Acemetacin polymorphs: a rare case of carboxylic acid catemer and dimer synthons†‡
Palash
Sanphui
a,
Geetha
Bolla
a,
Uday
Das
b,
Alok K.
Mukherjee
*b and
Ashwini
Nangia
*a
aSchool of Chemistry, University of Hyderabad, Prof. C. R. Rao Road,
, Gachibowli, Central University P.O., Hyderabad 500 046, India
bDepartment of Physics, Jadavpur University, 188 Raja S. C. Mullick Road, Kolkata 700 032, India. E-mail: akm_ju@rediffmail.com; ashwini.nangia@gmail.com
First published on 22nd October 2012
Abstract
Acemetacin is the first example of an API polymorph with a carboxylic acid catemer and dimer O–H⋯O synthons. The auxiliary stabilization to the catemer motif from stronger C–H⋯O and C–Cl⋯O interactions compared to those in the dimer structure is discussed for a polymorph pair. The crystal structure of the stable dimer polymorph was solved by structure determination from powder X-ray diffraction data (SDPD).
Acemetacin is a glycolic acid ester of indomethacin (Fig. 1a). They are non-steroidal anti-inflammatory drugs (NSAIDs). The active pharmaceutical ingredient (API) acemetacin reduces the gastric side effect better than its parent drug indomethacin.1a Five polymorphs of acemetacin were characterized by PXRD, IR and DSC,1b,c and a monohydrate by single crystal X-ray diffraction.1d No X-ray crystal structures are reported for acemetacin polymorphs. We now report the crystal structure of the stable polymorph in the centrosymmetric space group P21/n solved from high resolution powder X-ray diffraction data. The crystal structure of a metastable form was solved in the chiral space group P21 using single crystal X-ray diffraction. The basic difference between the two polymorphs is the dimer and catemer O–H⋯O synthon of the carboxylic acids (Fig. 1b).
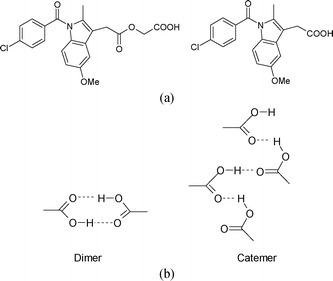 | ||
| Fig. 1 (a) Acemetacin (left) and indomethacin (right). (b) The carboxylic acid dimer R22(8) ring and catemer C(4) chain. | ||
Polymorphism2a is the existence of a compound in more than one crystalline arrangement of the same molecule in the solid-state. This phenomenon has gained immense importance in the pharmaceutical industry, particularly in the last two decades. Polymorphs differ in drug-like physicochemical properties,2b,c such as solubility, stability, bioavailability, compressibility, melting point, etc. An understanding of crystallization and control of a particular polymorph2d is of utmost importance. Polymorph screening of active pharmaceutical ingredients gives the best crystalline form with desirable physicochemical properties for oral formulation.
Carboxylic acids represent 6% of the organic entries in the Cambridge Structural Database (CSD).3 25 of the top 100 prescription drugs are acids. Carboxylic acids generally crystallize as dimers when heteroatoms are absent in the molecule. Hydrated and solvated structures are also possible. The catemer motif is, as such, rare in carboxylic acids.4 The graph set notation5 of the O–H⋯O hydrogen bond dimer and catemer synthons is R22(8) ring and C(4) chain, respectively. Among 6612 carboxylic acid structures archived in the CSD,6 35.9% (2377 hits) are dimer and only 3.2% (210 hits) are catemer. Generally, aliphatic and aromatic carboxylic acids adopt the dimer synthon in the solid-state because of better stability, higher packing efficiency, and the centrosymmetric motif is independent of the nature/size of the alkyl group in RCOOH. The rare catemer synthon appears to be present when the alkyl chain has flexibility and there is less steric hindrance in the molecule. Desiraju and coworkers7 have demonstrated that additional support from weak C–H⋯O interactions is necessary to stabilize the catemer motif, e.g. as in cubane carboxylic acids and phenyl propiolic acids. They showed that an additional C–H⋯O interaction from an activated, acidic CH donor to the COO acceptor is always present in the catemer synthon.
It is well known that the best systems to probe the role of weak interactions are polymorphic structures, because molecular effects are the same. However, neither cubane acids nor propiolic acids gave even a single example of polymorphs after extensive crystallization experiments.7 There are only three known examples of dimer–catemer polymorph pairs among carboxylic acids: tetrolic acid, oxalic acid, and 2,6-bis(CF3)benzoic acid.8 However, the role of the C–H⋯O interaction in stabilizing the catemer synthon cannot be examined in these systems because both oxalic acid and tetrolic acid are stand alone examples with their specific steric/electronic nature of the R group. Oxalic acid is devoid of CH donors and propiolic acid has a rigid propynyl group. The last case of 2,6-bis(CF3)benzoic acid is complicated by multiple molecules in the asymmetric unit. The steric demands of ortho-CF3 groups twist the phenyl ring almost perpendicular to the COOH, and this probably leads to the catemer motif. We discuss herein O–H⋯O dimer and catemer synthons and contribution from weak C–H⋯O and C–Cl⋯O interactions in acemetacin polymorphs.
Acemetacin is a polymorphic drug existing in as many as five polymorphs and one hydrate form.1b–d The monohydrate is the stable crystalline form (crystal structure CSD Refcode CIJYUQ).1d We found it difficult to crystallize acemetacin in the anhydrous state because of its high tendency to transform to hydrate/solvate irrespective of the solvent used. We therefore carried out crystallizations in dry organic solvents. Thin, long, needle-shaped crystals of a metastable form I were obtained from dry EtOAc or a CH3NO2–EtOH (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent mixture. The structure was solved and refined in the chiral space group P21 using single crystal X-ray diffraction data. The catemer chain is present in this structure (Fig. 2). The PXRD pattern of the commercial API did not match with the calculated XRD lines of the catemer crystal structure solved from single crystal X-ray diffraction (Fig. S1, ESI‡). There were considerable differences in PXRD peak positions of the bulk starting material and XRD lines calculated for the catemer crystal structure. The crystal structure of the stable form was solved by structure determination from powder data (SDPD) of the as received bulk material using high resolution powder X-ray diffraction data. Attempts to crystallize the bulk material under any conditions gave form I or hydrate. The XRD lines of the commercial material were different from that of form I (named form II hereafter). The crystal structure of form II was solved in the centrosymmetric space group P21/n from high resolution PXRD data using FOX and GSAS packages (see ESI‡ for details). ORTEP diagrams of acemetacin polymorphs are displayed in Fig. S2, ESI.‡ The crystal structure of form II shows the standard COOH dimer synthon (Fig. 3).§
:1) solvent mixture. The structure was solved and refined in the chiral space group P21 using single crystal X-ray diffraction data. The catemer chain is present in this structure (Fig. 2). The PXRD pattern of the commercial API did not match with the calculated XRD lines of the catemer crystal structure solved from single crystal X-ray diffraction (Fig. S1, ESI‡). There were considerable differences in PXRD peak positions of the bulk starting material and XRD lines calculated for the catemer crystal structure. The crystal structure of the stable form was solved by structure determination from powder data (SDPD) of the as received bulk material using high resolution powder X-ray diffraction data. Attempts to crystallize the bulk material under any conditions gave form I or hydrate. The XRD lines of the commercial material were different from that of form I (named form II hereafter). The crystal structure of form II was solved in the centrosymmetric space group P21/n from high resolution PXRD data using FOX and GSAS packages (see ESI‡ for details). ORTEP diagrams of acemetacin polymorphs are displayed in Fig. S2, ESI.‡ The crystal structure of form II shows the standard COOH dimer synthon (Fig. 3).§
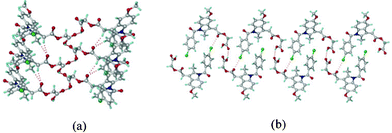 | ||
| Fig. 2 (a) O–H⋯O catemer chain propagating parallel to the b-axis (screw direction) in form I. (b) The C–Cl⋯O interactions (3.11 Å) together with O–H⋯O interactions make a sheet motif. | ||
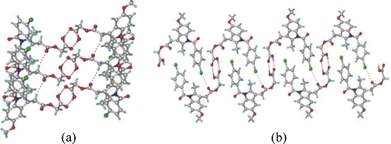 | ||
| Fig. 3 (a) O–H⋯O centrosymmetric carboxylic acid dimer in form II. (b) Supportive C–Cl⋯O interactions (3.25 Å) result in a sheet packing. | ||
Acemetacin molecules assemble via the carboxylic acid catemer synthon of O–H⋯O hydrogen bonds (O4–H4⋯O5: 1.81 Å, 2.728(3) Å, 157°) along the b-axis in polymorph I (Fig. 2a). There is a bifurcated C–H⋯O hydrogen bond (C9–H9A⋯O2: 2.37 Å, 3.434(4) Å, 168° and C18–H18A⋯O2: 2.56 Å, 3.534(4) Å, 157°) from aliphatic CH2 and CH3 donors of the indole ring to the ester carbonyl of acemetacin. The molecules in the C(4) chain are connected by a C–Cl⋯O interaction (3.11 Å) to the ester carbonyl oxygen (Fig. 2b). The stable dimer structure of acemetacin form II consists of a centrosymmetric O–H⋯O dimer (O4–H4⋯O5: 1.71 Å, 2.678(3) Å, 167°) (Fig. 3a). The dimers are linked to the next layer by a C–H⋯O interaction (C18–H18A⋯O2: 2.57 Å, 3.585(4) Å, 163°) through a R44(28) ring motif. Similar to the form I catemer, acemetacin dimer molecules are also connected through a C–Cl⋯O interaction (3.25 Å) to the ester carbonyl in form II (Fig. 3b). There is some closeness in the calculated X-ray diffraction lines of form I and II (Fig. S3, ESI‡). A few characteristic peaks are common for both polymorphs and there is some degree of overlap, but this is not uncommon for polymorphs.9 The characteristic X-ray diffraction peaks of form I are at 2θ = 22.45°, 25.28° and 29.72°, and form II at 21.24°, 22.05° and 23.46; other peaks are too close or overlapping. The experimental PXRD patterns of forms I and II in our study match with β and α forms of Yoneda et al.1b and form I and II of Burger et al.1c The reference PXRD patterns1b,c are reproduced in Fig. S4, ESI. ‡
The main issue to be addressed is why the catemer synthon is present in acemetacin polymorph I. Indomethacin crystallizes as dimorphs, with both polymorphs having the carboxylic acid dimer synthon.10 The CH2 hydrogens adjacent to the indole ring of acemetacin are acidic, since they are flanked by an electron-withdrawing ester group and indole aromatic ring. The natural charge11 of the CH2 carbon adjacent to the indole ring is more electronegative than that on the CH2 next to the carboxylic acid (−0.799 vs. −0.139 coulomb). C–H⋯O bond strength depends both on the acidity of the C–H donor and the basicity of the oxygen atom. The more acidic CHs will participate in stronger C–H⋯O interactions.12 The crystal structure of metastable form I has more C–H⋯O interactions and they are shorter than those in stable form II (Fig. 4). The O–H⋯O catemer synthon is stabilized by the dominance of the C–H⋯O interactions compared to weaker C–H⋯O interactions assisting the dimer hydrogen bond network. Moreover the C–Cl⋯O interaction is also shorter in the catemer structure (3.11 vs. 3.25 Å, Fig. 2b and 3b). The most acidic CH donors, i.e. those sandwiched between indole and ester, are engaged in these C–H⋯O motifs, instead of the less acidic CHs adjacent to the COOH group. Even in the case of cubane and propiolic acids,7 the acidic cubyl and phenyl CHs provide the C–H⋯O fortification to the catemer O–H⋯O chain. In acemetacin, the less acidic CH2 hydrogens adjacent to COOH do not participate in C–H⋯O interactions but instead make C–H⋯π interactions, which are nominally shorter in the catemer compared to the dimer (C–H⋯π: 2.91, 2.73 Å vs. 2.93 Å).
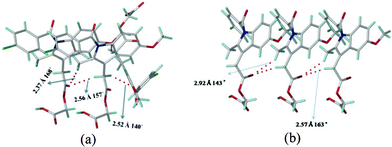 | ||
| Fig. 4 (a) C–H⋯O interactions present in (a) form I and (b) form II. The C–H⋯O interactions are stronger in form I (catemer, left) than form II (dimer, right). | ||
The contribution of all hydrogen bonds and interactions in a crystal structure may be quantified and displayed as Hirshfeld plots13 (Fig. 5), which suggest that the contribution of H⋯O (O–H⋯O + C–H⋯O) is 1.3% higher in the catemer structure (14.3% vs. 13.0%), even though the distance is shorter in the latter case (catemer di = 0.72 Å, de = 1.08 Å; dimer di = 0.64 Å, de = 1.02Å). On the other hand, the Cl⋯O contribution is slightly lower in the catemer (1.8% vs. 1.9%) but penetration is deeper (catemer di = 1.70 Å, de = 1.40 Å; dimer di = 1.78 Å, de = 1.44 Å). The dissimilarity index of polymorphs by the XPac method14 is 4.0, a low number that quantifies the kind of similarities noted in their PXRD line patterns. XPac is a program for comparing complete crystal structures based on the geometric conformations and positions of molecules. Out of 14 near-neighbor molecules in a cluster, 10 molecules were matched in a 2D supramolecular construct (Fig. S5a, ESI‡). Isostructurality was further supported by calculating the packing similarity of the polymorphs using mercury 3.0. Out of 15 surrounding molecules, 11 were common in the close packing arrangement (Fig. S5b, ESI‡).
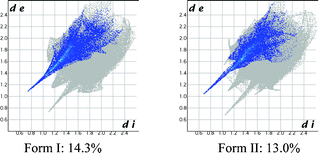 | ||
| Fig. 5 Hirshfeld surface 2D fingerprint plots for the form I and II crystal structures depicting the H⋯O percentage contribution. | ||
Lastly, the stability of polymorphs I and II was evaluated. The higher density (1.467, 1.434 g cm−3) and packing fraction (70.6%, 69.1%) suggest that form I (catemer) is stable, while lattice energy calculations (−54.71, −54.24 kcal mol−1; in Materials Studio, COMPASS force field) gave very close values. The thermodynamic nature of stable form II (dimer, matching with commercial material) was inferred by DSC (Fig. 6). The DSC endotherm for form I at Tonset 147.2 °C and Tpeak 149.2 °C is slightly broad (recorded at 5 °C heating rate), whereas the values for form II are at a higher temperature of 150.6 and 151.3 °C. The two polymorphs did not exhibit phase transformation before melting, consistent with the literature reports.1b,c The Hf values of form I and II are 46.2 and 49.0 kJ mol−1, which means a monotropic system according to the enthalpy of fusion rule.15 Thus, form II of acemetacin, which is the main polymorph in the commercial sample used for this study,¶ was assigned as the thermodynamic modification by crystallographic (dimer is the default synthon) and thermal measurements (higher melting point). The DSC at a faster heating rate of 20 °C and HSM snapshots of form I and II are shown in Fig. S6 and S7 (ESI‡).
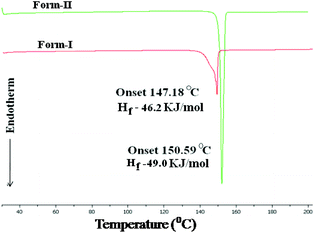 | ||
| Fig. 6 Monotropy of acemetacin polymorphs in DSC recorded at 5 °C heating rate and the thermodynamic stability (higher Hf) of the higher melting temperature form II. | ||
The bulk purity of each polymorph was confirmed by matching their PXRD with the calculated lines from the crystal structure. The absence of acemetacin hydrate in the bulk phases was ascertained by the absence of a peak at low 2θ of 6.62° (Fig. 7 and S8, ESI‡). Among the three acemetacin forms discussed herein, the hydrate is the most crystalline of the solids because of its high intensity X-ray diffraction lines and the metastable form I is the least crystalline as manifested by its broad peaks. The two polymorphs and hydrate were further characterized by FT-IR spectroscopy to correlate the crystal structure with hydrogen bonding (Fig. S9, ESI‡). The three carbonyl functional groups of carboxylic ester, acid and amide occur at 1751.2, 1726.5 and 1665.8 cm−1 in stable form II. The values in form I are at 1743.9, 1724.6 and 1672.3 cm−1 and for the hydrate at 1727.6 and 1694.2 cm−1. When the PXRD lines are very close, solid-state NMR is helpful to differentiate between polymorphs.9,1613C ss-NMR spectra of the acemetacin polymorphs (Fig. 8) indicate small differences in chemical shift values and peak shape. Normally, non-equivalent carbons should resonate as distinct peaks in the 13C NMR spectrum. Out of 21 carbon atoms in the molecule, there are 17 single peaks for form II and 19 for form I in the ss-NMR spectra. We know from the X-ray crystal structure that there is only one molecule in the asymmetric unit. In the present case, PXRD lines and NMR peaks are very close for form I and II.
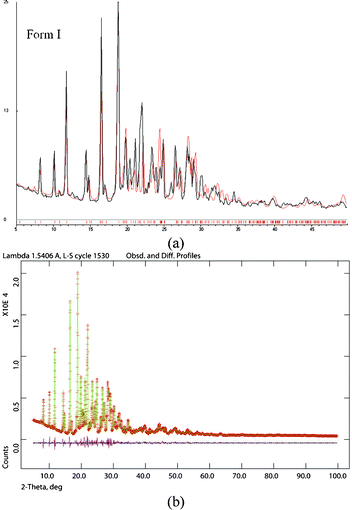 | ||
| Fig. 7 (a) Experimental PXRD of acemetacin form I (black trace) with the calculated line pattern from the X-ray structure (red trace) indicate bulk phase purity of the crystalline metastable form I. (b) The Rietveld refinement XRD pattern of form II (green points and trace) with the observed SDPD structure line profile (red) match nicely as indicated by the baseline difference (magenta line). | ||
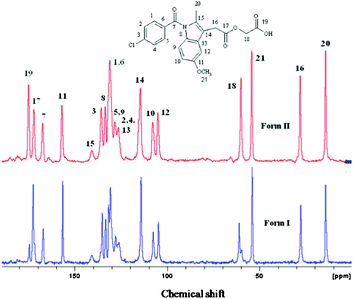 | ||
| Fig. 8 13C ss-NMR spectra of acemetacin polymorphs indicate significant overlap of peaks, similar to their PXRD lines. | ||
Acemetacin is a poorly aqueous soluble drug (0.8 g L−1 in pH 6 phosphate buffer at 37 °C).1b Intrinsic dissolution and solubility measurements were carried out at 37 °C in pH 7 phosphate buffer. Both polymorphs exhibited a similar dissolution rate up to 45 min and dissolved about 3.5 times faster than the monohydrate (Fig. 9). The maximum solubility Cmax occurred at 1.5–2 h, and after this point transformation to the hydrate limited further solubility. Both polymorphs transformed to acemetacin hydrate after 4 h in the dissolution medium as confirmed by IR spectroscopy and the powder XRD of the product solids. The thermodynamic nature of the hydrate was confirmed in slurry experiments at 24 h (Fig. S10, ESI‡). The metastable form I exhibited a slightly faster dissolution rate than stable form II at 2 h: IDR of form I, II and the hydrate are 2.2, 2.1 and 0.63 (mg cm−2) min−1. The apparent solubility of forms I and II are 18.7 and 17.5 g L−1.
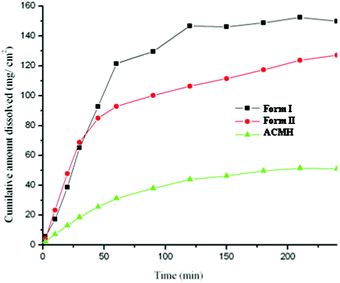 | ||
| Fig. 9 IDR experiments of acemetacin polymorphs I and II and hydrate ACMH in pH 7 phosphate buffer at 37 °C. | ||
Drug particle morphology was analyzed by SEM. The stable form II has block morphology (28.2 × 97.3 μm) and metastable form I crystallized as acicular/long fibers (2.83 × 51.1 μm; Fig. 10). Generally, block crystals dissolve faster because of higher surface area and a greater number of solute–solvent interactions than needle morphology. Here, both polymorphs exhibited a comparable dissolution rate initially and later on the metastable form (needle crystals) dissolved faster than the stable form (blocks). The stable form II transformed to acemetacin hydrate faster than metastable polymorph, perhaps due to a larger contact surface area with water.
 | ||
| Fig. 10 SEM image of acemetacin polymorphs (a) I and (b) II. | ||
In conclusion, the first example of carboxylic acid catemer and dimer synthon polymorphs for an API are reported in acemetacin. Due to the difficulty in crystallization of stable form II from common laboratory solvents, the crystal structure was solved from high resolution powder X-ray diffraction data. The catemer polymorph crystallized in a chiral space group while the dimer form is in a centrosymmetric space group. The conformation of the flexible acemetacin molecule is similar in both polymorphs but it is completely different from that in the hydrate structure. The polymorphs are stable at ambient conditions but transformed to the hydrate in aqueous slurry medium. The anhydrate polymorphs are 3.5 times faster dissolving than the hydrate in pH 7 buffer medium.
Acknowledgements
PS and GB thank the UGC for fellowship. We thank the DST (JC Bose fellowship SR/S2/JCB-06/2009) and DST (IRPHA) and UGC (PURSE grant) for providing instrumentation and infrastructure facilities.References
- (a) A. E. Chávez-Piña, W. McKnight, M. Dicay, G. Castañeda-Hernández and J. L. Wallace, Br. J. Pharmacol., 2007, 152, 930 Search PubMed; (b) M. Yoneda, Y. Ohkawa, Y. Watanabe, M. Ogawa and H. Nagai, Yakugaku Zasshi, 1981, 101, 939 Search PubMed; (c) A. Burger and A. Lettenbichler, Pharmazie, 1993, 48, 262 CAS; (d) T. Gelbrich, M. F. Haddow and U. J. Griesser, Acta Crystallographica Section C Crystal Structure Communications, 2007, 63, o451 Search PubMed.
- (a) J. Bernstein, Polymorphism in Molecular Crystals, Clarendon, Oxford, 2002 Search PubMed; (b) Polymorphism in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley-VCH, Weinheim, 2006 Search PubMed; (c) H. G. Brittan, Polymorphism in Pharmaceutical Solids, Marcel Dekker, New York, 1999 Search PubMed; (d) J. Chen, B. Sarma, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2011, 11, 887 CrossRef CAS.
- T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533 CrossRef CAS.
- (a) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS; (b) T. Beyer and S. L. Price, J. Phys. Chem. B, 2000, 104, 2647 CrossRef CAS; (c) R. J. Davey, G. Dent, R. K. Mughal and S. Parveen, Cryst. Growth Des., 2006, 6, 1788 CrossRef CAS.
- (a) M. C. Etter and J. C. Mcdonald, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256 CrossRef; (b) J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- Cambridge Structural Database, ver. 5.32. ConQuest 1.14, November 2011 release, May 2012 update, CCDC, www.ccdc.cam.ac.uk.
- (a) S. S. Kuduva, D. C. Craig, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1999, 121, 1936 CrossRef CAS; (b) D. Das, R. K. R. Jetti, R. Boese and G. R. Desiraju, Cryst. Growth Des., 2003, 3, 675 CrossRef CAS; (c) D. Das and G. R. Desiraju, Chem.–Asian J., 2006, 1, 231 CrossRef CAS.
- (a) V. Benghiat and L. J. Leiserowitz, J. Chem. Soc., Perkin Trans. 2, 1972, 1763 RSC; (b) A. Gavezzotti, G. Filippini, J. Kroon, B. P. van Eijick and P. Klewinghaus, Chem.–Eur. J., 1997, 3, 893 CrossRef CAS; (c) J. L. Derissen and P. H. Smit, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2240 CrossRef; (d) J. M. Tobin and J. D. Masuda, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o1217 CrossRef; (e) N. A. Giffin, A. D. Hendsbee and J. D. Masuda, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1054 Search PubMed.
- (a) L. Fábián, A. Kálmán, G. Argay, G. Bernáth and Z. Cs. Gyarmatib, Chem. Commun., 2004, 2114 RSC; (b) P. Sanphui, N. R. Goud, U. B. R. Khandavilli, S. Bhanoth and A. Nangia, Chem. Commun., 2011, 47, 5013 RSC.
- (a) T. J. Kistenmacher and R. E. Marsh, J. Am. Chem. Soc., 1972, 94, 1340 CrossRef CAS; (b) X. Chen, K. R. Morris, U. J. Griesser, S. R. Byrn and J. G. Stowell, J. Am. Chem. Soc., 2002, 124, 15012 CrossRef CAS.
- Natural population charges of the acemetacin molecule were calculated in Gaussian using DFT methods at B3LYP/6-311G++ (d,p) level. http://www.gaussian.com..
- (a) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, OUP, Oxford, 1999 Search PubMed; (b) G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal Engineering: A Textbook, World Scientific Publishing, Singapore, 2011, pp. 36–37 Search PubMed.
- (a) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC; (b) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC.
- (a) T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324 RSC; (b) T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2006, 8, 448 RSC; (c) T. Gelbrich, D. S. Hughes, M. B. Hursthouse and T. L. Threlfall, CrystEngComm, 2008, 10, 1328 RSC.
- A. Burger and R. Ramberger, Mikrochim. Acta II, 1979, 259 Search PubMed.
- (a) L. Seton, D. Khamar, I. J. Bradshaw and G. A. Hutcheon, Cryst. Growth Des., 2010, 10, 3879 CrossRef CAS; (b) M. B. Pranzo, M. Coruzzi, M. R. Caira and R. Bettini, J. Pharm. Sci., 2010, 99, 3731 CAS.
Footnotes |
| † This paper is dedicated to Prof. Gautam R. Desiraju on his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: CIF files and hydrogen bonding of acemetacin polymorphs. CCDC 900783 and 902230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ce26534f |
| § Crystal data for acemetacin polymorphs: C21H18ClNO6, Form I: M = 415.81, colorless needle, 0.40 × 0.10 × 0.18 mm3, monoclinic, space group P21, a = 11.858(3), b = 4.8452(9), c = 16.785(3) Å, β = 102.47(2)°, V = 941.6(3) Å3, Z = 2, Dc = 1.467 g cm–3, Flack = 0.0(2), Mo Kα radiation, λ = 0.71073 Å, T = 298(2) K, 3561 reflections collected, 1298 unique. Final GoF = 0.998, R1 = 0.0882, wR2 = 0.1598. Form II: M = 415.81, monoclinic, space group P21/n, a = 22.5733 (13), b = 4.8945 (2), c = 18.3874 (9) Å, β = 108.519 (3)°, V = 1926.3 (2) Å3, Z = 2, Dc = 1.434 g cm–3, Cu Kα1 radiation, λ = 1.5405 Å, T = 298(2) K, Rp = 0.044, Rwp = 0.062, Rexp = 0.028, R(F2) = 0.0616, χ2 = 5.336. Single crystal data of form I was collected on an OXFORD Gemini CCD diffractometer and solved using SHELX-TL and OLEX2 package. The PXRD data for stable form II was collected on Bruker D8 Advance powder X-ray diffractometer in the 2θ range 5–100° and step size 0.02°. The data was collected for 7 h at 25 °C. The structure was solved by FOX and GSAS packages (see ESI‡). Structure solution and refinement gave the final CIF files. |
| ¶ Acemetacin was purchased from Dalian HongRiDongSheng Import & Export Co. Ltd., China, http://dlhongridongsheng.guidechem.com/ and used as such without any purification for all experiments described in this paper. Acemetacin purchased from Sigma-Aldrich, USA was found to be different from the China sourced material based on PXRD and FT-IR analysis. Acemetacin (Aldrich) did not match with known the polymorphs or hydrate of acemetacin by PXRD peak picking and DSC-TGA (Fig. S11, ESI‡). A complete characterization of the Aldrich material is pending. |
| This journal is © The Royal Society of Chemistry 2013 |