and definition than reality. Overlapping an occupied orbital of the negative site with a σ* antibonding orbital involving the atom with the σ-hole is simply a mathematical technique for describing the physical phenomenon, the polarization of the negative site and of the atom with the σ-hole.
A σ-hole interaction is typically accompanied by changes in the length and vibration frequency of the covalent bond that is co-linear with the σ-hole bond, e.g. the F–S bond in F–(H)S⋯NH3. The changes may be an elongation of the bond and a decreased frequency (red shift) or a shortening and a frequency increase (blue shift).104,105 The elongation and red shift suggest weakening of the bond, and have sometimes been used as evidence for the idea of charge transfer into an antibonding orbital. However both red and blue shifts can be predicted, using the formalism of Hermansson,106 from the electric field created by the negative site and the permanent and induced dipole moments of the molecule containing the σ-hole.104,105 No orbitals need be invoked.
Finally, we would like to comment on the practice of decomposing noncovalent interaction energies into contributions from various supposed components. There is no rigorous basis for doing this, and accordingly a variety of arbitrary procedures have been proposed, which focus upon different subsets of a group of suggested components that has included electrostatics, exchange, Pauli exclusion, polarization, charge transfer, dispersion, induction, orbital interactions, electronic correlation, delocalization, deformation, etc. From a physical standpoint, however, these are not separate and independent contributions to an interaction, and the results may not be even qualitatively meaningful. For example, two different decomposition schemes were applied to the complexes H3C–X⋯OCH2 and F3C–X⋯OCH2 (X = Cl, Br and I). One predicted that the main stabilization comes from electrostatics and dispersion,93 while the other attributed it to charge transfer and polarization, with only a slight electrostatic contribution!107
Thermodynamic stability
Studies of σ-hole bonding have generally focused upon the energy or enthalpy change in forming the complex, ΔE or ΔH, as a measure of the strength of the interaction. While this is valid, it does not tell the whole story as far as thermodynamic stability is concerned, which depends upon the free energy change ΔG. At a given temperature T,| ΔG = ΔH − TΔS | (5) |
For the formation of complexes in the gas phase, ΔG is very often positive, despite ΔH being negative.108,109 This is due to the relatively large negative values of TΔS. For weakly- or moderately-bound complexes, TΔS usually dominates over ΔH in eqn (3), resulting in ΔG > 0 and the complex being thermodynamically unstable. Only relatively strong interactions, having large negative ΔH, are likely to be stable.
All of this is evident in Table 3, which gives the computed ΔE°, ΔH°, TΔS° and ΔG° at 298.15 K for a series of complexes involving σ-hole bonding by Group IV–VII atoms. While all of the ΔE° and ΔH° are negative, most of the ΔG° are positive; these complexes would be thermodynamically unstable in the gas phase at 298 K. This should however be kept in perspective; ΔG° > 0 does not totally preclude the interaction. It means that the equilibrium constant for the formation of the complex is less than one.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,b
a,b
| Complex | ΔE° | ΔH° | TΔS° | ΔG° | Ref. |
|---|---|---|---|---|---|
| a Properties computed at 298.15 K. b Computational levels: geometry optimizations and energy minima: M06-2X/aug-cc-pVTZ. Zero-point energies and thermal terms: M06-2X/6-311G(d). c Present work. | |||||
| F–F⋯NH3 | −0.1 | −0.6 | −6.3 | 5.7 | 109 |
| F3C–Cl⋯NCH | −0.1 | −0.7 | −6.4 | 5.6 | |
| Cl–CO–CO–Cl⋯1,4-dioxane | −0.5 | −1.1 | −7.0 | 6.0 | 109 |
| F3C–Cl⋯NH3 | −0.6 | −1.2 | −7.3 | 6.1 | 109 |
| Br2CCBr2⋯NCH |
−0.6 | −1.2 | −6.6 | 5.4 | 109 |
| Br–CC–Br⋯NCH |
−1.2 | −1.8 | −6.8 | 5.1 | 109 |
| F3C–Br⋯NH3 | −1.8 | −2.4 | −7.7 | 5.3 | 109 |
| Cl2Se⋯NCH | −2.4 | −3.0 | −7.9 | 4.9 | |
| FH3Si⋯NCH | −2.5 | −3.1 | −7.7 | 4.6 | |
| Br–CC–Br⋯NH3 |
−2.5 | −3.1 | −6.9 | 3.9 | 109 |
| F2S⋯NCH | −2.9 | −3.5 | −8.0 | 4.6 | |
| FH2P⋯NCH | −3.0 | −3.6 | −7.9 | 4.3 | |
| FH3Ge⋯NCH | −3.2 | −3.8 | −7.8 | 4.0 | |
| F–Br⋯CO | −3.3 | −3.8 | −8.3 | 4.4 | 109 |
| F–Cl⋯OH2 | −3.8 | −4.4 | −8.0 | 3.6 | 109 |
| FH2As⋯NCH | −4.1 | −4.7 | −8.1 | 3.4 | |
| F–Br⋯NCH | −5.5 | −6.1 | −8.4 | 2.3 | 109 |
| FH2As⋯NH3 | −6.3 | −6.9 | −8.6 | 1.6 | |
| (NC)2S⋯NH3 | −6.3 | −6.9 | −9.6 | 2.7 | |
| ClF3Si⋯NH3 | −7.6 | −8.1 | −10.6 | 2.5 | |
| F–Cl⋯NH3 | −8.2 | −8.8 | −8.7 | −0.1 | 109 |
| F–Br⋯NH3 | −12.4 | −13.0 | −8.9 | −4.1 | 109 |
| (NC)2S⋯Cl− | −27.5 | −28.1 | −6.9 | −21.2 | |
The reason for the large negative TΔS° is that the interaction of two molecules to form a complex introduces constraints upon their freedom of motion, with some lost or diminished rotational and translational degrees of freedom.110–112 This causes the total entropy of the system to be significantly reduced. There is some tendency (loosely obeyed) for the decrease in entropy to be greater as the formation of the complex is more exothermic. This is known as “enthalpy/entropy compensation”, and has been extensively analyzed and discussed,1,111–118 with some disagreements. It can be rationalized by arguing that a stronger interaction (more negative ΔH°) is likely to impose more severe constraints upon the complex, i.e. increased rigidity and less disorder, and therefore greater loss of entropy (more negative TΔS°).
What is most important, however, is that TΔS° has a relatively narrow range, just 4 kcal mol−1 in Table 3, which is much smaller than that of ΔH°. This means that TΔS° has an approximate limiting upper magnitude. For weak interactions, |TΔS°| > |ΔH°| (Table 3), and ΔG° > 0. However the dominance of TΔS° diminishes for stronger interactions having more negative ΔH°. Once |ΔH°| > |TΔS°|, then ΔG° < 0 and the complex is thermodynamically stable. This is the case for just two of the systems in Table 3; in one instance, ΔG° ≈ 0.
It is accordingly only the relatively strong gas-phase σ-hole interactions that are thermodynamically stable. In this context, note the very negative ΔH° (and negative ΔG°) for (NC)2S⋯Cl− (Table 3). This and other examples108 show how strong can be the σ-hole bonds to anions. This is because of their very negative electrostatic potentials. For instance, the uniform value of VS(r) over the 0.001 au surface of the chloride anion is −141 kcal mol−1, computed with the M06-2X/6-311G(d) procedure. Compare this with the VS,min of the NH3 and HCN lone pairs, −47 and −33 kcal mol−1, respectively.
So far, we have considered ΔG° only for gas phase systems (for a more extensive discussion, see Politzer and Murray109). In solution or in a solid phase, additional factors come into play that may result in ΔG° < 0 even when the gas phase complex has ΔG° > 0. This is shown by the numerous examples that are known of stable σ-hole interactions in condensed phases, as mentioned or cited earlier. For instance, one of the first halogen-bonded solids to be characterized crystallographically was Cl–C(O)–C(O)–Cl⋯1,4-dioxane;119 yet Table 3 predicts its gas phase ΔG° to be +6.0 kcal mol−1 at 298 K!
Simultaneous interactions
It is important to recognize the possibility that a particular noncovalent interaction may be accompanied by others – such as σ-hole, hydrogen bonding, etc.30,45,49,53,75,78,79,120 In the case of large or condensed-phase systems, this should be viewed as quite likely. It is a major cause of σ-hole interactions deviating from linearity. Thus, in the complex F2Se⋯NH3, the F–Se⋯N angle is 169° rather than 180°, due to the simultaneous H⋯F interaction, 6; the Se⋯N and H⋯F separations are both less than the sums of the respective van der Waals radii.44 See also the caption for Fig. 5.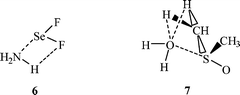
Simultaneous interactions are particularly prevalent in liquid and solid phases. It has been suggested that a major reason for the solution-forming capacities of dimethyl sulfoxide, (H3C)2SO, and dimethyl sulfone, (H3C)2SO2, are the possibilities for σ-hole bonding through σ-holes on the sulfurs plus hydrogen bonding,49 as shown for instance for dimethyl sulfoxide and water, 7. For examples in the solid state, in which fluorine σ-hole interactions co-exist with hydrogen bonding, see Chopra et al.,78,120 and Metrangolo et al.79
Stronger σ-hole and π-hole interactions
A typical gas phase σ-hole bond is linear or nearly so. The distance between the σ-hole atom and the negative site is less than the sum of the respective van der Waals radii, but usually by no more than 30%.53 The covalent bond on the extension of which is the σ-hole will have changed from its equilibrium length, but generally by ±0.03 Å or less.104,105 The interaction energy tends to be less negative than about −12 kcal mol−1 (for neutral bases).53It has been found, however, that some complexes that are ostensibly σ-hole-bonded differ from the above description in one or more respects that suggest stronger interactions. The separation of the σ-hole atom and the negative site may be as short as half of the sum of the van der Waals radii, the covalent bond to the σ-hole atom may be lengthened by as much as 0.3 Å, and ΔE may have a magnitude several times greater than that expected for a σ-hole interaction with a neutral base.
These seemingly-anomalous features were observed computationally in some, but not all, of the complexes of the types F–Cl⋯CN–Q121 and F–Cl⋯SiN–Q,122 where Q is an atom or group. For instance, F–Cl⋯CN–NO2 is a normal σ-hole interaction: the Cl⋯C distance is 70% of the sum of the carbon and chlorine van der Waals radii, the F–Cl bond is elongated by 0.035 Å and ΔE = −6.0 kcal mol−1. In contrast, F–Cl⋯CN–SiH3 has a Cl⋯C separation that is 49% of the sum of the van der Waals radii, the F–Cl bond is lengthened by 0.26 Å and ΔE = −15.5 kcal mol−1.
We have demonstrated in the present work that the strengths of normal σ-hole bonds can be represented quite well in terms of the VS,max of the σ-holes and the VS,min of the negative sites (Fig. 8). This supports the interpretation of these interactions as electrostatics–polarization plus dispersion. The density difference plots mentioned earlier show that there is some polarization of both the negative site and the σ-hole atom.47,91,92
In the case of the F–Cl⋯CN–Q and F–Cl⋯SiN–Q complexes, the σ-hole is the same in all cases, and it is fairly strong; the VS,max of the chlorine in F–Cl is 40 kcal mol−1 [B3PW91/6-31G(d,p)]. The marked gradations in interaction energies, from −1.9 to −33.4 kcal mol−1, presumably reflect the variation in the relevant properties of the negative sites. One of these properties can be assumed to be the VS,min, as is demonstrated by Fig. 8; the other, we have suggested,30,122 should be a measure of the local polarizability of the negative site. For the latter purpose, we have proposed the local ionization energy;123 the lower are its values in a given region of a molecule, the less tightly-held and more polarizable are the electrons in that region.
In the two complexes mentioned above, F–Cl⋯CN–NO2 and F–Cl⋯CN–SiH3, and focusing upon the negative sites, in CN–NO2 the VS,min and the local ionization energy of the carbon are −19 kcal mol−1 and 10.0 eV, respectively, while in CN–SiH3 they are −34 kcal mol−1 and 8.6 eV.122 Thus both the VS,min and the local ionization energies of the carbons suggest that the interaction with CN–SiH3 should be much stronger, as was already stated to be the case.
As a more stringent test, the ΔE for all 22 F–Cl⋯CN–Q and F–Cl⋯SiN–Q complexes was expressed by double regression analysis as a function of the negative site VS,min and their local ionization energies.122 For the entire range of ΔE values, from −1.9 to −33.4 kcal mol−1, the R2 for the relationship between the predicted and the computed ΔE was 0.99! Thus, apart from the normal dependence of ΔE upon the VS,max and VS,min, if the positive site has a very strong electric field (i.e. large VS,max) and if the negative site has relatively weakly-held and highly polarizable electronic charge, then its polarization may be to such an extent as to correspond to some degree of dative sharing of electrons (coordinate covalence) and a much stronger bonding interaction.30,122 Our use of the terms “dative sharing” and “coordinate covalence” should not be viewed as implying a fundamental change in the nature of the interaction or a “transfer” of electronic charge; they are simply meant to describe a greater extent of polarization.
We repeated the double regression analysis using Cl–Cl as the σ-hole-containing molecule.122 The σ-hole VS,max on the chlorines in Cl–Cl are much weaker than that in F–Cl, 24 kcal mol−1vs. 40 kcal mol−1. As anticipated, the very strong interactions were now quite rare, occurring only for Q = Li; the ΔE for 10 Cl–Cl⋯CN–Q and Cl–Cl⋯SiN–Q were between −1.3 and −15.0 kcal mol−1. The R2 was 0.96.
It is also notable that for 11 complexes between F–Cl and NC–Q molecules, the σ-hole bonds were all normal,124 despite the very positive VS,max of F–Cl. The most negative ΔE was −11.8 kcal mol−1, when Q = Li. We explain the absence of strong interactions by noting that the nitrogens in the NC–Q have considerably higher local ionization energies, by 1.3 to 2.0 eV, than do the carbons in the CN–Q.122 Thus they are less susceptible to strong polarization and dative sharing.
The evidence suggests, therefore, that there is in these systems a gradation from normal σ-hole bonding to a stronger form having a greater degree of polarization, which can be regarded as significant dative (coordinate covalent) character. The trend in interaction energies can be represented quite satisfactorily in terms of the VS,min of the negative site and its local polarizability. (If the negative site for a series of complexes is in all instances the same atom in the same valence state – e.g. just the CN–Q or just the SiN–Q – then VS,min correlates extremely well with the local ionization energy and only one of them need be used in representing ΔE.122)
A degree of coordinate covalent character is also believed to be present in certain of the noncovalent interactions known as π-hole bonding. A π-hole is a region of low electronic density that is perpendicular to an atom in a planar portion of a molecular framework,125 instead of being along the extension of a covalent bond to the atom as is a σ-hole. Obvious π-hole-containing atoms are the borons in the BX3 molecules (X = F, Cl, Br, I); their valence electrons are all involved in σ bonds in the molecular planes. Other examples include the sulfur in SO2 (8) and the nitrogen in FNO2 (9), the formal charges shown being indicative of electronic deficiencies.
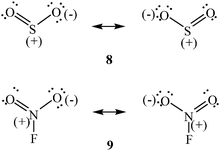
The interactions of positive π-holes with negative sites can lead to noncovalent bonding, the strength of which depends upon the magnitudes of the VS,max of the π-hole and the VS,min of the negative site and the polarizabilities of both. For example, ΔE for the π-hole complex F2(O)C⋯NCH is −4.6 kcal mol−1, and the C⋯N separation is 85% of the sum of the van der Waals radii (MP2/aug-cc-pVDZ).125 The O–C⋯N angle is 97°, slightly more than for a completely perpendicular interaction. For the F2CO π-hole complex with NH3, the structural data are essentially the same but ΔE is more negative, −6.1 kcal mol−1, due to the more negative VS,min of the nitrogen in NH3 (−47 kcal mol−1) compared to HCN (−33 kcal mol−1).
If we now proceed to HCN and NH3 interacting with F2SiO rather than F2CO, the results are quite different. The Si⋯N distances are only about 54% of the sums of the van der Waals radii, and the O–Si⋯N angles are approximately 108°, indicating considerable distortion of the F2SiO molecules. The ΔE are −22.6 kcal mol−1 for F2(O)Si⋯NCH and −42.4 kcal mol−1 for F2(O)Si⋯NH3. The interactions can now be described as involving significant coordinate covalence, as indicated by the short Si⋯N separations, the changes in the O–Si⋯N angles and the much more negative ΔE. The fact that coordinate covalence is a great deal more evident for the F2SiO complexes than for the F2CO presumably reflects the higher polarizability of the silicon compared to the carbon, which increases its capacity for accommodating dative sharing of the nitrogens' electronic charge126 (for another example of this, see Brinck et al.127). The NH3 nitrogen is better able to provide these electrons, having a lower local ionization energy than the HCN nitrogen, 7.3 vs. 10.7 eV.125
Overall, π-hole interactions are analogous to σ-hole, the key factors being electrostatics/polarization and dispersion. Both can show a marked gradation in strength, evolving into some degree of what might be termed coordinate covalence, but physically is simply a high degree of polarization.
Gibbs and Mendeleev
Our objective has been to provide a perspective on σ-hole interactions, but not a complete review. For example, we did not go into the arguments (persuasive to us) that hydrogen bonding can be viewed as a σ-hole interaction.29,30,46,89,90,125,128We have tried to demonstrate that the formation and properties of σ-hole-bonded complexes can be explained in a very satisfactory and straightforward manner in terms of electrostatics/polarization plus dispersion. We believe that this in the spirit of J. W. Gibbs, who has been quoted as saying, “One of the principal objects of theoretical research in any department of knowledge is to find the point of view from which the subject appears in its greatest simplicity”.129
Another goal (this one in the spirit of Mendeleev) is to unify rather than to compartmentalize. Whether the σ-hole is on an atom of Group IV, V, VI or VII, and whether the negative site is a lone pair, a negative ion, π electrons, etc., it is all σ-hole bonding (as is possibly also hydrogen bonding). While not ignoring differences in detail, we feel that it is important to stress the fundamental similarities.
References
- H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS.
- P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12, 5835–5838 CAS.
- F. Guthrie, J. Chem. Soc., 1863, 16, 239–244 RSC.
- I. Remsen and J. F. Norris, Am. Chem. J., 1896, 18, 90–96 CAS.
- O. Hassel and C. Rømming, Q. Rev., Chem. Soc., 1962, 16, 1–18 RSC.
- H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- M.-C. Bernard-Houplain and C. Sandorfy, Can. J. Chem., 1973, 51, 3640–3647 CrossRef CAS.
- T. Di Paolo and C. Sandorfy, Can. J. Chem., 1974, 52, 3612–3622 CrossRef CAS.
- P. Murray-Rust and W. D. S. Motherwell, J. Am. Chem. Soc., 1979, 101, 4374–4376 CrossRef CAS.
- P. Murray-Rust, W. C. Stallings, C. T. Monti, R. K. Preston and J. P. Glusker, J. Am. Chem. Soc., 1983, 105, 3206–3214 CrossRef CAS.
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308–4314 CrossRef CAS.
- J.-M. Dumas, M. Kern and J. L. Janier-Dubry, Bull. Soc. Chim. Fr., 1976, 1785–1787 CAS.
- J.-M. Dumas, H. Peurichard and M. J. Gomel, Chem. Res. (S), 1978, 54–57 CAS.
- T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1992, 44(Suppl. 19), 57–64 CrossRef.
- T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1993, 48(Suppl. 20), 73–88 CrossRef CAS.
- J. S. Murray, K. Paulsen and P. Politzer, Proc. Indian Acad. Sci. (Chem. Sci.), 1994, 106, 267–275 CAS.
- P. Auffinger, F. A. Hays, E. Westhof and P. Shing Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem.–Eur. J., 2006, 12, 8952–8960 CrossRef CAS.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS.
- Y. Lu, J. Zou, H. Wang, Q. Yu, H. Zhang and Y. Jiang, J. Phys. Chem. A, 2005, 109, 11956–11961 CrossRef CAS.
- F.-F. Wang, J.-H. Hou, Z.-R. Li, D. Wu, Y. Li, Z.-Y. Lu and W.-L. Cao, J. Chem. Phys., 2007, 126, 144301 CrossRef (1–5).
- P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2008, 14, 659–665 CrossRef CAS.
- T. Imakubo, H. Sawa and R. Kato, Synth. Met., 1995, 73, 117–122 CrossRef CAS.
- V. Amico, S. V. Meille, E. Corradi, M. T. Messina and G. Resnati, J. Am. Chem. Soc., 1998, 120, 8261–8262 CrossRef CAS.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS.
- K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC.
- A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC.
- J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63, 1598–1607 CrossRef CAS.
- P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278–294 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, Int. J. Quantum Chem., 2007, 107, 2286–2292 CrossRef CAS.
- R. E. Rosenfield, Jr., R. Parthasarathy and J. D. Dunitz, J. Am. Chem. Soc., 1977, 99, 4860–4862 CrossRef.
- T. N. Guru Row and R. Parthasarathy, J. Am. Chem. Soc., 1981, 103, 477–479 CrossRef.
- R. S. Drago, N. Wong and D. C. Ferris, J. Am. Chem. Soc., 1991, 113, 1970–1977 CrossRef CAS.
- J. A. Kapecki and J. E. Baldwin, J. Am. Chem. Soc., 1969, 91, 1120–1123 CrossRef CAS.
- C. Cohen-Addad, M. S. Lehmann, P. Becker, L. Párkányi and A. Kálmán, J. Chem. Soc., Perkin Trans. 2, 1984, 191–196 RSC.
- M. Widhalm and C. Kratky, Chem. Ber., 1992, 125, 679–689 CrossRef CAS.
- F. Carré, C. Chuit, R. J. P. Corriu, P. Monforte, N. K. Nayyar and C. Reyé, J. Organomet. Chem., 1995, 499, 147–154 CrossRef.
- N. W. Mitzel, A. J. Blake and D. W. H. Rankin, J. Am. Chem. Soc., 1997, 119, 4143–4148 CrossRef CAS.
- U. Losehand, N. W. Mitzel and D. W. H. Rankin, J. Chem. Soc., Dalton Trans., 1999, 4291–4297 RSC.
- M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 1902–1909 CrossRef CAS.
- A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190 CrossRef CAS.
- C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666–2674 CrossRef CAS.
- J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033–1038 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 723–729 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- P. Politzer, K. E. Riley, F. A. Bulat and J. S. Murray, Comput. Theor. Chem., 2012, 998, 2–8 CrossRef CAS.
- P. Politzer and J. S. Murray, in Concepts and Methods in Modern Theoretical Chemistry, Vol. 1: Electronic Structure and Reactivity, ed. K. Ghosh and P. Chattaraj, Taylor & Francis, New York, 2013, ch. 9, pp. 181–199 Search PubMed.
- T. Clark, J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2008, 14, 689–697 CrossRef CAS.
- T. M. Klapötke, B. Krumm and M. Scherr, Inorg. Chem., 2008, 47, 7025–7028 CrossRef.
- J. S. Murray, P. Lane, A. Nieder, T. M. Klapötke and P. Politzer, Theor. Chem. Acc., 2010, 127, 345–354 CrossRef CAS.
- P. Politzer, J. S. Murray, P. Lane and M. C. Concha, Int. J. Quantum Chem., 2009, 109, 3773–3780 CrossRef CAS.
- A. Mohajeri, A. H. Pakiari and N. Bagheri, Chem. Phys. Lett., 2009, 467, 393–397 CrossRef CAS.
- O. V. Shishkin, I. V. Omelchenko, A. L. Kalyuzhny and B. V. Paponov, Struct. Chem., 2010, 21, 1005–1011 CrossRef CAS.
- L. Junming, L. Yunxiang, Y. Subin and Z. Weiliang, Struct. Chem., 2011, 22, 757–763 CrossRef.
- Q. Zhao, D. Feng, Y. Sun, J. Hao and Z. Cai, Int. J. Quantum Chem., 2011, 111, 3881–3887 CrossRef CAS.
- S. Zahn, R. Frank, E. Hey-Hawkins and B. Kirchner, Chem.–Eur. J., 2011, 17, 6034–6038 CrossRef CAS.
- B. Jing, Q. Li, B. Gong, R. Li, Z. Liu, W. Li, J. Cheng and J. Sun, Int. J. Quantum Chem., 2012, 112, 1491–1498 CrossRef CAS.
- Q.-Z. Li, R. Li, P. Guo, H. Li, W.-Z. Li and J.-B. Cheng, Comput. Theor. Chem., 2012, 980, 56–61 CrossRef CAS.
- F. T. Burling and B. M. Goldstein, J. Am. Chem. Soc., 1992, 114, 2313–2320 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, Int. J. Quantum Chem., 2008, 108, 2770–2781 CrossRef CAS.
- G. Delgado-Barrio and R. F. Prat, Phys. Rev. A: At., Mol., Opt. Phys., 1975, 12, 2288 CrossRef CAS.
- K. D. Sen and P. Politzer, J. Chem. Phys., 1989, 90, 4370–4372 CrossRef CAS.
- E. D. Stevens, Mol. Phys., 1979, 37, 27–45 CrossRef CAS.
- S. C. Nyburg and W. Wong-Ng, Proc. R. Soc. London, 1979, A367, 29–45 CrossRef.
- S. L. Price, A. J. Stone, J. Lucas, R. S. Rowland and A. E. Thornley, J. Am. Chem. Soc., 1994, 116, 4910–4918 CrossRef CAS.
- V. G. Tsirelson, P. F. Zou, T.-H. Tang and R. F. W. Bader, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 143–153 CrossRef.
- S. L. Price, J. Chem. Soc., Faraday Trans., 1996, 92, 2997–3008 RSC.
- E. Bilewicz, A. J. Rybarczyk-Pirek, A. T. Dubis and S. J. Grabowski, J. Mol. Struct., 2007, 829, 208–211 CrossRef CAS.
- R. F. W. Bader, M. T. Carroll, J. R. Cheeseman and C. Chang, J. Am. Chem. Soc., 1987, 109, 7968–7979 CrossRef CAS.
- R. F. Stewart, Chem. Phys. Lett., 1979, 65, 335–342 CrossRef CAS.
- Chemical Applications of Atomic and Molecular Electrostatic Potentials, ed. P. Politzer and D. G. Truhlar, Plenum Press, New York, 1981 Search PubMed.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1693 CrossRef CAS.
- A. Bundhun, P. Ramasami, J. S. Murray and P. Politzer, J. Mol. Model., 2012 DOI:10.1007/s00894-012-1571-4.
- K. E. Riley, J. S. Murray, J. Fanfrlík, J. Řezáč, R. J. Solá, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2011, 17, 3309–3318 CrossRef CAS.
- P. Politzer, J. S. Murray and P. Lane, Int. J. Quantum Chem., 2007, 107, 3046–3052 CrossRef CAS.
- P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2007, 13, 643–650 CrossRef CAS.
- D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 2175–2186 RSC.
- P. Metrangolo, J. S. Murray, T. Pilati, P. Politzer, G. Resnati and G. Terraneo, Cryst. Growth Des., 2011, 11, 4238–4246 CAS.
- J. S. Murray and P. Politzer, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2011, 1, 153–163 CrossRef CAS.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504–1518 CrossRef CAS.
- P. Dobeš, J. Řezáč, J. Fanfrlík, M. Otyepka and P. Hobza, J. Phys. Chem. B, 2011, 115, 8581–8589 CrossRef.
- M. A. A. Ibrahim, J. Comput. Chem., 2011, 32, 2564–2574 CrossRef CAS.
- M. Kolař and P. Hobza, J. Chem. Theory Comput., 2012, 8, 1325–1333 CrossRef.
- M. Carter, A. K. Rappé and P. Shing Ho, J. Chem. Theory Comput., 2012, 8, 2461–2473 CrossRef CAS.
- W. L. Jorgensen and P. Schyman, J. Chem. Theory Comput., 2012, 8, 3895–3901 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- K. E. Riley, J. S. Murray, P. Politzer, M. C. Concha and P. Hobza, J. Chem. Theory Comput., 2009, 5, 155–163 CrossRef CAS.
- M. Hennemann, J. S. Murray, K. E. Riley, P. Politzer and T. Clark, J. Mol. Model., 2012, 18, 2461–2469 CrossRef CAS.
- T. Clark, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2012, 3, 13–20 CrossRef.
- M. Solimannejad, M. Malokani and I. Alkorta, J. Phys. Chem. A, 2010, 114, 12106–12111 CrossRef CAS.
- S. Scheiner, J. Chem. Phys., 2011, 134, 164313 CrossRef (1–9).
- K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2008, 4, 232–242 CrossRef CAS.
- K. E. Riley, J. S. Murray, J. Fanfrlík, J. Řezáč, R. J. Solá, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2012 DOI:10.1007/s00894-012-1428-x.
- A. C. Legon and D. J. Millen, Chem. Soc. Rev., 1987, 16, 467–498 RSC.
- A. J. Stone and S. L. Price, J. Phys. Chem., 1988, 92, 3325–3335 CrossRef CAS.
- A. C. Legon and D. J. Millen, Faraday Discuss. Chem. Soc., 1982, 73, 71–87 RSC.
- A. D. Buckingham and P. W. Fowler, J. Chem. Phys., 1983, 79, 6426–6428 CrossRef CAS.
- A. D. Buckingham and P. W. Fowler, Can. J. Chem., 1985, 63, 2018–2025 CrossRef CAS.
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- W. A. Sokalski and S. M. Roszak, THEOCHEM, 1991, 234, 387–400 CrossRef.
- J. Chen and T. J. Martínez, Chem. Phys. Lett., 2007, 438, 315–320 CrossRef CAS.
- W. Wang, N.-B. Wong, W. Zheng and A. Tian, J. Phys. Chem. A, 2004, 108, 1799–1805 CrossRef CAS.
- J. S. Murray, M. C. Concha, P. Lane, P. Hobza and P. Politzer, J. Mol. Model., 2008, 14, 699–704 CrossRef CAS.
- K. Hermansson, J. Phys. Chem. A, 2002, 106, 4695–4702 CrossRef CAS.
- M. Palusiak, THEOCHEM, 2010, 945, 89–92 CrossRef CAS.
- X. Lu, H. Li, X. Zhu, W. Zhu and H. Liu, J. Phys. Chem. A, 2011, 115, 4467–4475 CrossRef.
- P. Politzer and J. S. Murray, CrystEngComm, 2013 10.1039/c2ce26883c.
- D. H. Williams, J. P. L. Cox, A. J. Doig, M. Gardner, U. Gerhard, P. J. Kaye, A. R. Lal, L. A. Nicholls, C. J. Salter and R. C. Mitchell, J. Am. Chem. Soc., 1991, 113, 7020–7030 CrossRef CAS.
- M. S. Searle and D. H. Williams, J. Am. Chem. Soc., 1992, 114, 10690–10697 CrossRef CAS.
- M. S. Searle, D. H. Williams and U. Gerhard, J. Am. Chem. Soc., 1992, 114, 10697–10704 CrossRef CAS.
- J. D. Dunitz, Chem. Biol., 1995, 2, 709–712 CrossRef CAS.
- M. S. Westwell, M. S. Searle, J. Klein and D. H. Williams, J. Phys. Chem., 1996, 100, 16000–16001 CrossRef CAS.
- L. Liu and Q.-X. Guo, Chem. Rev., 2001, 101, 673–695 CrossRef CAS.
- A. Cornish-Bowden, J. Biosci., 2002, 27, 121–126 CrossRef.
- A. G. Grechin, H.-J. Buschmann and E. Schollmeyer, Thermochim. Acta, 2006, 449, 67–72 CrossRef CAS.
- E. B. Starikov and B. Nordén, Chem. Phys. Lett., 2012, 538, 118–120 CrossRef CAS.
- E. Damm, O. Hassel and C. Romming, Acta Chem. Scand., 1965, 19, 1159–1165 CrossRef CAS.
- D. Chopra, V. Thiruvenkatam, S. G. Manjunath and T. N. Guru Row, Cryst. Growth Des., 2007, 7, 868–874 CAS.
- J. E. Del Bene, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2010, 114, 12958–12962 CrossRef CAS.
- P. Politzer and J. S. Murray, Theor. Chem. Acc., 2012, 131, 1114 CrossRef (1–10).
- P. Politzer, J. S. Murray and F. A. Bulat, J. Mol. Model., 2010, 16, 1731–1742 CrossRef CAS.
- J. E. Del Bene, I. Alkorta and J. Elguero, Chem. Phys. Lett., 2011, 508, 6–9 CrossRef CAS.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS.
- P. Politzer, J. E. Huheey, J. S. Murray and M. Grodzicki, THEOCHEM, 1992, 259, 99–120 CrossRef.
- T. Brinck, J. S. Murray and P. Politzer, Inorg. Chem., 1993, 32, 2622–2625 CrossRef CAS.
- Z. P. Shields, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 2010, 110, 2823–2832 CrossRef CAS.
- J. W. Gibbs, quoted in: Josiah Willard Gibbs, The History of a Great Mind, ed. L. P. Wheeler, Yale University Press, New Haven, CT, 1962, p. 88 Search PubMed. (This is cited by Reed et al.97).
| This journal is © the Owner Societies 2013 |