Structural diversity and luminescent properties of lanthanide 2,2- and 2,3-dimethylsuccinate frameworks†
Paul J.
Saines
a,
Mark
Steinmann
a,
Jin-Chong
Tan
ab,
Hamish H.-M.
Yeung
a and
Anthony K.
Cheetham
*a
aDepartment of Materials Science and Metallurgy, The University of Cambridge, Cambridge CB2 3QZ, United Kingdom. E-mail: akc30@cam.ac.uk; Fax: +44(0) 1223 334567; Tel: +44(0) 1223 767061
bDepartment of Engineering Science, University of Oxford, Parks Rd. Oxford OX1 3PJ, United Kingdom
First published on 19th September 2012
Abstract
The structures of fourteen new lanthanide frameworks (La, Ce, Eu, Tb, Y and Lu) containing the 2,2- or 2,3-dimethylsuccinate ligands are reported. While the majority of the known 2,2-dimethylsuccinate frameworks feature two dimensionally bonded layers, capped by hydrophobic methyl groups, several of these new frameworks adopt quite different architectures. These include one dimensional inorganically connected chains (La and Ce) with only non-covalent interactions in the other two dimensions, and three dimensional covalently bonded frameworks (Eu and Lu) with spaces in their structure to accommodate the bulky methyl groups. The new 2,3-dimethylsuccinate frameworks (La and Y) adopt three-dimensional covalently bonded frameworks. The factors affecting the formation of structures with different dimensionalities are examined and compared to previously reported transition metal frameworks. In addition, the sequence of phases formed with changing lanthanide size, concentrations and temperatures are rationalised. The luminescent properties of several 1- and 2-D frameworks doped with Eu and Tb are reported, with the Y host exhibiting the most intense emission.
1. Introduction
Inorganic-organic (hybrid) framework materials have attracted significant and sustained interest for over a decade due to their fascinating range of structures and wide variety of interesting properties.1,2 The structures of these hybrid materials are influenced significantly by both their cation and ligand building blocks, and they can adopt either porous or dense structures with covalent connectivity of different dimensions. Porous frameworks, often referred to as metal–organic frameworks (MOFs), are of significant interest due to their catalytic, gas storage and separation properties,2,3 while dense frameworks attract interest for their ability to exhibit cooperative behaviour more commonly associated with purely inorganic materials, such as magnetic order, semiconductivity and multiferroicity.4 Such frameworks can also exhibit a wide range of structures with different dimensionalities, having covalent connectivity in one, two or three dimensions; furthermore, this covalent connectivity can either be organic in nature (metal–ligand–metal) or inorganic (typically metal–O–metal).5Most studies of hybrid frameworks have focused on the synthesis of single crystals and powders with micron-sized particles, but significant attention has recently been drawn to the possibility of making nano-sized particles of these materials, principally as a means of incorporating them into thin films for technological applications.6 There have been a number of topical reports of exfoliation, via ultrasonication, of layered frameworks with weak intra-layer interactions. This “top–down” approach produces nanosheets that are only a few nanometres thick but have lateral dimensions on the order of microns.7,8–10 Dense frameworks with simple linear dicarboxylate ligands that feature bulky substituent groups appear to encourage the formation of such layered phases and are therefore a particularly versatile platform from which nanosheets can be exfoliated. A number of such compounds feature the 2,2-dimethylsuccinate (2,2-DMS) and 2,3-dimethylsuccinate (2,3-DMS) ligands in combination with either monovalent alkali or divalent transition metal cations.8–10 All 2,2-DMS frameworks have been found to adopt layered structures suitable for exfoliation. The 2,3-DMS compounds, however, appear to be a mixture of layered and three dimensionally covalently bonded structures, depending on whether chiral or meso-ligands are incorporated; this is due to the preference for these isomers to adopt gauche or trans-arrangements, respectively.
There have been a large number of studies of simple, linear dicarboxylate frameworks in which the various factors influencing the formation of different structures have been probed.8,9,11–13 The subtle role that the different DMS isomers play in directing the formation of framework structures with very different dimensionalities, however, highlights the need to further explore this interesting family of compounds. This will enable a better understanding of the effects of reaction conditions, ionic radius and charge on the formation of low dimensional structures. Additionally, the development of novel, layered frameworks that are suitable for exfoliation is an important area to on which to focus. The lanthanide 2,2-DMS and 2,3-DMS frameworks are of particular interest since they provide a ready means of examining the effect of ionic radii on the dimensionalities of the structure formed as well as a route towards frameworks with interesting luminescence behaviour. In this work, therefore, we explore the synthesis and structures of a number of lanthanide (Ln) 2,2-DMS and 2,3-DMS frameworks and report the luminescence properties of a number of doped frameworks with one- and two-dimensional covalent connectivity.
2. Experimental
The compounds examined in this work were made hydrothermally, from commercially available starting materials, in 23 mL Teflon lined Paar autoclaves heated at or below 200 °C for three days. In total it was possible to grow single crystals suitable for structure determination for 13 compounds. Synchrotron X-ray diffraction data were collected in order to determine the structure of framework 9, Lu2(C6H8O4)3(H2O)4, which is isostructural with one of the Y containing phases (see Table 1 for chemical formula and connectivity). The synthetic conditions used to produce the crystals used for structure determination and, where possible, pure phase samples, are described in the supplementary information alongside experimental details of the single crystal X-ray structure determinations. The crystallographic parameters of phases studied using single crystal X-ray diffraction are presented in Tables S1–S3, ESI† and the cation-anion bond distances of phases 1––––13 are listed in Tables S4–6, ESI.†| Structure | Formula | Connectivity | Ln coordination | O/Ln |
|---|---|---|---|---|
| 1 | La(C6H8O4)(C6H9O4)(H2O)2 | I1O0 | 10 | 10 |
| 2 | Ce(C6H8.5O4)2(H2O)2 | 11O0 | 10 | 10 |
| 3 | Ce2(C6H8O4)3(H2O)2 | I1O1 | 9 | 7 |
| 4 | Eu2(C6H8O4)3(H2O)3 | I0O3 | 8 | 7.5 |
| 5 | Tb2(C6H8O4)3(H2O)3 | I0O3 | 8 | 7.5 |
| 6 | Y2(C6H8O4)3(H2O)4·H2O | I0O2 | 8 | 8.5 |
| 7 | Y2(C6H8O4)3(H2O)4 | I0O2 | 8 | 8 |
| 8 | Y4(C6H8O4)6(H2O) | I0O2 | 6, 7, 8 | 6.25 |
| 9 | Lu2(C6H8O4)3(H2O)4 | I0O2 | 8 | 8 |
| 10 | Lu2(C6H8O4)3(H2O) | I1O1 | 8 | 6.5 |
| 11 | Lu3(C6H8O4)4(OH)(H2O)2 | I0O2 | 7, 8 | 6.3 |
| 12 | Lu3(C6H8O4)4(OH) | I0O3 | 7 | 5.7 |
| 13 | La2(C6H8O4)3(H2O)2 | I1O2 | 9 | 7 |
| 14 | Y2(C6H8O4)3 | I1O2 | 8 | 6 |
Powder X-ray diffraction (PXRD) patterns were collected of all samples prepared in this study using Cu Kα radiation on a Bruker D8 Advance diffractometer equipped with a position sensitive linear detector. To indicate the high level of purity to which samples of 1––6, 13 and 14 can be made, Le Bail fits to PXRD patterns of these frameworks are presented in Fig. S1–8, ESI.† Microanalysis results, obtained from the Department of Chemistry at the University of Cambridge, are presented in Table S7, ESI.† A synchrotron X-ray diffraction pattern of compound 9 was collected using beamline I11 at the Diamond Light Source UK.14 A wavelength of 0.827153(1) Å and the Mythen position sensitive detector were used, and the structure refined by use of the program GSAS with appropriate C–O and C–C bond distance and angle restraints to retain typical dicarboxylate geometry15 (see supplementary information for further details of the refinement, Fig. S9, ESI† for fit and Table S8, ESI† for crystallographic details).
Thermogravimetric analysis (TGA) of the bulk frameworks was performed in air on a TA Instruments Q500 using a heating rate of 10 °C min−1; experimental details and results from infra-red spectroscopy are described in the ESI.† Nitrogen sorption isotherms were measured at 77 K on a Micromeritics ASAP 2020 instrument. Samples were degassed in vacuum for at least 5 h at 100 °C before starting the gas uptake measurements. The surface area was estimated using the Brunauer–Emmett–Teller (BET) equation for the relative pressure range (P/P0) of 0.002 to 0.3; the saturation pressure, P0, corresponds to 103.88 kPa. Fluorescence spectra were measured using a custom made Horiba Fluorolog-3 fitted with a 450 W xenon lamp, a double monochromator in the emission channel, and a red-sensitive R928 photomultiplier tube detector.
3. Results and discussion
3.1. Synthesis and structures of the Ln 2,2-DMS frameworks
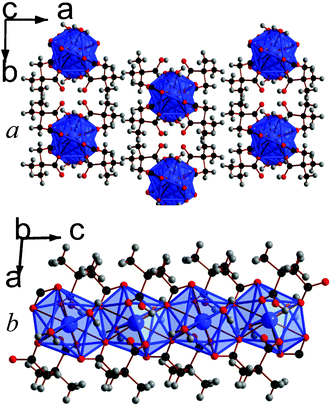 | ||
| Fig. 1 Crystal structure of 1 showing a) the arrangement of neighbouring chains and b) the chain of edge-sharing polyhedra. The La atoms and polyhedra are blue and the carbon, oxygen and hydrogen atoms are black, red and gray, respectively. | ||
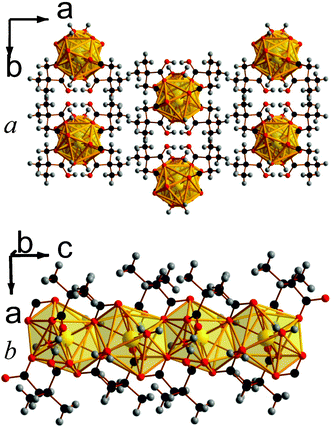 | ||
| Fig. 2 Crystal structure of 2 showing a) the arrangement of neighbouring chains and b) the chain of edge-sharing polyhedra. The Ce atoms and polyhedra are yellow and all other colours are as in Fig. 1. | ||
The structure of 3 features 1 nm thick covalently bonded layers that are hydrophobically capped by the methyl groups of 2,2-DMS ligands, typical for all the 2,2-DMS structures reported previously (see Fig. 3).9,10 The architecture of the layers of 3, however, is novel and features sinusoidal chains of CeO9 polyhedra, which are connected by alternating edge- and face-sharing connectivity. These chains are bridged by the backbone of the carboxylate groups at the points where neighbouring chains are closest, leading to I1O1 connectivity overall.5 The asymmetric unit of 3 has two Ce cations, three 2,2-DMS ligands and two water molecules, one of which is involved in the edge-sharing connectivity (see Fig. S12, ESI†). The two Ce cations bond to one and two water molecules and have bond valencies of 3.28 and 2.92, respectively.16 The backbones of all carboxylate ligands adopt the gauche arrangement and two of the 2,2-DMS ligands have 1212 connectivity while the third has (0111) connectivity.13 All of the aquo hydrogen atoms have intralayer hydrogen bonds with carboxylate oxygen atoms.
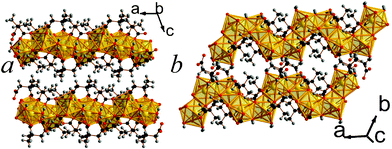 | ||
| Fig. 3 Crystal structure of 3 showing a) the arrangement of neighbouring layers and b) the layer architecture. The colours are as in Fig. 2. | ||
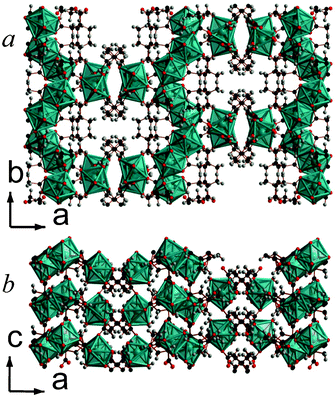
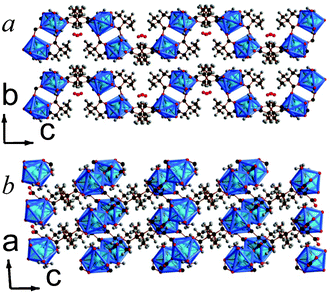 | ||
| Fig. 5 Crystal structure of 6 showing a) the arrangement of neighbouring layers and b) the layer architecture. Both components of the disordered 2,2-DMS ligand are displayed. The Y cations and YO8 polyhedra are blue and all other colours are as in Fig. 1. | ||
When reactions are carried out using higher concentrations of reagents and lower temperatures, framework 7, Y2(C6H8O4)3(H2O)4, forms along with compound 6. 7 is another example of a 2,2-DMS framework with covalently bonded layers capped by methyl groups, although these layers are more corrugated than found in frameworks 3 and 6 (see Fig. 6). Similarly to 6, the architecture of the layers features dimers of YO8 polyhedra bridged by two carboxylate groups; dimers within a layer are coupled through the backbone of the ligand, giving rise to I0O2 connectivity.5 The arrangement of the polyhedra and 2,2-DMS ligand within a layer are, however, subtly different, such that the pores found in 6 do not exist in 7. Consequently the asymmetric unit of 7 features one Y cation, two coordinated water molecules, and one and a half 2,2-DMS ligands (the methyl groups on the half ligand are disordered with 50% occupancy, see Fig. S16, ESI†). The Y cations bond to two water molecules, with the remainder of its coordination completed by carboxylate oxygen atoms; they have a bond valency of 3.18.16 The 2,2-DMS ligands both have (1111) connectivity to Y cations and their carbon backbones adopt arrangements close to gauche and trans, the latter being required by the symmetry of the structure.13 Each of the hydrogen atoms from the water molecules has one hydrogen bond with an oxygen atom from a nearby carboxylate group. An isostructural Er phase has previously been reported by Bernini et al.,17 although they suggest it adopts non-centrosymmetric P1 symmetry in which the methyl groups of the ligand that adopts the trans-structure are not disordered. In the case of the Y compound, however, attempts to solve the structure in the non-centrosymmetric space group indicated that there is equivalent electron density in all four possible positions for the methyl carbon atoms, suggesting that the disorder is genuine, at least in the case of the Y framework.
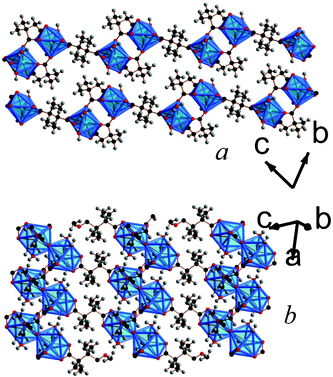 | ||
| Fig. 6 Crystal structure of 7 showing a) the arrangement of neighbouring layers and b) the layer architecture. Both components of the disordered methyl substituents are displayed. The colours are the same as Fig. 5. | ||
Reactions carried out with higher concentrations and elevated temperatures lead to the formation of a mixture of frameworks 6 and 8, Y4(C6H8O4)6(H2O). 8 is another compound that features hydrophobically capped covalently bonded layers that only interact with each other through van der Waals forces (see Fig. 7). Similarly to 7, it does not feature any pore solvent but the surfaces of its layers are somewhat smoother. The architecture of the layers of 8 are quite complex, consisting of tetramers of edge-sharing YO8 polyhedra with the outer polyhedra sharing corners with YO7 polyhedra, giving rise to inorganically connected discrete chains of six YOx polyhedra. The YO7 polyhedra are also connected to two YO6 octahedra, via one and three carboxylate groups. It is through these linkages that neighbouring chains are connected, giving rise to a structure with I0O2 connectivity overall.5 To accommodate this structural complexity, the asymmetric unit of 8 features four distinct Y cations, six distinct carboxylate groups, and one water molecule that coordinates to one of the YO8 polyhedra (see Fig. S17, ESI†). The Y cations all have bond valencies between 3.11 and 3.30 and the backbone of the 2,2-DMS ligands adopt an arrangement close to gauche.16 The oxygen atoms in the carboxylate ligand adopt different coordination modes to the Y cations, with three having (1111) connectivity, two having (1112) connectivity, and the remaining ligand adopting (1212) connectivity.13 All of the hydrogen bonds from the hydrogen atoms in the water molecule are with oxygen atoms from the carboxylate groups.
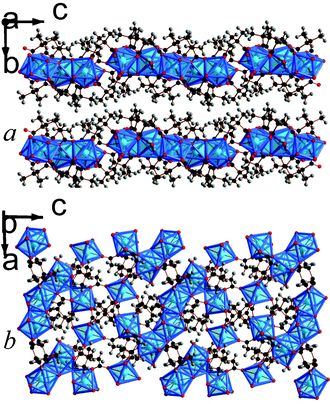 | ||
| Fig. 7 Crystal structure of 8 showing a) the arrangement of neighbouring layers and b) the layer architecture. The colours are the same as Fig. 5. | ||
Increasing the temperature to 180 °C leads to the formation of a two phase mixture of frameworks 10, Lu2(C6H8O4)3(H2O), and 11, Lu3(C6H8O4)4(OH)(H2O)2, and increasing the reaction temperature further to 200 °C leads to the formation of 12, Lu3(C6H8O4)4(OH), instead of 10. Attempts were made to alter the conditions and temperatures to obtain each of these phases in pure form. This did not fundamentally change the reaction mixtures obtained but merely confirmed that 10 and 12 form sequentially, alongside 11, as temperature is increased above 150 °C. Similarly to 9, frameworks 10 and 11 both feature covalently bonded layers capped with methyl groups with only van der Waals interactions acting between layers, although their layers are smoother. (see Fig. 8 and 9). The layer architecture of 10 features edge-sharing dimers of LuO8 polyhedra, which are connected to neighbouring dimers by their corners to form a sinusoidal shaped chain. Neighbouring chains are bridged by the backbones of the dicarboxylate ligands giving rise to a structure with I1O1 connectivity.5 It is isostructural to an Er phase that was previously reported by Bernini et al.,17 and the asymmetric unit of 10 features two Lu cations, three 2,2-DMS ligands, and a water molecule that coordinates to one of the Lu cations (see Fig. S18, ESI†). The Lu cations have bond valencies of 3.16 and 3.21, the latter being the Lu cation that bonds to the water molecule.16 One of the 2,2-DMS ligands has a carbon backbone that adopts gauche-geometry. The other two have intermediate arrangements and are the first two ligands in the structures reported in this study to have torsion angles more than 15° away from an ideal gauche or trans-arrangement (77.8(6)° and 83.4(6)°). The ligand adopting a gauche-like arrangement adopts (1212) connectivity while other two ligands adopt (1112) and (1111) type-arrangements.13 The aquo-hydrogen form hydrogen bonds with oxygen atoms from the carboxylate groups.
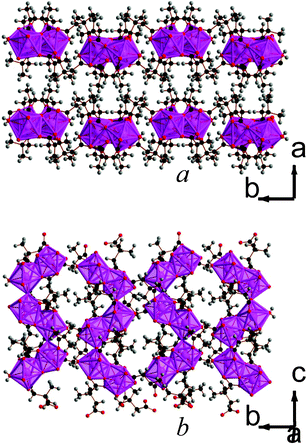 | ||
| Fig. 8 Structure of compound 10 showing a) the arrangement of neighbouring layers and b) the architecture of an individual layer, highlighting the sinusoidal inorganic chains. The Lu cations and LuO8 polyhedra are pink and the other colours are as in Fig. 1. | ||
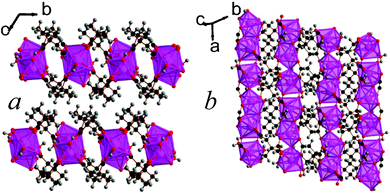 | ||
| Fig. 9 The structure of framework 11 showing a) the arrangement of neighbouring layers and b) the architecture of an individual layer. The colours are the same as in Fig. 8. | ||
The asymmetric unit of 11 features three Lu cations, four 2,2-DMS ligands, two water molecules and a hydroxide group (see Fig. S19, ESI†). The three Lu are connected inorganically into trimers with each Lu1O7 polyhedra connected to a Lu2O7 polyhedra via a corner, and Lu2O7 and Lu3O8 polyhedra sharing edges. The closest oxygen to the Lu2O7 polyhedra that is bonded to Lu1 but not Lu2 is disordered over two sites that are very close to each other and are each fixed at 50% occupancy. This disorder along with the displacement parameter of the carbon atom in the carboxylate group suggests that there may be some rotation of the Lu1O7 polyhedra towards that of the Lu2O7 groups. These LuOx trimers are connected into rows along the a-axis by two carboxylate groups with connectivity within a layer being completed through the backbone of the dicarboxylate ligand, giving rise to a structure that has I0O2 connectivity overall.5
In 11, Lu1 coordinates to both water molecules, the hydroxide anion, and four oxygen atoms from different carboxylate groups; Lu2 bonds on one hydroxide anion and six oxygens from five different carboxylate groups, and Lu3 bonds only to oxygen atoms from carboxylate groups. The Lu cations have bond valencies of 3.19, 3.09 and 3.17 for Lu1-3, respectively.16 The coordinated water and hydroxide groups bond to one and two Lu cations, respectively, and the 2,2-DMS ligands, which all adopt arrangements that are close to gauche, either adopt (1111) or (1112) connectivity, with two of each type of coordination found in the structure.13 Three of the four hydrogen atoms from the water molecules have hydrogen bonds with carboxylate oxygen atoms and the fourth has one with an oxygen atom from the crystallographically identical water molecule.
12 is the second 2,2-DMS framework architecture to feature three dimensional covalent connectivity. Like compounds 4 and 5, 12 has I0O3 connectivity.5 It features trimers of LuO7, which all bond to a central hydroxide group, and two of the polyhedra in a trimer share edges with the third (see Fig. 10). The trimers are connected to each other along the a- and c-axis via carboxylate groups and the offset of trimers by half a unit cell down the c-axis creates space between a grid of four trimers that accommodates the bulky methyl groups (see Fig. 10a). The asymmetric unit of 12 features three Lu cations, four 2,2-DMS ligands, and a single hydroxide group (see Fig. S20, ESI†). The bond valencies of the Lu cations are all between 3.15 and 3.21 and the 2,2-DMS ligands, whose carbon backbones all adopt an arrangement close to gauche, either bond to Lu cations in a (1111) or (1112) fashion, with half adopting each configuration.13,16 The hydrogen from the hydroxide anion has hydrogen bonds with a carboxylate oxygen from a neighbouring trimer (Odonor–Oacceptor distance of 2.974(7) Å).
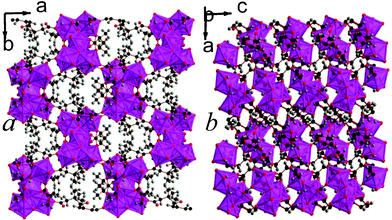 | ||
| Fig. 10 The structure of framework 12 viewed in the a) ab and b) bc plane. The colours are the same as in Fig. 8. | ||
3.2. Synthesis and structures of the Ln 2,3-DMS frameworks
Both of the frameworks synthesized with the 2,3-DMS ligands feature three dimensional covalent connectivity, although this may be related to the apparent necessity to use high temperatures to obtain crystals suitable for structure determinations in these La and Y frameworks. Both frameworks can also be made from either a mixture of 2,3-DMS isomers or using the pure meso-ligand. Framework 13, La2(C6H8O4)3(H2O)2, features corner sharing chains of LaO9 polyhedra aligned along the c-axis (see Fig. 11). Neighbouring chains are connected along the b-axis through three atom carboxylate groups bridges and along the a-axis by the backbone of the 2,3-DMS ligands, leading to a structure with I1O2 connectivity.5 Viewed down the c-axis there are hexagonal pores, which are occupied by the methyl groups, enabling them to be incorporated into a phase with three-dimensional covalent connectivity. These pores, however, unlike those in other three dimensional 2,2-DMS and 2,3-DMS frameworks, are not fully occupied, leading to a structure featuring 4.2% solvent accessible voids according to the SOLV routine in the program PLATON.18 Nitrogen sorption measurements indicate a surface area of only 1.6 m2 g−1, possibly due to the tortuous nature of the cavity which renders the pores inaccessible at atmospheric pressures (see Fig. S21, ESI†).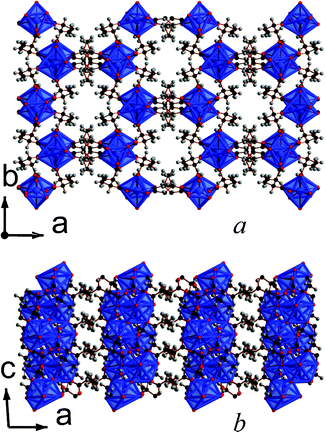 | ||
| Fig. 11 The structure of 13 showing the a) ab and b) ac plane. The colours are the same as in Fig. 1 and both components of the disorder ligand are displayed. | ||
The asymmetric unit of 13 has one La cation, one and a half 2,3-DMS ligands, and one water molecule that coordinates to one La (see Fig. S22, ESI†). The 2,3-DMS ligand, whose two halves are crystallographically identical, provides the connectivity along the a-axis and is disordered over two sites. Both the 2,3-DMS ligands in the structure adopt the meso-isomer, but the backbone of the disordered ligand adopts a trans-configuration and features (1212) connectivity to the La cations; the other distinct ligand adopts a gauche-configuration and has 1112 connectivity.13 The La cations have bond valencies of 3.19 and the hydrogen atoms from the water molecule each have a hydrogen bond with a nearby carboxylate group.16
Framework 14 is the only phase found in the present study where the dicarboxylate ligand is the only ligand present. Y2(C6H8O4)3 features chains of edge-sharing YO8 polyhedra, which are bridged in the other two dimensions via the backbone of the ligand, giving rise to a structure with I1O2 connectivity overall (see Fig. 12).5 The YO8 chains are arranged into a square array with space inside the square to accommodate the methyl groups of the ligands. The asymmetric unit of 14 includes three Y sites, two of which are on special positions, and three ligands, all of which are disordered over two sites with approximately half occupancy of each component present. The high apparent level of disorder in the structure means that the details of the structure must be examined carefully, but two of the three ligands clearly coordinate to Y in an (1112) fashion, while the third adopts a (1212) arrangement.13 All the ligands are the meso-isomer with carbon backbones close to a trans-arrangement.
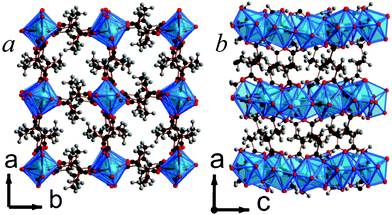 | ||
| Fig. 12 The structure of 14 illustrating a) the ab and b) the ac planes. The colours are the same as Fig. 5 and, for clarity, only half of the disordered ligands are shown. | ||
3.3. Structural trends in the Ln 2,2-DMS and 2,3-DMS frameworks
There are a number of significant changes in the structures adopted by the Ln frameworks with changing cation size, synthesis temperature and reagent concentration. All previously reported 2,2-DMS phases have adopted layered structures, which is attributed to the methyl groups always being on the same side and closer together in this isomer, making for formation of such structures energetically favourable.8–10 In this study, however, a wider range of dimensionalities is reported for the 2,2-DMS phases, although the geometry of the carboxylate groups of the ligand is generally close to a gauche-geometry where it is not required by symmetry to be trans.The ionic radii of the lanthanide cation clearly plays a significant role in the dimensionality of the structure adopted, although the trend is not as simple as with some alkaline earth frameworks, such as those formed by the thiazolothiazole dicarboxylates reported by Falcão et al.19 The 10-fold coordination spheres found with the largest Ln cations, La and Ce, form frameworks with two 2,2-DMS ligands per cation, with one of these ligands retaining its acidic proton. This leads to the formation of the first 2,2-DMS frameworks with one-dimensional covalent connectivity, which is necessary to accommodate the additional methyl groups. The second Ce phase, 3, features 9 coordinate cations, and as the lanthanide ionic radius decreases 8-fold coordination becomes dominant. Several of the Y and Lu phases have cations with 6- and 7-fold coordination, consistent with trends observed amongst the LnCl3·xH2O and some lanthanide dicarboxylate frameworks.20 Structures 3–12 have a maximum of 1.5 ligands per cation, reducing the number of methyl groups that need to be accommodated in the structure; in most cases this results in the anticipated two-dimensional layered structures.8–10 The intermediate sized Ln, Eu and Tb, however, adopt 2,2-DMS structures with three dimensional covalent connectivity. As with 13 (see below), the formation of architectures where their larger polyhedra are arranged with three-dimensional connectivity creates spaces that are suitable for accommodating the methyl groups.
Almost all of the 2,2-DMS structures featuring the smaller Y and Lu cations have two-dimensional covalent connectivity, although the architectures of their layers are quite distinct. In the Y case, low reagent concentrations lead to the formation of 6, which features pore water within a two dimensional framework. As might be expected, however, increasing the reagent concentrations results in phases from which the pore water is eliminated. Structure 8 has higher condensation, as measured by its lower O/Ln ratio, and is more dehydrated than 7. Its formation at higher temperatures is therefore consistent with previous studies of succinate-related frameworks.9,11
Five Lu 2,2-DMS phases were observed during this study and the trends among them are quite complex. The lowest temperature phase is a Lu analogue of 6, which forms in combination with 9, a Lu analogue of 7. The fraction of 9 in the mixtures increases with increasing temperature, as expected for the more dehydrated phase 9. Above 150 °C, however, 10, 11 and 12 form with increasing temperature; all of these phases are significantly more dehydrated and condensed than 9. Bernini et al.17 have previously suggested that the Er analogue of 10 is more thermodynamically stable than the corresponding analogue of 9, with the latter being favoured kinetically. In the present work, this is consistent with 10 forming at higher temperatures than 9. The similar degree of condensation in 10, 11 and 12 may be related to issues encountered in obtaining these phases in pure form. Condensation increases in the order that these three phases appear, consistent with expected behaviour with increasing temperature. The overall connectivity of the anhydrous, high temperature phase, 12, is three dimensional, while all the other Lu phases are two dimensional and hydrated, consistent with previously reported studies.9,11 It should, however, be noted that 10 is the only Lu phase that has any inorganic connectivity. In addition, it has fewer water molecules in its structure than we find in 11 (1/2 per cation compared to 2/3), despite preferring to form at lower temperatures. This is the opposite of the behaviour expected from previous studies, but seems to be compensated by the inclusion of a hydroxide ligand in 11, which is not found in 10; this ensures that 11 has slightly higher condensation overall, along with higher density.9,11 It is likely that the overall O/Ln ratio (Table 1) is a more accurate insight into which phases will form with increasing temperature in cases where the extent of hydration is similar.
While the three dimensional structures formed by compounds 13 and 14 are consistent with those formed by the other meso-2,3-DMS frameworks synthesized to date, 13 does this despite two thirds of its ligands adopting gauche-arrangements.8 Amongst the transition metal 2,2-DMS frameworks, gauche-geometry was only adopted by the D- and L- ligands, which prefer to form two dimensional, covalently bonded structures. The meso-ligand, however, always adopts the trans-geometry in the transition metal systems. 13 defies this trend by forming a 3-D structure which contains both conformations, suggesting that the larger lanthanide cations can form structures with larger voids that are suitable for accommodating methyl groups on the same side of the ligand. This enables three dimensional connectivity and gauche-arrangements to be simultaneously achieved.
3.4. Thermal stabilities of the 2,2-DMS and 2,3-DMS phases
The thermal stabilities of the frameworks that could be obtained with high purity were examined using thermogravimetric analysis (TGA). The results from this are summarized in Table 2, with the details of their thermal stabilities and decomposition being presented in Fig. S23–31, ESI.† The first noticeable result is that the 1-D frameworks, compounds 1 and 2, lose their coordinated water at lower temperatures and then begin to decompose at much lower temperatures than the other compounds examined. Interestingly, particularly in the case of the La compound, it seems that one of the 2,2-DMS ligands begins to decompose before the other. Compounds 6 and 13 also lose their water molecules at lower temperatures than the other compounds, which is likely due to the presence of pore water in 6 and the elongated bond distance between the water molecule and the La cation in 13. Framework decomposition, excluding any rearrangement caused by the loss of coordinated water, occurs for most two and three dimensional compounds at around 350 °C. However, 9 appears to be slightly more stable, only beginning to decompose at 380 °C, while 3 and 13 decompose at significantly lower temperatures than this. The particularly low decomposition temperature of 3 is likely related to the loss of the coordinated water at lower temperatures, which plays an important role in the inorganic connectivity in this structure.| Compound | Connectivity | Water liberated (°C) | Framework decomposition (°C) |
|---|---|---|---|
| 1 | I1O0 | 100 | 150 |
| 2 | 11O0 | 80 | 150 |
| 3 | I1O1 | 150 | 280 |
| 4 | I0O3 | 150 | 350 |
| 5 | I0O3 | 140 | 350 |
| 6 | I0O2 | 110 | 350 |
| 9 | I0O2 | 130 | 380 |
| 13 | I1O2 | 110 | 320 |
| 14 | I1O2 | N/A | 360 |
3.5. Luminescence properties
Compounds 1, 6 and 9 were doped with Ce3+, Eu3+ and Tb3+ to investigate their potential suitability as low dimensional, florescence materials. Ce doping did not result in any significant fluorescent behaviour, and nominal doping with Eu and Tb above 3% led to the formation of phases isostructural with the three dimensional architectures, 4 and 5. Only samples of 1, 6 and 9 doped with 3% nominal Eu or Tb were characterized further as they retained pure phases with one or two dimensional architectures that can potentially be made into fluorescent nanosheets or chains.Florescence measurements of the samples doped with a nominal percentage of 3% Eu3+ revealed that the most efficient excitation wavelength above 350 nm for all samples is at 394 nm, which excites a transition from the 7F0 ground state to the 5D3 (see Fig. 13).21 Non-radiative, energy transfer to the 5D0 state via multi-phonon relaxation then occurs, with subsequent sharp emission from the 5D0 level to the 7FJ, J = 0, 1, 2, 3 and 4, states. The strongest emission is to the 7F2 state, which results in the red emission at around 615–620 nm. The emission from the doped sample of 6 is strongest and is clearly perceptible to the naked eye. The sample of 9 has about half the emission intensity of 6, and the doped sample of 1 emits only very weakly with about a quarter of the intensity of 6. The excitation of these samples with lower wavelength radiation does not qualitatively change the emission spectra, showing that emission occurs from the 5D0 level regardless of the energy level to which the Eu is initially excited.
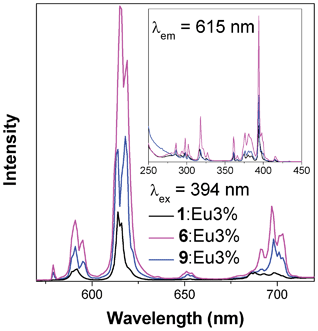 | ||
| Fig. 13 Emission (main) and excitation (insert) spectra of samples of compounds 1, 6 and 9 doped with nominal 3% Eu. | ||
378 nm excitation of samples of 1, 6 and 9 doped with 3% nominal Tb3+ leads to an excitation from the 7F6 ground state of Tb to the 5D3 state (see Fig. 14).21 Subsequent emission occurs from the 5D4 state to the 7FJ, J = 6, 5, 4 and 3 states, with distinct green luminescence to the 7F5 level being observed at 544 nm. As with the case of Eu doping, emission is strongest from the doped sample of 6, with the emission of 9 being slightly over 50% of that seen from 6 and 1 emitting approximately half that of 9. Excitation with higher energy ultraviolet radiation does not qualitatively change the spectra, indicating that all emission occurs from the 5D4 state with any additional energy again lost via multi-phonon relaxation.
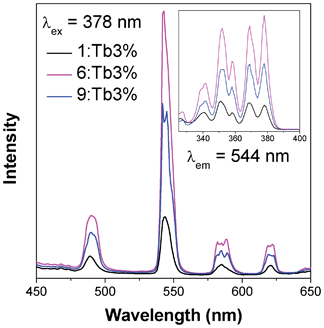 | ||
| Fig. 14 Emission (main) and excitation (insert) spectra of samples of compounds 1, 6 and 9 doped with nominal 3% Tb. | ||
4. Conclusions
This work reports the synthesis and structures of fourteen new Ln 2,2-DMS and 2,3-DMS frameworks, and shows that the Ln compounds exhibit more structural diversity than is encountered amongst the analogous transition metal hybrids. While both the 2,3-DMS frameworks are three dimensional, as expected for frameworks incorporating the meso-isomer, the 2,2-DMS frameworks exhibit covalent bonding in one, two or three dimensions.In contrast to the clear correlation between cation radius and inorganic connectivity that has been seen in alkaline earth frameworks,19 the trends in the Ln-DMS systems are more complex. The 2,2-DMS frameworks containing the largest lanthanides form structures with one-dimensional inorganically connected chains and inclusion of two DMS ligands per metal. As the ionic radius of the lanthanides decreases the coordination number of the lanthanide also decreases significantly and the ligand to metal ratio reduces to 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. These frameworks form structures with two- or three-dimensional covalent connectivity, with the lanthanides with intermediate ionic radii favouring the latter. The compounds featuring the smallest cations, Y and Lu, appear to exhibit more structural diversity, despite predominantly adopting two dimensional layered structures; increasing reactant concentrations and temperature favour more condensed and, generally, more dehydrated structures. Samples of 1, 6 and 9 doped with Eu3+ and Tb3+ at the 3% level were also made. These exhibit characteristic red and green emissions, respectively, which was strongest in all cases for compound 6.
:1. These frameworks form structures with two- or three-dimensional covalent connectivity, with the lanthanides with intermediate ionic radii favouring the latter. The compounds featuring the smallest cations, Y and Lu, appear to exhibit more structural diversity, despite predominantly adopting two dimensional layered structures; increasing reactant concentrations and temperature favour more condensed and, generally, more dehydrated structures. Samples of 1, 6 and 9 doped with Eu3+ and Tb3+ at the 3% level were also made. These exhibit characteristic red and green emissions, respectively, which was strongest in all cases for compound 6.
Acknowledgements
The authors would like to thank the European Research Council for financial support. We would like to thank Diamond Light Source for access to Beamline I11, which, contributed to the results presented here and to Alistair Lennie and Stephen Thompson for their assistance in the use of the beamline. P.J.S. would like to thank Jack Clegg for his advice with regards to disorder modelling.References
- (a) A. K. Cheetham and C. N. R. Rao, Science, 2007, 318, 58–59 CrossRef CAS; (b) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC; (c) J. C. Tan and A. K. Cheetham, Chem. Soc. Rev., 2011, 40, 1059–1080 RSC.
- (a) A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491–2510 CrossRef CAS; (b) G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- (a) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (b) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- (a) C. N. R. Rao, A. K. Cheetham and A. Thirumurugan, J. Phys.: Condens. Matter, 2008, 20, 083202 CrossRef; (b) P. J. Saines, J. R. Hester and A. K. Cheetham, Phys. Rev. B, 2010, 82, 144435 CrossRef; (c) P. J. Saines, B. C. Melot, R. Seshadri and A. K. Cheetham, Chem.–Eur. J., 2010, 16, 7579–7585 CrossRef CAS; (d) P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS; (e) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- (a) L.-G. Qiu, Z.-Q. Li, Y. Wu, W. Wang, T. Xu and X. Jiang, Chem. Commun., 2008, 3642–3644 RSC; (b) W. Lin, W. J. Rieter and K. M. L. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650–658 CrossRef CAS; (c) A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218–1227 RSC; (d) B. W. Jacobs, R. J. T. Houk, M. R. Anstey, S. D. House, I. M. Robertson, A. A. Talin and M. D. Allendorf, Chem. Sci., 2011, 2, 411–416 RSC; (e) X. Zhang, M. A. Ballem, Z.-J. Hu, P. Bergman and K. Uvdal, Angew. Chem., Int. Ed., 2011, 50, 5729–5733 CrossRef CAS; (f) A. Sachse, R. Ameloot, B. Coq, F. Fajula, B. Coasne, D. De Vos and A. Galarneau, Chem. Commun., 2012, 48, 4749–4751 RSC.
- (a) P. Amo-Ochoa, L. Welte, R. Gonzalez-Prieto, P. J. Sanz Miguel, C. J. Gomez-Garcia, E. Mateo-Marti, S. Delgado, J. Gomez-Herrero and F. Zamora, Chem. Commun., 2010, 46, 3262–3264 RSC; (b) P.-Z. Li, Y. Maeda and Q. Xu, Chem. Commun., 2011, 47, 8436–8438 RSC.
- P. J. Saines, M. Steinmann, J.-C. Tan, W. Li, P. T. Barton and A. K. Cheetham, J. Am. Chem. Soc.. Search PubMed Submitted.
- P. J. Saines, J.-C. Tan, H. H.-M. Yeung, P. T. Barton and A. K. Cheetham, Dalton Trans., 2012, 41, 8585–8593 RSC.
- J.-C. Tan, P. J. Saines, E. G. Bithell and A. K. Cheetham, ACS Nano, 2012, 6, 615–621 CrossRef CAS.
- (a) P. M. Forster, A. R. Burbank, C. Livage, G. Férey and A. K. Cheetham, Chem. Commun., 2004, 368–369 RSC; (b) P. M. Forster, N. Stock and A. K. Cheetham, Angew. Chem., Int. Ed., 2005, 44, 7608–7611 CrossRef CAS.
- C. Livage, P. M. Forster, N. Guillou, M. M. Tafoya, A. K. Cheetham and G. Férey, Angew. Chem., Int. Ed., 2007, 46, 5877–5879 CrossRef CAS.
- (a) P. J. Saines, P. Jain and A. K. Cheetham, Chem. Sci., 2011, 2, 1929–1939 RSC; (b) P. J. Saines, P. T. Barton, P. Jain and A. K. Cheetham, CrystEngComm, 2012, 14, 2711–2720 RSC.
- (a) C. C. Tang, S. P. Thompson, T. P. Hill, G. R. Wilkin and U. H. Wagner, Z. Kristallogr., Suppl., 2007, 26, 153–158 CrossRef; (b) S. P. Thompson, J. E. Parker, J. Potter, T. P. Hill, A. Birt, T. M. Cobb, F. Yuan and C. C. Tang, Rev. Sci. Instrum., 2009, 80, 075107 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Strucuture Analysis System (GSAS), Los Alamos National Laboratory, Los Alamos, 1994 Search PubMed.
- (a) I. D. Brown and D. Altermatt, Acta Crystallogr., 1985, B41, 244–247 CAS; (b) N. E. Brese and M. O'Keeffe, Acta Crystallogr., 1991, B47, 192–197 CAS.
- M. C. Bernini, V. A. de la Peña-O'Shea, M. Iglesias, N. Snejko, E. Gutierrez-Puebla, E. V. Brusau, G. E. Narda, F. Illas and M. Á. Monge, Inorg. Chem., 2010, 49, 5063–5071 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- E. H. L. Falcão Naraso, R. K. Feller, G. Wu, F. Wudl and A. K. Cheetham, Inorg. Chem., 2008, 47, 8336–8342 CrossRef.
- (a) A. Thirumurugan and S. Natarajan, Eur. J. Inorg. Chem., 2004, 762–770 CrossRef CAS; (b) D.-X. Hu, F. Luo, Y.-X. Che and J.-M. Zheng, Cryst. Growth Des., 2007, 7, 1733–1737 CrossRef CAS.
- M. D. Chambers and D. R. Clarke, Annu. Rev. Mater. Sci., 2009, 39, 325–359 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, single crystal X-ray diffraction experimental and crystallographic information, including CIF files, results from microanalysis, infrared and detailed thermogravimetric analysis results and figures of powder X-ray diffraction patterns, assymetric units, sorption results and thermogravimetric analysis. CIFs for structures 1–8 and 10–14 have been deposited with the CCDC with deposit numbers 895574–895586. See DOI: 10.1039/c2ce26279g |
| This journal is © The Royal Society of Chemistry 2013 |