DOI:
10.1039/C2CE26072G
(Paper)
CrystEngComm, 2013,
15, 78-85
Discrepant gas adsorption in isostructural heterometallic coordination polymers: strong dependence of metal identity†
Received
5th July 2012
, Accepted 13th September 2012
First published on 14th September 2012
Abstract
Two isostructural porous 3d–4d materials [M(cyclam)Pd(2,4-pydc)2·4H2O]n (2,4-H2pydc = pyridine-2,4-dicarboxylate acid) (1, M = Ni and 2, M = Zn) and five other isostructural complexes {[M′(cyclam)]2[M(2,5-pydc)3]·xH2O}n (2,5-H2pydc = pyridine-2,5-dicarboxylate acid) (3, M = Co, M′ = Ni; 4, M = M′ = Ni; 5, M = Zn, M′ = Ni; 6, M = Ni, M′ = Zn and 7, M = M′ = Zn) were synthesized. Both complexes 1′ and 2′ show high selective CO2 capture over CH4 and N2. The comparisons of different gas sorption properties between 1′ and 2′ strongly suggest that metal identity has a great influence on the polarity of frameworks, but just the polar molecules respond to the polarity changes of different frameworks. The comparisons of CO2 sorption properties between five other isostructural complexes 3′–7′ also strongly suggest that metal identity has great influence on polarity of frameworks and further the CO2 uptake.
Introduction
In order to reduce air pollution and therefore improve human health, our society pays increasing attention to the capture and separation of greenhouse gas – CO2.1 Compared with traditional porous materials such as carbon nanostructures, zeolites and mesoporous silica, porous coordination polymers (PCPs) represent a new class of crystallized porous materials, and they have exhibited great potential for gas storage and gas separation.2 Because CO2 capture and separation are mainly based on adsorbate–surface interactions between CO2 and frameworks, great efforts thus have been made to synthesize new PCPs with enhanced CO2 affinity.3 One of the ongoing endeavors to effectively study the mechanism for gas capture and adsorptive gas separation is to systematically compare isostructural PCPs with various building units (organic linkers or metal ions).4,5 Isostructural coordination polymers with the same metals but different organic linkers are easily obtained, and gas sorption or separation property mutation induced by the different organic linkers have been studied thoroughly.4 Nevertheless, metal ions, as the cores of coordination complexes, have a significant effect on their physical and chemical properties (for example, photoluminescence,6 catalysis7 and gas adsorption5). Moreover, due to the uncontrollable coordination chemistry of metal ions in the synthetic reaction, isostructural coordination polymers with different metals are rare. Due to their different quadrupole moment and polarizability of gases, selective capture of one specific gas will mainly effected by the interaction between the gas and frameworks.8 To our knowledge, there has been no previous report that unambiguously studies the gas adsorption properties of various polar frameworks via metal substitution.
The chemical attributes of organic ligands, such as coordination ability, geometry and relative orientation of the donor groups, play a very important role in constructing polymer framework topology. Perhaps the most ubiquitous functional groups used for the synthesis of coordination polymers are the pyridyl group9 and carboxylic acid.10 It is worth stressing that the coexistence of pyridyl and carboxylate groups (dual functionality) imparts these ligands with special networking abilities.11 Especially, the presence of the pyridyl donor and the carboxylate group in the adjacent position (i.e. pyridine-2,n-dicarboxylate and pyrazine-2-carboxylate) furnishes an additional functionality, the chelating effect, which enables these ligands to be used for constructing stable metallotectons that can further coordinate to other metal centers.12 These metallotectons have two kinds of Lewis-base coordination groups: 2-carboxylate (group B) and n-carboxylate (n = 3, 4 or 5; group A) moieties (Scheme 1). Group A has a stronger bridging capability than group B because of its strong electron-donating and electrostatic power. Using these metallotectons has resulted in the formation of a very large number of heterometallic coordination polymers with intriguing structures and functional properties.13 In the assembly of coordination polymer networks, utilization of a macrocyclic complex [for example, M(cyclam)(ClO4)2] as a metal building block will simplify the coordination chemistry of metal ions and make extending directions of the network easily controllable. Especially, following change of metal ions embedded in the macrocyclic ligand, the isostructural complexes with different metals will be easily obtained. Furthermore, macrocyclic complexes are able to prevent the interpenetration of networks due to their bulkiness.14 Thus, we mainly focused on using macrocyclic complexes as metal source for the construction of TMCPs.
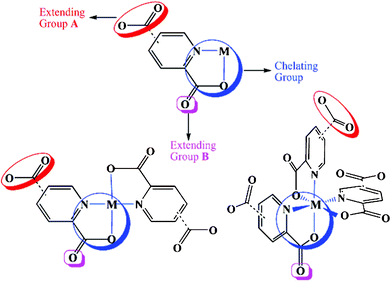 |
| | Scheme 1 Observed coordination modes of (2,n-pydc)–metal complexes and polymers. | |
Experimental
Materials and physical measurements
All reagents and solvents employed were commercially available and used as supplied without further purification. Powder XRD patterns were recorded with Cu Kα radiation by using a PANalytical X'Pert PRO diffractometer. TGA analyses of the complexes were performed on a NETZSCH STA 409 PC Simultaneous Thermal Analyzer under an N2 atmosphere at a scan rate of 10 °C min−1. Elemental analysis was performed on an Elementar vario EL III analyzer. IR (KBr) spectra were recorded on the Nicolet FT-IR spectrophotometer. All the gas sorption experiments were measured with an IGA-002 gas adsorption instrument.
Synthesis
[Zn(cyclam)Pd(2,4-pydc)2·4H2O]n2. Pd(2,4-Hpydc)2 (1.00 mmol, 0.44 g) was added to an aqueous solution of NaOH (2.00 mmol, 0.08 g). After stirring for half an hour, an aqueous solution of [Zn(cyclam)](ClO4)2 (1.00 mmol, 0.46 g) was slowly added. A colourless precipitate was formed immediately. The mixture was stirred for a further 2 h to ensure completion of the reaction. The solid was collected by filtration, washed with H2O and THF, and dried in air to afford a pale yellow powder. A solution of [Zn(cyclam)](ClO4)2 (0.1 mmol) in H2O (2.5 mL) was carefully layered on the top of a solution of [Pd(2,4-Napydca)2)] (0.1 mmol) in H2O–ethylene glycol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2.5 mL). Colourless needle-shaped crystals suitable for X-ray crystallographic analysis were obtained after few days. Yield: 93%. Anal. Calcd for C24H38N6O12ZnPd: C, 37.22; H, 4.95; O, 24.79. Found: C, 37.15; H, 5.03; O, 24.86. IR (KBr, cm−1): 3451 (m), 3215 (w), 1664 (s), 1624 (w), 1382 (s), 1316 (s), 1100 (s), 930 (m), 871 (w), 778 (w), 746 (w), 688 (w).
:1, 2.5 mL). Colourless needle-shaped crystals suitable for X-ray crystallographic analysis were obtained after few days. Yield: 93%. Anal. Calcd for C24H38N6O12ZnPd: C, 37.22; H, 4.95; O, 24.79. Found: C, 37.15; H, 5.03; O, 24.86. IR (KBr, cm−1): 3451 (m), 3215 (w), 1664 (s), 1624 (w), 1382 (s), 1316 (s), 1100 (s), 930 (m), 871 (w), 778 (w), 746 (w), 688 (w).
{[Ni(cyclam)]2[Co(2,5-pydc)3]·4H2O}n3. Co[Co(2,5-Hpydc)3]2·6H2O (0.50 mmol, 0.64 g) was added to an aqueous solution of NaOH (3.00 mmol, 0.12 g). After stirring for half an hour, filtering and an aqueous solution of [Ni(cyclam)](ClO4)2 (2.00 mmol, 0.92 g) was added and stirred for an hour. The resulting solution was evaporated, and block-shaped crystals suitable for X-ray crystallographic analysis were obtained after few days. The solid was collected by filtration, washed with H2O and dried in air to afford a pink powder. Yield: 83%. Anal. Calcd for C41H65N11O16CoNi2: C, 43.03; H, 5.73; O, 22.37. Found: C, 42.96; H, 5.83; O, 22.44.
{[Ni(cyclam)]2[Ni(2,5-pydc)3]·4H2O}n4. The synthesis of 4 was carried out similarly to that of 3 except for using Ni[Ni(2,5-Hpydc)3]2·6H2O (0.50 mmol, 0.64 g) instead of Co[Co(2,5-Hpydc)3]2·6H2O. Yield: 91%. Anal. Calcd for C41H65N11O16Ni3: C, 43.04; H, 5.73; O, 22.37. Found: C, 42.95; H, 5.82; O, 22.46.
{[Ni(cyclam)]2[Zn(2,5-pydc)3]·4H2O}n5. The synthesis of 5 was carried out similarly to that of 3 except for using Zn[Zn(2,5-Hpydc)3]2·6H2O (0.50 mmol, 0.65 g) instead of Co[Co(2,5-Hpydc)3]2·6H2O. Yield: 91%. Anal. Calcd for C41H65N11O16ZnNi2: C, 42.79; H, 5.69; O, 22.24. Found: C, 42.76; H, 5.60; O, 22.18.
{[Zn(cyclam)]2[Ni(2,5-pydc)3]·7H2O}n6. The synthesis of 6 was carried out similarly to that of 3 except for using Ni[Ni(2,5-Hpydc)3]2·6H2O (0.50 mmol, 0.64 g) instead of Co[Co(2,5-Hpydc)3]2·6H2O and [Zn(cyclam)](ClO4)2 (2.00 mmol, 0.92 g) instead of [Ni(cyclam)](ClO4)2. Yield: 91%. Anal. Calcd for C41H71N11O19NiZn2: C, 40.65; H, 5.91; O, 25.09. Found: C, 40.60; H, 6.01; O, 25.01.
{[Zn(cyclam)]2[Zn(2,5-pydc)3]·5H2O}n7. The synthesis of 7 was carried out similarly to that of 3 except for using Zn[Zn(2,5-Hpydc)3]2·6H2O (0.50 mmol, 0.65 g) instead of Co[Co(2,5-Hpydc)3]2·6H2O and [Zn(cyclam)](ClO4)2 (2.00 mmol, 0.92 g) instead of [Ni(cyclam)](ClO4)2. Yield: 91%. Anal. Calcd for C41H67N11O17Zn3: C, 41.65; H, 5.71; O, 23.01. Found: C, 41.58; H, 5.79; O, 23.10.
Crystal structure analysis
Diffraction data of 2 were collected on a Bruker APEX DUO diffractometer equipped with triumph-monochromated Cu Kα radiation (λ = 1.54178 Å). Diffraction data of 3–7 and A1–A3 were collected on a Bruker Smart APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). These data were collected at 173 K (A3, 293 K) temperature and the structures were solved by direct methods and subsequently refined on F2 by using full-matrix least-squares techniques (SHELXL).15 SADABS16 absorption corrections were applied to the data. There are disordered water molecules in the asymmetric unit of 2, which cannot be refined properly, so the SQUEEZE algorithm was run to remove them before the structures were refined to convergence. According to elemental analysis (EA), IR spectra and TGA analyses, the solvent molecules were confirmed to be H2O. All the non-hydrogen atoms were refined anisotropically, and hydrogen atoms were located at calculated positions. However, hydrogen atoms of water molecules cannot be found. In the asymmetric unit of A2, a water molecule coordinated to Cu1 was disordered and it was divided into two parts (81:19). A summary of the crystallographic data are given in Table 1.
| Compounds |
2
|
3
|
4
|
5
|
6
|
7
|
A1
|
A2
|
A3
|
|
R
1 = Σ||Fo| − |Fc||/Σ|Fo|, wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2.
|
| Formula |
C24H30N6O8PdZn |
C41H65N11O16CoNi2 |
C41H65N11 O16Ni3 |
C41H65N11O16ZnNi2 |
C41H71N11O19NiZn2 |
C41H67N11O17Zn3 |
C24H36N6O11CuNi |
C24H34N6O10Cu2 |
C24H50N6O18CuNi |
| Formula weight |
702.31 |
1144.39 |
1144.17 |
1150.83 |
1211.54 |
1182.17 |
706.84 |
693.65 |
832.95 |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P |
P21/c |
P |
P |
|
a, Å |
23.8588(12) |
11.4952(9) |
11.4505(8) |
11.4882(8) |
11.5109(14) |
11.5056(8) |
13.8978(8) |
8.2345(9) |
9.706(4) |
|
b, Å |
18.6112(9) |
11.5504(8) |
11.5123(8) |
11.5521(8) |
11.5916(14) |
11.6391(8) |
15.0595(9) |
13.4315(15) |
10.184(4) |
|
c, Å |
8.4514(5) |
21.6426(16) |
21.6191(14) |
21.6308(16) |
21.717(3) |
21.8286(15) |
15.5605(9) |
13.4515(15) |
10.607(4) |
|
α, ° |
90 |
94.3510(10) |
94.2070(10) |
94.3610(10) |
91.170(2) |
91.6950(10) |
90 |
65.4050(10) |
62.712(4) |
|
β, ° |
100.598(3) |
91.2180(10) |
91.5310(10) |
91.3170(10) |
94.449(2) |
94.3000(10) |
115.0970(10) |
81.539(2) |
85.470(5) |
|
γ, ° |
90 |
113.6270(10) |
113.6120(10) |
113.5840(10) |
113.834(2) |
113.4320(10) |
90 |
85.676(2) |
69.691(4) |
|
V, Å3 |
3688.8(3) |
2620.9(3) |
2599.3(3) |
2618.8(3) |
2638.3(6) |
2668.9(3) |
2949.3(3) |
1337.9(3) |
869.8(6) |
|
Z
|
4 |
2 |
2 |
2 |
2 |
2 |
4 |
2 |
1 |
|
D
c, g cm−3 |
1.265 |
1.450 |
1.462 |
1.459 |
1.525 |
1.471 |
1.592 |
1.722 |
1.590 |
|
μ, mm−1 |
5.111 |
1.099 |
1.151 |
1.241 |
1.337 |
1.413 |
1.427 |
1.659 |
1.237 |
|
θ range/° |
3.03–62.49 |
0.95–27.54 |
0.95–28.25 |
0.95–27.01 |
0.94–28.28 |
0.94–27.51 |
1.62–27.73 |
1.67–28.26 |
2.17–25.03 |
| GOF on F2 |
1.088 |
0.995 |
1.081 |
1.065 |
1.066 |
1.081 |
0.942 |
1.146 |
1.023 |
|
R
1
a [I > 2σ(I)] |
0.0424 |
0.0519 |
0.0569 |
0.0492 |
0.0497 |
0.0528 |
0.0362 |
0.0390 |
0.0648 |
| wR2a (all data) |
0.1312 |
0.1947 |
0.1977 |
0.1527 |
0.1611 |
0.1935 |
0.0867 |
0.1589 |
0.1773 |
| Δρmax, Δρmin, e Å−3 |
0.822, −1.310 |
0.774, −0.734 |
1.609, −1.025 |
0.621, −0.522 |
1.138, −0.973 |
1.209, −0.948 |
0.407, −0.334 |
0.916, −0.765 |
1.012, −1.468 |
Results and discussion
Synthesis and design
Recently, the porous TMCPs complexes [Ni(cyclam)Pd(2,4-pydc)2·4H2O]n have been reported by us.17 Importantly, the varied metal ions have been confirmed to have a great influence on the synthesis of these complexes. Our further efforts to extend this series to other first-row transition metals {Ni(II) ions of [Ni(cyclam)Pd(2,4-pydc)2·4H2O]n may be substituted by other cations} using a similar synthesis protocol, resulted in either noncrystalline substances (Mn2+, Co2+) or nonporous precipitates with different crystal structures (Cu2+, Fig. S1, ESI†), with Zn2+ being the only exception. This is not surprising due to the different coordination chemistry of various metal cations. It is well known that Cu(II) favors a square planar arrangement similar to Pd(II). If the Pd(II) ions of Pd(2,4-pydc)2 were replaced by Cu(II), whether the complexes with the structures similar to [M(cyclam)Pd(2,4-pydc)2]n (M = Ni, Zn) will be obtained. In view of this, the metallotectons Cu(2,4-Hpydc)2, Cu(2,5-Hpydc)2 and their further constructing complexes [Ni(cyclam)Cu(2,5-pydc)2·3H2O] (A1), [Cu(cyclam)Cu(2,5-pydc)2·2H2O] (A2) and [Ni(cyclam)Cu(2,4-pydc)2·10H2O]n (A3) were synthesized, but the extending structures were largely different from [M(cyclam)Pd(2,4-pydc)2]n. In complexes A1 and A2, both group A and group B moieties of [Cu(2,5-pydc)2]2− did not connect with [M(cyclam)]2+, which resulted in the formation of their discrete structures (Fig. S2 and S3, ESI†). Nevertheless, in complex A3, group B connected with [Ni(cyclam)]2+ and group A remained idle. Compared with complexes 1 and 2, complex A3 also holding the 1D linear chain structure, but axial positions of Cu(II) were occupied by H2O molecules and no M⋯M interaction existed between the two chains, which resulted in its nonporous structure. Based on the preferred octahedral coordination environment of Co(II), Ni(II) and Zn(II), we also developed a series metallotectons M[M(2,5-Hpydc)3]2·6H2O [M = Co(II), Ni(II), Zn(II)], in which the M2+ is octahedrally coordinated with three N and three O atoms from three 2,5-Hpydc− moieties.
Crystal structures
Crystal structures of 1–2
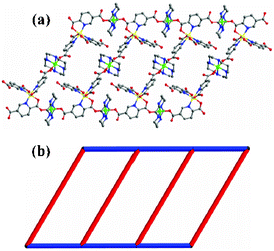
In the current work, we mainly focus on these two isostructural structure types: type I and type II. Complexes [M(cyclam)Pd(2,4-pydc)2·4H2O]n (1–2) have the type I structure, and complexes {[M′(cyclam)]2[M(2,5-pydc)3]·xH2O}n (3–7) have the type II structure. Both complexes 1–2 were constructed from the metallotectons Pd(2,4-Hpydc)2 and M(cyclam)(ClO4)2. The powder X-ray diffraction patterns (PXRD) (Fig. 1c) for complexes 1 and 2 accord well with each other, confirming that the two 3d–4d coordination polymers are indeed isostructural and they may be an ideal system to study the impact of metal cation exchange on polarity changes of frameworks and their gas adsorption properties. In such structures, all crystallize in the monoclinic space group C2/c with the Pd(II) and M(II) (M = Ni or Zn) atoms lying on independent inversion centres, and with each asymmetric unit containing one complete 2,4-pydc2− ligand and half a cyclam ligand (crystallographic data are summarized in Table 1). Each Pd(II) cation lies in a square planar environment with the two oxygen atoms and the two nitrogen atoms of 2,4-pydc2− ligands in the equatorial plane, leaving two vacant axial positions. Nevertheless, M(II) takes the classical octahedral coordination environment [MN4O2] formed by four nitrogen atoms from cyclam and two carboxylate oxygen atoms of 2,4-pydc2−, respectively. The carboxylate group at the 2-position together with the nitrogen atom chelate to the Pd(II) cation and the 4-position carboxylate group is coordinated to M(II), which results in the formation of linear chains which come from two different directions to cross each other through Pd⋯Pd interaction and form rhombus grid with dihedral angle 74.430° for 1 and 76.871° for 2 (Fig. 1a). The packing of these linear chains generates a 1D channel with petal-like cylindrical pores (Fig. 1b).
![(a) View showing the stacking of linear chains. (b) View of 1D channels present in the structure [M(cyclam)Pd(2,4-pydc)2]n (H atoms were omitted for clarity, C, gray; O, red; N, blue; Pd, orange; M, green.) (c) PXRD for 1 and 2 depicting the isostructural relationship.](/image/article/2013/CE/c2ce26072g/c2ce26072g-f1.gif) |
| | Fig. 1 (a) View showing the stacking of linear chains. (b) View of 1D channels present in the structure [M(cyclam)Pd(2,4-pydc)2]n (H atoms were omitted for clarity, C, gray; O, red; N, blue; Pd, orange; M, green.) (c) PXRD for 1 and 2 depicting the isostructural relationship. | |
Crystal structures of 3–7
The one-pot reaction of [M(2,5-pydc)3]4− [M = Co(II), Ni(II), Zn(II)] and [M′(cyclam)](ClO4)2 [M′ = Ni(II), Zn(II)] in solvent H2O at room temperature yielded type II complexes. The block-shaped crystals suitable for X-ray crystallographic analysis were obtained in high yield after few days. All the structures of 3, 4, 5, 6 and 7 crystallize in the triclinic space group P (crystallographic data are summarized in Table 1), and their asymmetric units contain one [M(2,5-pydc)3]4− and two [M′(cyclam)]2+. The coordination modes of [M(2,5-pydc)3]4− in all the complexes are given in Fig. S5, ESI.† Each [M(2,5-pydc)3]4− acts as a μ3 bridge to link three [M′(cyclam)]2+ ions in which one 2,5-pydc2− moiety remains idle, the 5-carboxylate groups of one 2,5-pydc2− and the 2-, 5-carboxylate groups from the other 2,5-pydc2− moieties are coordinated with [M′(cyclam)]2+ with monodentate η1:μ2 bridging modes. [Corresponding bond lengths (Å) of complexes 3–7 are shown in Table S1, ESI†]. Although [M(2,5-pydc)3]4− acts as a μ3 building block, the directions of coordinated 2-carboxylate and 5-carboxylate groups from the same 2,5-pydc2− moieties are in the inverse positions. So it finally resulted in the formation of 1D ladder-shaped coordination polymers (Fig. 2). The 1D ladder frameworks extended to 2D frameworks through H bonds between the 1D ladder frameworks and [M′(cyclam)]2+ (Fig. 3).
 |
| | Fig. 2 (a and b) View showing the 1D ladder shapes (the H atoms were omitted for clarity, C, gray; O, red; N, blue; M, yellow; M′, green.) | |
![H bonds between the 1D ladder frameworks and [M′(cyclam)]2+.](/image/article/2013/CE/c2ce26072g/c2ce26072g-f3.gif) |
| | Fig. 3 H bonds between the 1D ladder frameworks and [M′(cyclam)]2+. | |
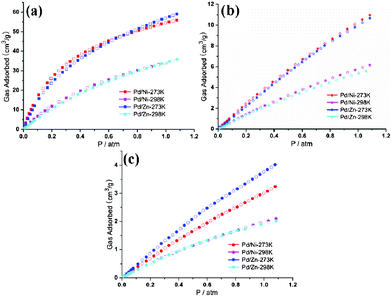
![(a) Gas sorption curves [273 K (red) and 298 K (purple)] of 1′. (b) Gas sorption curves [273 K (blue) and 298 K (green)] of 2′. Square, CO2; star, CH4, round, N2.](/image/article/2013/CE/c2ce26072g/c2ce26072g-f4.gif) |
| | Fig. 4 (a) Gas sorption curves [273 K (red) and 298 K (purple)] of 1′. (b) Gas sorption curves [273 K (blue) and 298 K (green)] of 2′. Square, CO2; star, CH4, round, N2. | |
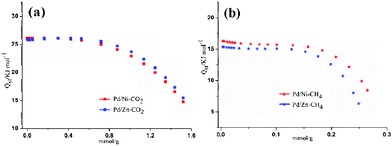 |
| | Fig. 5 Isosteric heats of gas adsorption (Qst) values for 1′ (blue) and 2′ (red): (a) CO2 and (b) CH4. | |
All CO2 isotherms are of type I according to the IUPAC classification.18 The CO2 sorption isotherms for complexes 1′ and 2′ measured at 298 K show that CO2 uptake of 2′ is always slightly less than 1′, which may be ascribed to the lighter atomic weight of Ni than Zn (Fig. 6a). Nevertheless, at 273 K, CO2 uptake of 2′ is also lower than 1′ below 0.7 atm, but with higher uptake above 0.7 atm. N2 sorption capacity of 1′ and 2′ are almost the same at 298 K. Furthermore, at 273 K, the N2 sorption capacity of 2′ is obviously superior to 1′ (Fig. 6c). The varied CO2 and N2 uptake of 1′ and 2′ is interpreted as specific interactions between the quadrupolar CO2, N2 molecules and the polar groups in the pore channels of different frameworks [the N–H distance of Zn-cyclam (0.907 Å) in 2 is much longer than that of Ni-cyclam (0.880 Å)] in 1]. The CH4 isotherms show that 1′ shows higher CH4 uptake than 2′ under all conditions (Fig. 6b), which may be ascribed to the fact that CH4 is non-polar (the non-polar molecules don't respond to the polarity changes of different frameworks) and here just the varied atom weight played a key role.
 |
| | Fig. 6 The comparisons of different gas sorption properties between 1′ and 2′: (a) CO2, (b) CH4 and (c) N2. | |
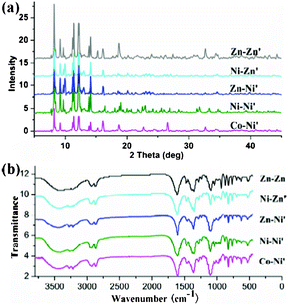 |
| | Fig. 7 (a) PXRD patterns for complexes 3–7; (b) IR spectrum of complexes 3–7. | |
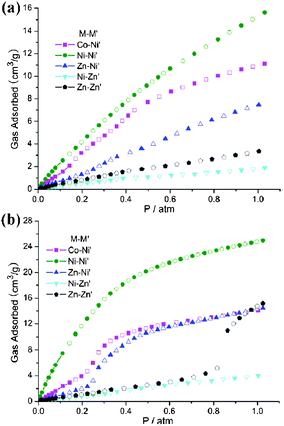 |
| | Fig. 8 The comparisons of CO2 sorption properties between 3′–7′: (a) 298 K, 1 atm; (b) 273 K, 1 atm. | |
Unlike CO2, CH4 and N2 gas were minimally absorbed in 4′ up to 1 atm (2.62 cm3 g−1 for CH4 and 0.69 cm3 g−1 for N2 at 298 K, 1 atm; 3.37 cm3 g−1 for CH4 and 1.20 cm3 g−1 for N2 at 273 K, 1 atm). Besides the adsorption capacity, the selectivity is a principal property relevant to adsorptive gas separation. Fig. 9 shows that complex 4′ can preferentially adsorb CO2 over CH4 and N2 and providing separation capability of CO2/N2 and CO2/CH4. The single-component CO2/CH4 separation ratios of 4′ at 273 K and 298 K (1 atm) are 7.3:1 (v:v), 5.8:1 (v:v), respectively, and for CO2/N2 they are 20.7:1 (v:v), 22.0:1 (v:v) at 273 K and 298 K (1 atm), respectively.
![Gas sorption curves [298 K (red) and 273 K (blue)] of 4′. Square, CO2; star, CH4, round, N2.](/image/article/2013/CE/c2ce26072g/c2ce26072g-f9.gif) |
| | Fig. 9 Gas sorption curves [298 K (red) and 273 K (blue)] of 4′. Square, CO2; star, CH4, round, N2. | |
Conclusions
In summary, two type I complexes [M(cyclam)Pd(2,4-pydc)2·4H2O]n (1–2) and five type II complexes {[M′(cyclam)]2[M(2,5-pydc)3]·xH2O}n (3–7) were synthesized and characterized. The high selective capture of CO2 over CH4 and N2 indicates that both complexes 1′ and 2′ are promising materials for the separation and purification of CO2 from CO2/CH4 or CO2/N2 mixtures. In addition, the comparisons of different gas sorption properties between 1′ and 2′ strongly suggest that metal identity has great influence on polarity of frameworks and further the adsorption of polar gas molecules, but the non-polar molecules don't respond to the polarity changes of different frameworks. The non-polar molecules uptake of isostructural materials with different metals will just be influenced by the varied atom weight. The comparisons of CO2 sorption properties between five type II complexes also strongly suggest that metal identity has a great influence on the polarity of frameworks and further on CO2 uptake. In addition, the high selective capture of CO2 over CH4 and N2 indicates that complex 4′ is also promising material for the separation and purification of CO2 from CO2/CH4 or CO2/N2 mixtures.
Acknowledgements
This work was supported by Shanghai Science and Technology Committee (08DZ2270500 and 08DJ1400103), Shanghai Leading Academic Discipline Project (B108) and the National Basic Research Program of China (2009CB825300 and 2010DFA41160).
References
-
(a) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC;
(b) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef CAS;
(c) G. Ferey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Liewellyn, G. D. Weireld, A. Vimont, M. Daturi and J. S. Chang, Chem. Soc. Rev., 2011, 40, 550 RSC;
(d) Y. Inubushi, S. Horike, T. Fukushima, G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Commun., 2010, 46, 9229 RSC;
(e) J. M. Zhang, H. H. Wu, T. J. Emge and J. Li, Chem. Commun., 2010, 46, 9152 RSC;
(f) Z. R. Herm, J. A. Swisher, B. Smit, R. Krishna and J. R. Long, J. Am. Chem. Soc., 2011, 133, 5664 CrossRef CAS;
(g) S. Bourrelly, P. L. Liewellyn, C. Serre, F. Millange, T. Loiseau and G. Ferey, J. Am. Chem. Soc., 2005, 127, 13519 CrossRef CAS.
-
(a) P. D. C. Dietzel, R. E. Johnsen, H. Fjellvaag, S. Bordiga, E. Groppo and R. Blom, Chem. Commun., 2008, 5125 RSC;
(b) Z. J. Liang, M. Marshall and A. L. Chaffee, Energy Fuels, 2009, 23, 2785 CrossRef CAS;
(c) Y. E. Cheon and P. M. Suh, Chem. Commun., 2009, 2296 RSC;
(d) D. Britt, H. Furukawa, B. Wang and T. G. Glover, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637 CrossRef CAS;
(e) H. H. Wu, R. S. Reali, D. A. Smith, M. Trachtenberg and J. Li, Chem.–Eur. J., 2010, 16, 13951 CrossRef CAS.
-
(a) E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382 CrossRef CAS;
(b) S. R. Miller, G. M. Pearce, P. A. Wright, F. Bonino, S. Chavan, S. Bordiga, I. Margiolaki, N. Guillou, G. Ferey, S. Bourrelly and P. L. Liewellyn, J. Am. Chem. Soc., 2008, 130, 15967 CrossRef CAS;
(c) Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Ferey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS;
(d) B. Arstad, H. Fjellvaag, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755 CrossRef CAS;
(e) Y. H. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650 CrossRef CAS;
(f) T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022 RSC;
(g) Z. X. Chen, S. C. Xiang, H. D. Arman, P. Li, D. Y. Zhao and B. L. Chen, Eur. J. Inorg. Chem., 2011, 2227 CrossRef CAS;
(h) Z. X. Chen, S. C. Xiang, H. D. Arman, P. Li, S. Tidro, D. Y. Zhao and B. L. Chen, Eur. J. Inorg. Chem., 2010, 3745 CrossRef CAS.
-
(a) Y. Hijikata, S. Horike, M. Sugimoto, H. Sato, R. Matsuda and S. Kitagawa, Chem.–Eur. J., 2011, 17, 5138 CrossRef CAS;
(b) Y. G. Zhao, H. H. Wu, T. J. Emge. Q. H. Gong, N. Nijem, Y. Chabal, L. Z. Kong, D. C. Langreth, H. Liu, H. P. Zeng and J. Li, Chem.–Eur. J., 2011, 17, 5101 CrossRef CAS;
(c) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS;
(d) Y. F. Han, W. G. Jia, W. B. Yu and G. X. Jin, Chem. Soc. Rev., 2009, 38, 3419 RSC;
(e) Y. F. Han, H. Li and G. X. Jin, Chem. Commun., 2010, 46, 6879 RSC;
(f) Y. F. Han, W. G. Jia, Y. J. Lin and G. X. Jin, Angew. Chem., Int. Ed., 2009, 48, 6234 CrossRef CAS;
(g) F. M. Conrady, R. Froehlich, C. Schulte-Brinke, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2011, 133, 11496 CrossRef CAS.
-
(a) W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268 CrossRef CAS;
(b) S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870 CrossRef CAS.
-
(a) S. L. Huang, X. X. Li, X. J. Shi, H. W. Hou and Y. T. Fan, J. Mater. Chem., 2010, 20, 5695 RSC;
(b) J. Y. Zhang, Q. Yue, Q. X. Jia and E. Q. Gao, CrystEngComm, 2008, 10, 1443 RSC;
(c) N. Y. Li, Z. G. Ren, D. Liu and J. P. Lang, Dalton Trans., 2010, 39, 4213 RSC;
(d) T. L. Hu, R. Q. Zou, J. R. Li and X. H. Bu, Dalton Trans., 2008, 1302 RSC;
(e) C. S. Liu, J. J. Wang, C. Ze and X. H. Bu, CrystEngComm, 2010, 12, 1833 RSC;
(f) L. Yan, Q. Yue, Q. X. Jia and E. Q. Gao, Cryst. Growth Des., 2009, 9, 2984 CrossRef CAS;
(g) Y. Chen, H. X. Li, D. Liu and J. P. Lang, Cryst. Growth Des., 2008, 8, 3810 CrossRef CAS.
- J. A. Zhao, L. W. Mi, J. Y. Hu, H. W. Hou and Y. T. Fan, J. Am. Chem. Soc., 2008, 130, 15222 CrossRef CAS.
-
(a) A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS;
(b) S. Couck, J. F. M. Denayer, G. V. Baron, T. Remy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326 CrossRef CAS;
(c) A. Vimont, A. Travert, P. Bazin, J. C. Lavalley, M. Daturi, C. Serre, G. Ferey, S. Bourrelly and P. L. Llewellyn, Chem. Commun., 2007, 3291 RSC.
-
(a) W. Steffen, B. Kohler, M. Altmann, U. Scherf, K. Stitzer, H. C. zur Loye and U. H. F. Bunz, Chem.–Eur. J., 2001, 7, 117 CrossRef CAS;
(b) N. G. Pschirer, D. M. Ciurtin, M. D. Smith, U. H. F. Bunz and H. C. zur Loye, Angew. Chem., Int. Ed., 2002, 41, 583 CrossRef CAS;
(c) Y. B. Dong, X. Zhao, B. Tang, H. Y. Wang, R. Q. Huang, M. D. Smith and H. C. zur Loye, Chem. Commun., 2004, 220 RSC;
(d) Y. B. Dong, X. Zhao, R. Q. Huang, M. D. Smith and H. C. zur Loye, Inorg. Chem., 2004, 43, 5603 CrossRef CAS;
(e) D. M. Ciurtin, N. G. Pschirer, M. D. Smith, U. H. F. Bunz and H. C. zur Loye, Chem. Mater., 2001, 13, 2743 CrossRef CAS;
(f) C. L. Chen, A. M. Goforth, M. D. Smith, C. Y. Su and H. C. zur Loye, Inorg. Chem., 2005, 44, 8762 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
-
(a) S. K. Ghosh, G. Savitha and P. K. Bharadwaj, Inorg. Chem., 2004, 43, 5495 CrossRef CAS;
(b) C. L. Chen, A. M. Goforth, M. D. Smith, C. Y. Su and H. C. zur Loye, Angew. Chem., Int. Ed., 2005, 44, 6673 CrossRef CAS;
(c) S.-I. Noro, H. Miyasaka, S. Kitagawa, T. Wada, T. Okubo, M. Yamashita and T. Mitani, Inorg. Chem., 2005, 44, 133 CrossRef CAS;
(d) J. Leciejewicz, H. Ptasiewicz-Bak, T. Premkumar and S. Govindarajan, J. Coord. Chem., 2004, 57, 97 CrossRef CAS;
(e) C. L. Chen, J. M. Ellsworth, A. M. Goforth, M. D. Smith, C. Y. Su and H. C. zur Loye, Dalton Trans., 2006, 5278 RSC.
-
(a) L. M. Zheng, X. Q. Wang and A. J. Jacobson, J. Solid State Chem., 2000, 152, 174 CrossRef CAS;
(b) D. M. Ciurtin, M. D. Smith and H. C. zur Loye, Polyhedron, 2003, 22, 3043 CrossRef CAS;
(c) Y. B. Dong, J. P. Ma, R. Q. Huang, F. Z. Liang and M. D. Smith, Dalton Trans., 2003, 1472 RSC;
(d) J. H. Luo, B. Alexander, T. R. Wagner and P. A. Maggard, Inorg. Chem., 2004, 43, 5537 CrossRef CAS;
(e) C. L. Ma, Y. F. Han, R. F. Zhang and D. Q. Wang, Dalton Trans., 2004, 1832 RSC;
(f) Y. C. Liang, M. C. Hong, J. C. Liu and R. Cao, Inorg. Chim. Acta, 2002, 328, 152 CrossRef CAS.
-
(a) Y. L. Bai, J. Tao, R. B. Huang and L. S. Zheng, Angew. Chem., Int. Ed., 2008, 47, 5344 CrossRef CAS;
(b) X. L. Wang, C. Qin, S. X. Wu, K. Z. Shao, Y. Q. Lan, S. Wang, D. X. Zhu, Z. M. Su and E. B. Wang, Angew. Chem., Int. Ed., 2009, 48, 5291 CrossRef CAS;
(c) J. R. Li, D. J. Timmons and H. C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368 CrossRef CAS;
(d) J. R. Li and H. C. Zhou, Nat. Chem. Biol., 2010, 2, 893 CAS;
(e) K. Otsubo, Y. Wakabayashi, J. Ohara, S. Yamamoto, H. Matsuzaki, H. Okamoto, K. Nitta, T. Uruga and H. Kitagawa, Nat. Mater., 2011, 10, 291 CrossRef CAS.
-
(a) E. Y. Lee and M. P. Suh, Angew. Chem., Int. Ed., 2004, 43, 2798 CrossRef CAS;
(b) H. J. Choi, T. S. Lee and M. P. Suh, Angew. Chem., Int. Ed., 1999, 38, 1405 CrossRef CAS;
(c) M. P. Suh, H. J. Choi, S. M. So and B. M. Kim, Inorg. Chem., 2003, 42, 676 CrossRef CAS;
(d) M. P. Suh, H. R. Moon, E. Y. Lee and S. Y. Jang, J. Am. Chem. Soc., 2006, 128, 4710 CrossRef CAS;
(e) Y. E. Cheon and M. P. Suh, Chem.–Eur. J., 2008, 14, 3961 CrossRef CAS;
(f) H. Kim and M. P. Suh, Inorg. Chem., 2005, 44, 810 CrossRef CAS.
-
G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, Universität Gtötingen, Germany, 1997 Search PubMed.
-
G. M. Sheldrick, SADABS (2.01), Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS, Madison, WI, 1998 Search PubMed.
- S. L. Huang and G. X. Jin, CrystEngComm, 2011, 13, 6013 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Mouscou, J. Rouquerol and I. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures of complexes A1, A2 and A3; powder XRD patterns, TGA curves and gas sorption curves. CCDC 851502 (2), 889537 (3), 889538 (4), 889539 (5), 889540 (6), 889541 (7), 889542 (A1), 889543 (A2), –889544 (A3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ce26072g |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.