Point defect engineering strategies to retard phosphorous diffusion in germanium
Received
24th August 2012
, Accepted 5th November 2012
First published on 5th November 2012
Abstract
The diffusion of phosphorous in germanium is very fast, requiring point defect engineering strategies to retard it in support of technological application. Density functional theory corroborated with hybrid density functional calculations are used to investigate the influence of the isovalent codopants tin and hafnium in the migration of phosphorous via the vacancy-mediated diffusion process. The migration energy barriers for phosphorous are increased significantly in the presence of oversized isovalent codopants. Therefore, it is proposed that tin and in particular hafnium codoping are efficient point defect engineering strategies to retard phosphorous migration.
Introduction
Forming p-type doped regions in germanium (Ge)-devices can be achieved by using boron (B) but n-type dopants such as phosphorous (P) diffuse quickly and consequently the formation of ultra-shallow junctions, with high active dopant concentrations, is a challenge.1
It has previously been established that P diffuses via a vacancy (V) mechanism, which is the dominant intrinsic point defect in Ge.2–4 It was determined by Brotzmann and Bracht2 that P diffusion increases with the square of the free electron concentration. This strong doping dependence is explained by the formation of negatively charged phosphorous-vacancy, (PV)−, pairs via the reaction
Where P
s+ denotes the singly positively charged P substitutional atom and V
2− the doubly negatively charged vacancy. The key to controlling n-type dopant diffusion in Ge is the annihilation or confinement of the vacancies, as they are the vehicles for diffusion.
5 The diffusion of P in Ge can be affected not only by the intrinsic point defects but also by codopants. For example, in recent studies it has been verified that the introduction of small isovalent codopants such as carbon (C) can retard n-type dopant diffusion in Ge as C traps mobile PV pairs thereby forming stable CPV complexes.
6,7
The introduction of larger isovalent codopants can affect dopant–defect interactions in group IV semiconductors.8–10 The aim of the present contribution is to discover point defect engineering strategies based upon isovalent doping that will retard the V-mediated diffusion of P in Ge. In particular using density functional theory (DFT) calculations we investigate the impact of Sn and Hf on the migration and binding energies of (PV)−1 pairs in Ge.
Methodology
DFT as implemented in the Vienna ab initio Simulation Package (VASP)11 was used to calculate the total energies of the defects and their migration (defined here as the largest relative energy barrier along the ring mechanism) energies in Ge. The generalised gradient approximation (GGA) with electron exchange and correlation is described according to the Perdew–Burke–Ernzerhof (PBE)12 formalism. Pseudopotentials generated according to the projector augmented wave (PAW)13 method were used, which treats electrons as core except for the 4s24p2, 3s23p3, 5s25p2 and 6s25d2 orbitals of Ge, P, Sn and Hf respectively. The reciprocal space was sampled according to the Monkhorst–Pack scheme14 using a grid of 4 × 4 × 4 k-points. The calculations were done in a 64 atom supercell. Self-consistency was achieved by iterating the electronic cycles so that the energy differences were not larger than 1 × 10−5 eV. Similarly the forces on each atom were restricted to no more than 1 × 10−3 eV Å−1 at the end of the simulation. Migration energies were calculated using the climbing image nudged elastic band (CI-NEB) method,15 keeping the supercell under constant volume allowing only the atoms’ positions to relax during the simulation. All calculations were performed so as to account for the spin-polarization of the two unpaired electrons in Ge and Hf and the three unpaired electrons of P and because the introduction of a vacancy leaves behind dangling bonds, (i.e. more unpaired electrons). We employ the GGA + U approach discussed in previous work.16 In order to verify this methodology, we used the Heyd–Scuseria–Ernzerhof17 (HSE06) hybrid functional by setting the non-local Hartree–Fock potential to its default value of 25% and the screening parameter, μ = 0.207 Å−1. We used the same k-points grid and the same energy and forces convergence criteria as above. This hybrid functional gives a fairly accurate description of the electronic structure of many solids18 including Ge, for which a band gap of ∼0.85 eV is reproduced (compared to the experimental value of 0.74 eV).
In this study we will investigate the migration energies and mechanisms of vacancies in bulk Ge, which has been codoped with P and Sn or Hf. This is achieved through studying the interactions of both the dopant atoms with the host lattice and with each other, by analyzing their migration energies from one configuration to another and by considering the charge densities for selected geometries.
Results and discussion
As was previously reported oversized species will occupy the space between two semi-vacant lattice sites; this is known as the split-vacancy configuration.19 Substitutional Sn and Hf species next to a vacancy will relax to this split-vacancy configuration. In accordance with eqn (1) we investigated the migration of vacancies as constitutive elements of the singly negatively charged clusters (PSnV)−1 and (PHfV)−1. Previous studies have shown that the V-mediated diffusion of dopants in Ge proceeds via the ring mechanism.2–4 Following this, Fig. 1(a) shows the migration barriers, associated with Sn, obtained as the vacancy hops from one site to another. The migration energy, which corresponds to the step along the ring with the highest energy barrier, is 1.54 eV. This energy is comparable to the migration energy of a vacancy in the presence of Sn alone (i.e. 1.47 eV).16 Conversely, Hf dramatically increases the migration energy to about 3.04 eV (see Fig. 1(b)). To understand the origin of the high migration energy we calculated the binding energies for (PSnV)−1 and (PHfV)−1 clusters for all the configurations shown at the top of Fig. 1(a) and (b). The idealized ring mechanism of diffusion is shown schematically in Fig. 2. The binding energy is a measure of the stability of the cluster with respect to its constituent components (i.e. P, Sn and V−2 or P, Hf and V−2). For example, the binding energy of a substitutional P atom to a Hf atom and a V to form a PHfV cluster in Ge is given by:| | | Eb(PHfVGeN−3) = E(PHfVGeN−3) − E(PGeN−1) − E(HfGeN−1) − E(VGeN−1) + 2E(GeN) | (2) |
where E(PHfVGeN−3) is the energy of a N lattice site supercell (here N = 64) containing N − 3 Ge atoms, a P atom, one Hf atom and a V; E(PGeN−1) is the energy of a supercell containing one P atom and N − 1 Ge atoms; E(HfGeN−1) is the energy of a supercell containing one Hf atom and N − 1 Ge atoms; E(VGeN−1) is the energy of a supercell containing a V and N − 1 Ge atoms; and E(GeN) is the energy of the N Ge atom supercell. Therefore, here a negative binding energy corresponds to a defect cluster that is stable with respect to its constituent point defect components.
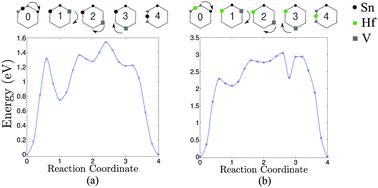 |
| | Fig. 1 Diffusion path of the PV pairs in the presence of (a) Sn and (b) Hf, calculated using GGA + U. On the top of the figures is the ring mechanism of diffusion for the PV pair in the presence of Sn and Hf, respectively, projected onto the (111) surface of Ge. The squares represent the V, smaller circles the P atom and larger circles the Sn (or Hf) atoms. In configurations 0 and 4 the Sn (or Hf) atoms are surrounded by two semi-vacant sites in what is known as the split-vacancy configuration. | |
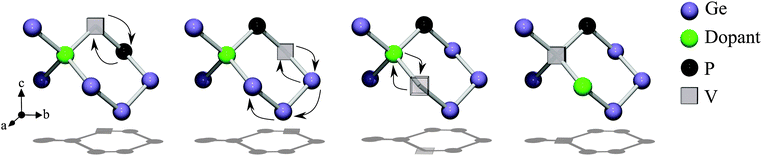 |
| | Fig. 2 A schematic diagram of diffusion via the ring mechanism. | |
The binding energies were calculated using GGA, GGA + U and the HSE06 functional as shown in Table 1. A comparison of these three modes of calculation reveals a good agreement between the GGA + U and HSE06. Since the magnitude of the migration energies of a species are reflected by how bound the clusters are together, and based on the values given in Table 1 it is safe to assume that the GGA + U (which is at least an order of magnitude computationally faster than HSE06) is a valid model and that the approach is justifiable for this type of studies. To further validate this model we calculated the densities of states (DOS) of bulk Ge and for supercells containing one Sn or Hf atom using GGA + U and HSE06 as shown in Fig. 3. It is evident that in both cases the defects do not introduce states into the band gap, which can be attributed to the isovalent nature of these defects. Furthermore, the energies and natures of the orbitals (s, p and d) are nearly identical with these two different functionals. We therefore proceed in analysing the clusters formed using GGA + U. The values show that those configurations which involve Hf are more bound than the corresponding configurations that incorporate Sn. For the starting configuration when Sn or Hf occupies the split-vacancy configuration next to a P atom, the binding energies are −1.49 and −3.55 eV respectively. As the V exchanges position with the P atom the binding energies drop to −0.75 eV and −1.48 eV for the pairs containing Sn or Hf respectively, which are nearly half the equivalent original energies in each case. The charge densities of these two particular configurations for the (PSnV)−1 and (PHfV)−1 clusters are shown in Fig. 4(a) and (b), respectively. As can be seen in the left panel of Fig. 4(a), when Sn occupies the split-V configuration (configuration 0) it tends not to share electrons with the P atom, which in turn draws additional electron density to its vicinity. This is to be expected because P has a high electronegativity, 2.19, compared to 1.96 for Sn and 2.01 for Ge. The right panel shows the charge distribution when V and P species exchange positions (configuration 1). Now P and Sn share electrons, which implies that they are bonded. Equivalently, in Fig. 4(b) the two configurations for Hf are shown, but it is clear that Hf still forms a bond even when it is in the split-V configuration. At first, configuration 1 might appear to be more bound as the Sn/Hf atoms form bonds with the P atom, but as the calculations show, the binding energies of configuration 0 are nearly twice those of configuration 1 for both Sn or Hf. To better understand this we analyzed the neighbours of each species forming these clusters and their bond lengths. The bond lengths of the nearest neighbors (NN) to P are always shorter than those formed between Sn/Hf and their neighboring Ge atoms. The left panel of Fig. 5(a) shows there are two NN Ge atoms with an average P–Ge bond length equal to 2.38 Å, whereas the five Ge atoms form Sn–Ge bonds with lengths varying between 2.69 Å to 2.95 Å. For configuration 2 the P atom is only surrounded by 2 NN Ge atoms with bond lengths of about 2.37 Å and the Sn atom forms bonds with 3 Ge atoms each being 2.59 Å. This reflects the much greater binding energy of configuration 0 compared to configuration 1 even when the P and Sn atoms do not share electrons (see Fig. 4(a)). Similar atomic arrangements are exhibited by the (PHfV)−1 cluster (see Fig. 5(b)), however, the P and Hf species come close enough to form a bond, which is about 2.96 Å, resulting in a much higher binding energy compared to the (PSnV)−1 cluster. In order to verify the role that Sn or Hf play in retarding the diffusion of P, we calculated the binding energies of SnV and HfV clusters. These were found to be −1.09 eV and −2.70 eV respectively as compared to the binding energy of the (PV)−1 cluster, which is −0.54 eV. These high binding energies, in particular for Hf, mean that as the vacancy migrates across the lattice and encounters an oversized atom, it is trapped, reducing the mobility of these mediating species, even before they become part of the larger clusters.
Table 1 Binding energies of the different configurations forming the (PSnV)−1 and (PHfV)−1 clusters calculated using GGA, GGA + U and HSE06
| Configuration |
Binding energy (eV) |
| (PSnV)−1 |
(PHfV)−1 |
| GGA |
GGA + U |
HSE06 |
GGA |
GGA + U |
HSE06 |
| 0 |
−0.58 |
−1.49 |
−1.20 |
−2.33 |
−3.55 |
−3.51 |
| 1 |
−0.43 |
−0.75 |
−0.81 |
−1.06 |
−1.48 |
−1.52 |
| 2 |
−0.16 |
−0.22 |
−0.43 |
−0.76 |
−0.70 |
−0.95 |
| 3 |
−0.30 |
−0.28 |
−0.47 |
−0.80 |
−0.62 |
−0.91 |
| 4 |
−0.58 |
−1.49 |
−1.20 |
−2.33 |
−3.55 |
−3.51 |
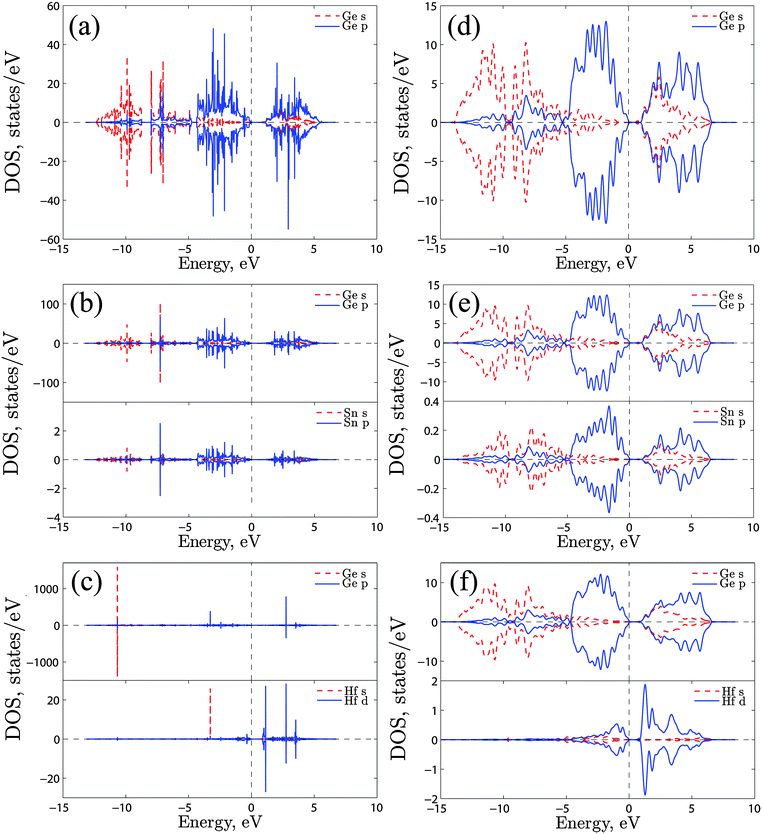 |
| | Fig. 3 The densities of states of (a) a perfect Ge crystal, (b) one Sn atom in Ge, (c) one Hf atom in Ge using GGA + U. (d) A perfect Ge crystal, (e) one Sn atom in Ge, (f) one Hf atom in Ge using HSE06. | |
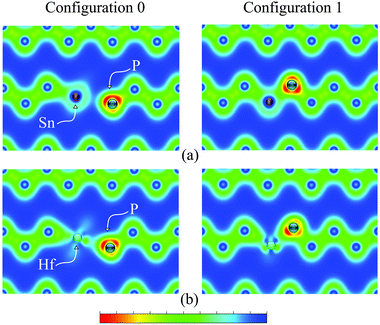 |
| | Fig. 4 The charge density plots of (a) configuration 0 which shows the Sn atom in the split-V configuration and configuration 1 and (b) similar plots for the (PHfV)−1 cluster. The projection is onto the (101) plane of Ge. | |
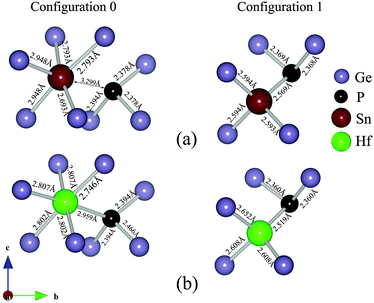 |
| | Fig. 5 The nearest neighbours species surrounding (a) the P and Sn atoms and (b) the P and Hf atoms. The number of nearest neighbours and their bond lengths determines the stability of the cluster. | |
Conclusions
In summary, we used a GGA + U approach to investigate the impact of oversized isolated co-dopants on the migration of P in Ge. We predict that codoping with Hf and to some extent with Sn, significantly increases the migration energy barriers of P via a V-mediated mechanism. In particular, the formation of strongly bound (PSnV)−1 and (PHfV)−1 clusters, in essence trap the migrating (PV)−1 pair. Thus, the introduction of oversized isovalent codopants is proposed as an efficient point defect engineering strategy to limit P migration in Ge. Finally, isovalent dopants have to be introduced at concentrations that exceed their solubility limit, probably via implantation. High concentrations will be required to suppress the diffusion of P. In turn the damage caused by implantation of Ge could lead to high concentrations of vacancies and interstitials that could participate in more complex defect reactions.
Acknowledgements
This publication was based on research supported by King Abdullah University for Science and Technology (KAUST). HT would like to acknowledge the useful discussions with Dr Samuel Murphy and his help in rendering some of the graphics in this publication. AC acknowledges financial support from the EU FP7-PEOPLE-2010-IEF project REACT-273631. Computing resources were provided by the Shaheen supercomputer at KAUST (http://www.hpc.kaust.edu.sa/) and the High Performance Computing (HPC) facility of Imperial College London (http://www3.imperial.ac.uk/ict/services/highperformancecomputing).
References
-
C. Claeys and E. Simoen, Germanium-based technologies: from materials to devices, Elsevier, 2007 CrossRef; E. Napolitani, G. Bisognin, E. Bruno, M. Mastronatteo, G. G. Scapellato, S. Boninelli, D. De Salvador, S. Mirabella, C. Spinella, A. Carnera and F. Priolo, Appl. Phys. Lett., 2010, 96, 201906 CrossRef; J. Kim, S. W. Bedell and D. K. Sadana, Appl. Phys. Lett., 2011, 98, 082112 CrossRef.
- S. Brotzmann and H. Bracht, J. Appl. Phys., 2008, 103, 033508 CrossRef.
- A. Chroneos, H. Bracht, R. W. Grimes and B. P. Uberuaga, Appl. Phys. Lett., 2008, 92, 172103 CrossRef.
- H. Tahini, A. Chroneos, R. W. Grimes, U. Schwingenschlögl and H. Bracht, Appl. Phys. Lett., 2011, 99, 072112 CrossRef.
- A. Chroneos, R. W. Grimes and H. Bracht, J. Appl. Phys., 2009, 106, 063707 CrossRef; G. Impellizzeri, S. Boninelli, F. Priolo, E. Napolitani, C. Spinella, A. Chroneos and H. Bracht, J. Appl. Phys., 2011, 109, 113527 CrossRef.
- S. Brotzmann, H. Bracht, J. Lundsgaard Hansen, A. Nylandsted Larsen, E. Simoen, E. E. Haller, J. S. Christensen and P. Werner, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235207 CrossRef.
- A. Chroneos, R. W. Grimes, B. P. Uberuaga and H. Bracht, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235208 CrossRef.
- A. Chroneos, C. Jiang, R. W. Grimes, U. Schwingenschlögl and H. Bracht, Appl. Phys. Lett., 2009, 94, 252104 CrossRef.
- A. Chroneos, C. A. Londos, E. N. Sgourou and P. Pochet, Appl. Phys. Lett., 2011, 99, 241901 CrossRef.
- A. Chroneos and A. Dimoulas, J. Appl. Phys., 2012, 111, 023714 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- P. E. Blöchel, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- H. Tahini, A. Chroneos, R. W. Grimes and U. Schwingenschlögl, Appl. Phys. Lett., 2011, 99, 162103 CrossRef CAS; H. Tahini, A. Chroneos, R. W. Grimes, U. Schwingenschlögl and H. Bracht, J. Phys.: Condens. Matter, 2012, 24, 195802 CrossRef.
- J. Heyd, E. G. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- M. Marsman, J. Paier, A. Stroppa and G. Kresse, J. Phys.: Condens. Matter, 2008, 20, 064201 CrossRef CAS.
- H. Höhler, N. Atodiresei, K. Schroeder, R. Zeller and P. Dederichs, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 35212 CrossRef.
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.