 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRationale for the sluggish oxidative addition of aryl halides to Au(I)†
Madeleine
Livendahl
a,
Charles
Goehry
a,
Feliu
Maseras
*ab and
Antonio M.
Echavarren
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: aechavarren@iciq.es
bDepartament de Química, Universitat Autonoma de Barcelona, 08193 Bellaterra, Spain
cDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, C/Marcel·li Domingo s/n, 43007 Tarragona, Spain
First published on 4th December 2013
Abstract
The oxidative addition of Csp2–Br or Csp2–I bonds to gold(I) does not take place even under very favorable intramolecular conditions that could form five- or six-membered gold(III) metallacycles. DFT calculations reveal that although this process could be feasible thermodynamically, it is kinetically very sluggish.
Gold(I) complexes activate unsaturated substrates in catalytic processes that are characterized by the invariance of the oxidation state of the metal.1 Although it was proposed that the Sonogashira2–5 and Suzuki coupling could be catalysed by gold,6,7 it seems now clear that at least in some cases the catalytic role is played by either small amounts of palladium contaminants8,9 or by gold nanoparticles,10–14 which probably mediate these couplings by mechanisms very different from those occurring under homogeneous conditions.15,16
Organogold(I) complexes transmetallate with Pd(II), which has been used in their coupling with aryl iodides with palladium catalysts.17–19 However, the oxidative addition of aryl halide ArX to a gold(I) complex [AuXL], a necessary step for a cross-coupling catalysed by this type of d10 complexes is unprecedented. Indeed, an alkenyl gold(I) complex with a pending aryl iodide has been structurally characterized as a stable complex, although it could have undergone intramolecular oxidative addition through a six-membered transition state.20 Complexes [AuMePR3] behave as ordinary SN2 nucleophiles upon adding slowly with alkyl iodides, following the expected order of reactivity: CH3I > EtI > i-PrI.21–23 Disulphides undergo oxidative addition reactions with gold(I) dithiolate complexes.24 Interestingly, the oxidative addition of a relatively weak Si–Si bond to gold(I) is a very favourable process.25 DFT calculations show that the oxidative addition of iodobenzene to complex [AuI(PMe3)] has a high activation barrier (31.6 kcal mol−1 in potential energy, likely higher in free energy).10a However, bisphosphine gold clusters [Au3L5]+ and [Au3L6]+ react in the gas phase by C–I bond activation.26
We decided to study the oxidative addition of Ar–X bonds to [AuXL] by examining systems of type I in which the metal coordinates the phosphorous ligand of a phosphine or phosphite bearing an ortho-halogenated aryl group (Scheme 1). Under these very favourable conditions, the oxidative addition could occur intramolecularly to form gold(III) metallacycles of type II. We have performed DFT calculations to clarify the origin of the sluggishness, thermodynamic or kinetic, of the oxidative addition of aryl halides to gold(I) complexes.
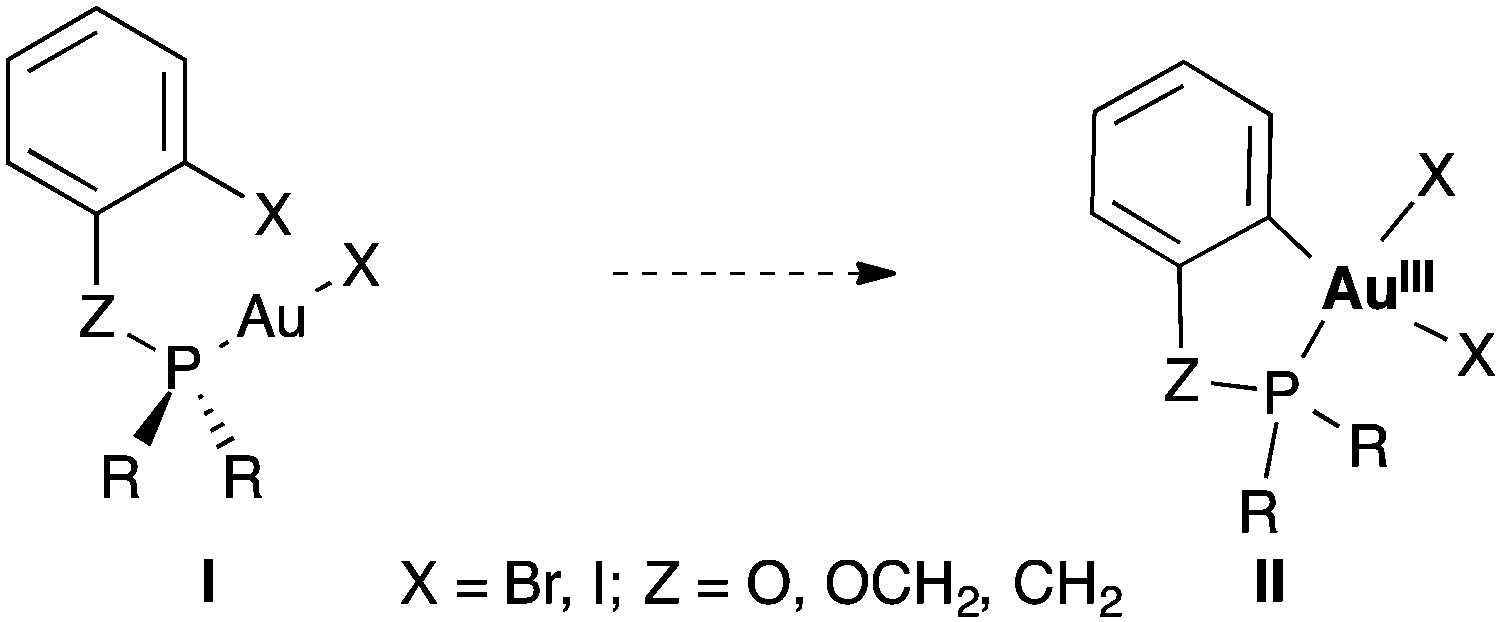 | ||
| Scheme 1 Hypothetical intramolecular oxidative addition of Csp2–X bonds to Au(I) to form Au(III) metallacycles II. | ||
Reaction of phosphites (o-IC6H4O)3P (1a) and (o-IC6H4CH2O)3P (1b) with [AuCl(THT)] (THT = tetrahydrothiophene) in CH2Cl2 led to complexes 2a (17%) and 2b (10%) as white solids. Their structures were determined using single crystal X-ray diffraction (Fig. 1 and 2).27 These complexes show non-C3-symmetrical structures in the solid state with the phosphite ligands adopting a syn-conformation.28 The Au–P (2.19–2.20 Å) and Au–Cl (2.28 Å) bond distances are similar in both complexes. In the case of complex 1a, two of the o-iodophenyl rings have the C–I bonds pointing towards the Au(I) centre, with a closest Csp2–Au distance of 3.795 Å (Fig. 1a). In contrast, in complex 2b the C–I bonds of the three aryl rings are anti-oriented with respect to the P–Au–Cl bond (Fig. 1b).
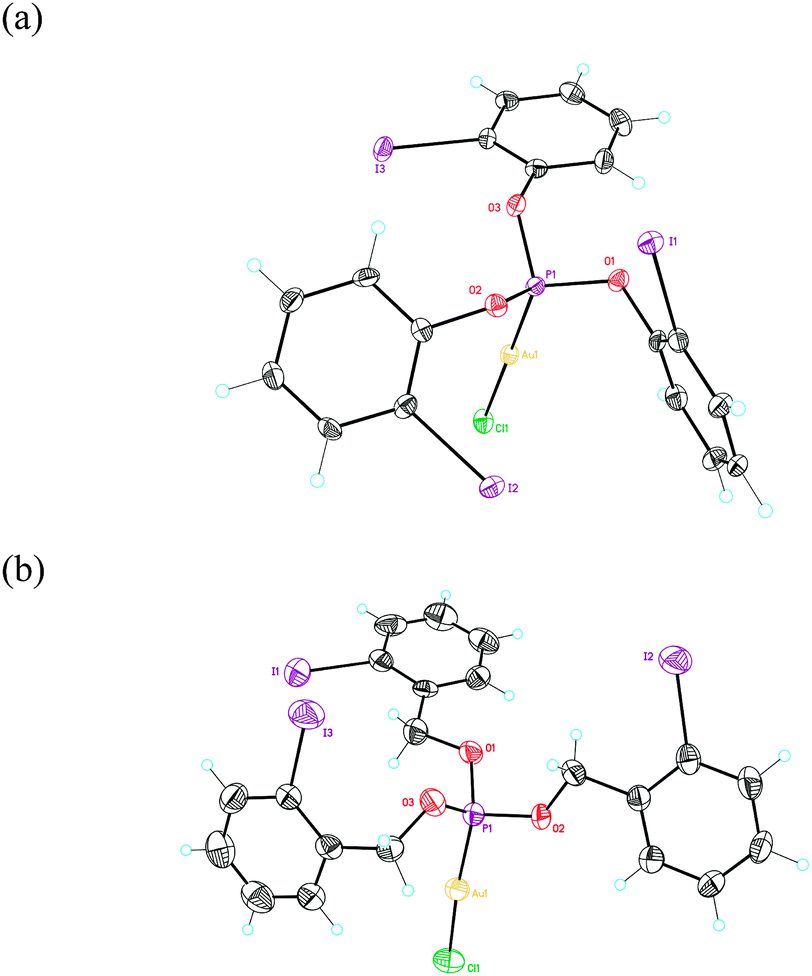 | ||
| Fig. 1 ORTEP plot (50% thermal ellipsoids) of the crystal structure of complex 2a (a) and 2b (b). | ||
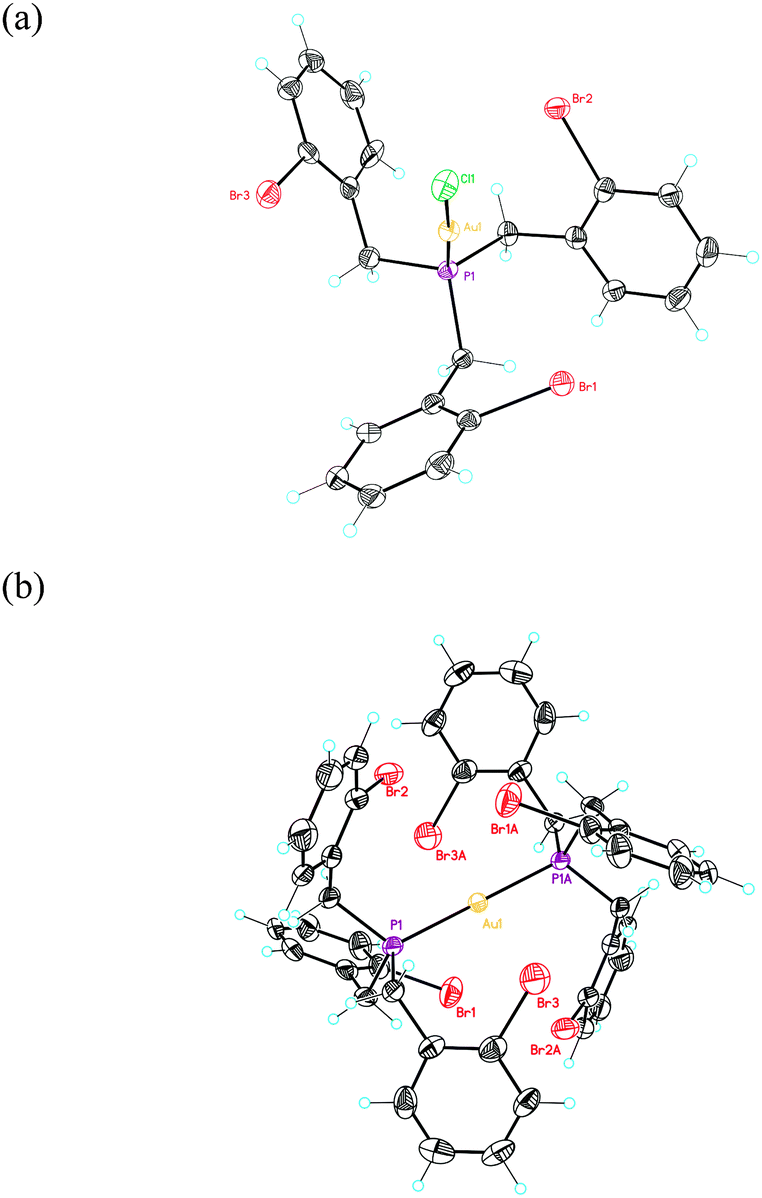 | ||
| Fig. 2 ORTEP plot (50% thermal ellipsoids) of the crystal structure of complexes 4a (a) and 5 (b). | ||
The related phosphine complex 4a was obtained in 60% yield by reacting (o-BrC6H3)3P (3) with [AuCl(THT)] in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. When the reaction was carried out in a 2:1 ligand to Au(I) ratio, bisphosphine gold(I) complex 5 was isolated in 70% yield. The structures of 4a and 5 were confirmed by X-ray diffraction (Fig. 2).27 The Au–P distance in 4a (2.22 Å) is much shorter than those in more crowded complex 5 (2.30 Å). Complex 4a displays an almost C3-symmetrical structure with Csp2(Br)–Au distances between 3.61 and 3.86 Å. We also prepared an analogous complex 4b from (2-bromobenzyl)diphenylphosphine.29 Complex 5 shows a D3d-symmmetrical structure with Csp2(Br)–Au distances of 4.1 Å and the six Br atoms in a non-octahedral arrangement around the metal centre (Br–Au distances of 3.7–3.8 Å).
:1 ratio. When the reaction was carried out in a 2:1 ligand to Au(I) ratio, bisphosphine gold(I) complex 5 was isolated in 70% yield. The structures of 4a and 5 were confirmed by X-ray diffraction (Fig. 2).27 The Au–P distance in 4a (2.22 Å) is much shorter than those in more crowded complex 5 (2.30 Å). Complex 4a displays an almost C3-symmetrical structure with Csp2(Br)–Au distances between 3.61 and 3.86 Å. We also prepared an analogous complex 4b from (2-bromobenzyl)diphenylphosphine.29 Complex 5 shows a D3d-symmmetrical structure with Csp2(Br)–Au distances of 4.1 Å and the six Br atoms in a non-octahedral arrangement around the metal centre (Br–Au distances of 3.7–3.8 Å).
Complexes 2a and 2b did not undergo oxidative addition to form the corresponding metallacycles of type II in CD2Cl2 solution. Similarly, more robust complex 4 was recovered unchanged after being heated in toluene solution at 60 °C for several days. Furthermore, bisphosphine gold(I) complex 5 failed to form any gold(III) metallacycle after being heated in DMSO at 100 °C for more than 5 days and was fully recovered without any sign of decomposition.
In order to better understand the reasons for the sluggish reactivity towards oxidative addition of this set of Au(I) complexes, we carried out computational studies (DFT calculations at the M06 level including solvation effects, values reported in text are free energies).30 The relative energies of all transition states and products are collected in Table 1. A representative transition state, 2t1ts, is shown in Fig. 3. There is simultaneous formation of the Au–C and Au–Br bonds, with distances of 2.273 and 2.587 Å, respectively. It is thus a concerted transition state that leads to complex [PhAuClBr(PMe3)] with the phosphine cis to Ph, in contrast to that reported from the oxidative addition of PhI to [Au(PMe3)I], which leads to trans-[PhAuI2(PMe3)].10a The alternative SN2-like transition states31 with the initial departure of a bromide anion were also located in a number of cases but had always higher energies than the concerted ones. The endergonic character of the oxidative addition processes computed in Table 1 is consistent with the facile reductive elimination of R–R from trialkyl [R3AuL] complexes.32
| Transition state | Product | |||||
|---|---|---|---|---|---|---|
| PMe3 | PPh3 | Other | PMe3 | PPh3 | Other | |
| 1t | — | — | 41.5 | — | — | 17.4 |
| 2t | 42.6 | 43.0 | — | 17.5 | 23.3 | — |
| 3t | 47.3 | 46.8 | — | 4.1 | 6.5 | — |
| 4t | 48.5 | 45.3 | — | 5.1 | 7.5 | — |
| 5t | — | — | 11.7 | — | — | 4.5 |
| 6t | 23.3 | 21.6 | — | 12.1 | 12.7 | — |
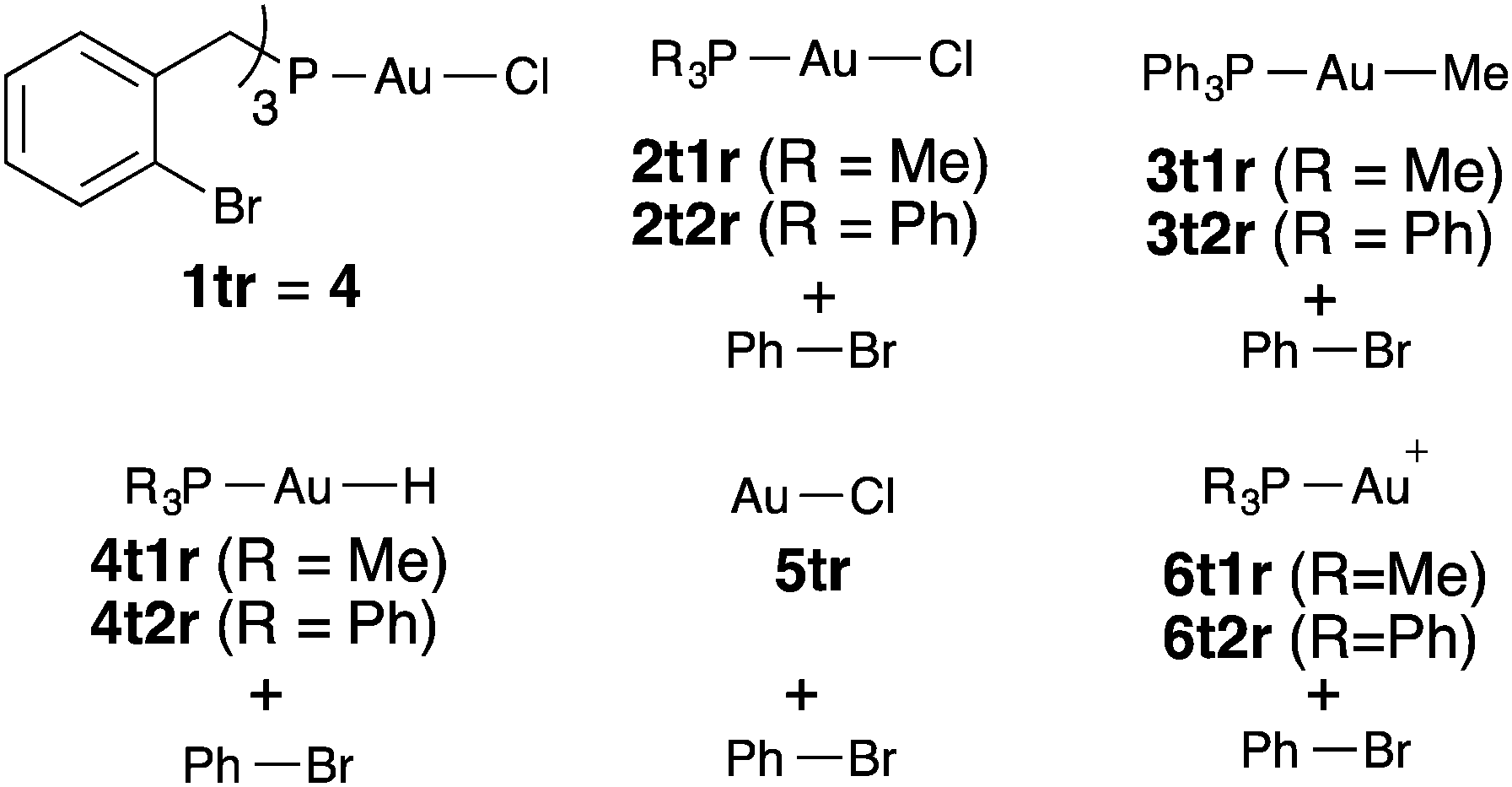
|
||||||
 | ||
| Fig. 3 M06 optimized structure of transition states 2t1ts and 1tts colour codes: red = Br, purple = P, yellow = Au, green = Cl. | ||
The X-ray structure of 4 was used a starting point in the geometry optimization of 1tr to avoid time-consuming conformational searches,33 and the same conformation was used in the calculations for 1tts and 1tp. The associated transition state 1tts (Fig. 3) has a relative free energy of 41.5 kcal mol−1 above the reactant. This is in full agreement with the lack of reactivity under the experimental conditions. The relative free energy of intermediate 1tp, which is only 17.4 kcal mol−1 higher than the starting Au(I) complex 1tr, could be accommodated into a catalytic cycle, but not that of the transition state. This hints at a kinetic rather than thermodynamic origin for the lack of reactivity of Au(I) complexes in oxidative addition processes.
Further calculations showed that the more electron depleted the aryl halide was rendered, the lower the barrier for the oxidative addition became.30 However, even in the most favourable case, 2,4,6-trinitro-bromobenzene, the activation energy is still relatively high (28.0 kcal mol−1).30
We also analysed the origin of this high barrier. One possible reason could be the strain associated with the fact that the intramolecular oxidative addition process leads to the trans arrangement of phenyl and bromide in the product. The geometrical arrangement in 1tts with the C–Br bond “above” gold, instead of the more usual in-plane arrangement must have some energy penalty. To clarify its importance we carried out additional calculations for the intermolecular reaction on systems 2t1, 2t2, 3t1, 3t2, 4t1, 4t2, (Table 1) where the oxidatively added C–Br bond is in a bromobenzene unit not previously connected to the metal. A variety of systems were considered that differed in the nature of the phosphine (PMe3 or PPh3) and of the spectator ligand (chloride, methyl or hydride). The free energy barriers for these intermolecular processes, leading to a cis arrangement of phenyl and bromide, were in a narrow span between 42.6 kcal mol−1 (for 2t1ts) and 48.5 kcal mol−1, (for 4t1ts). These barriers are also close to the 41.5 kcal mol−1 computed for 1tts. This means that the cost associated with the trans nature of 1t1ts is nearly identical to the entropic penalty for bringing two separate molecules together. In any case, these results mean that the high barrier for the oxidative addition to Au(I) reflects an intrinsic reluctance to undergo oxidative reaction in these systems. It must be mentioned that there is a much wider dispersion in the relative energies of oxidative addition products, from 4.1 kcal mol−1 for 3t1p to 23.3 kcal mol−1 for 2t2p. This seems to be related to different combinations of trans influences in the computed systems. It is however important to remark that although in some cases the reaction is too endergonic to take place, this is not the general behaviour. The high kinetic barrier is on the other hand present in all tested systems.
A possible origin for the high barrier in systems 1t to 4t is the well-known strong preference of Au(I) complexes for a linear 2-coordination,34 which is challenged by the simultaneous formation of two new bonds at the transition state. We tested this hypothesis through additional calculations on systems 5t, 6t1, and 6t2 (Table 1). The resulting barriers were 11.7, 23.3 and 21.6 kcal mol−1, respectively. It is clear that the reaction becomes much easier when starting from a mono-coordinated Au(I) complex, thus providing evidence in favour of our hypothesis that coordination number is the key.
Our joint experimental and computational study demonstrates that linear 2-coordinate d10 Au(I) complexes are much less reactive towards oxidative addition than the related Pd(0) complexes. The Au(III) species that would result from the reaction have in most cases reasonable energies, but the barriers to access them are prohibitively high for moderate temperatures. Oxidative addition is a key step in cross-coupling, and Au(I) will be thus unable to replace Pd(0) in most of these reactions. The sluggishness of the reaction in the Au(I) system seems to be related to its strong preference for coordination 2. This is a specific characteristic of Au(I) that is not shared by Pd(0), and explains why, despite having the same number of valence d electrons, these two metals have significantly different reactivity. Our results hint to a possible solution, since the oxidative addition would be much easier when starting from a [LAu(I)]+ complex. Although this type of mono-coordinated complexes are unknown, complexes [LAu(I)L′]+A− with a very weakly coordinated ligand L′ might undergo the required oxidative addition reaction under sufficiently mild conditions acting genuine catalysts for cross-coupling reactions of aryl halides. Work towards achieving this goal is in progress.
We thank the MINECO (CTQ2010-16088/BQU, CTQ2011-27033), the AGAUR (2009SGR47, 2009SGR0259), the European Research Council (Advanced Grant No. 321066), and the ICIQ Foundation for the support of this work. We also thank the ICIQ X-Ray Diffraction unit for the X-ray structures.
Notes and references
- (a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed; (b) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed; (c) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed; (d) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (e) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed; (f) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC; (g) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef CAS PubMed; (h) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536–6544 RSC; (i) C. Obradors and A. M. Echavarren, Chem. Commun., 2015, 50, 16–28 RSC; (j) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2013 DOI:10.1021/ar400174p.
- (a) C. González-Arellano, A. Abad, A. Corma, H. Garcia, M. Iglesias and F. Sánchez, Angew. Chem., Int. Ed., 2007, 46, 1536–1538 CrossRef PubMed; (b) A. Corma, C. González-Arellano, M. Iglesias, S. Pérez-Ferreras and F. Sánchez, Synlett, 2007, 1771–1774 CrossRef CAS PubMed; (c) C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Eur. J. Inorg. Chem., 2008, 1107–1115 CrossRef.
- P. Li, L. Wang, M. Wang and F. You, Eur. J. Org. Chem., 2008, 5946–5951 CrossRef CAS.
- O. M. A. de Souza, M. S. Bittar, L. V. P. Mendes, C. Michele and F. da Silva, Synlett, 2008, 1777–1780 Search PubMed.
- P. Yao, J. Chem. Res., 2013, 37, 174–176 CrossRef CAS.
- J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060–2061 CrossRef CAS PubMed.
- (a) S. Carrettin, A. Corma, M. Iglesias and F. Sánchez, Appl. Catal., A, 2005, 291, 247–252 CrossRef CAS PubMed; (b) C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, J. Catal., 2006, 238, 497–501 CrossRef PubMed; (c) A. Corma, E. Gutiérrez-Puebla, M. Iglesias, A. Monge, S. Pérez-Ferreras and F. Sánchez, Adv. Synth. Catal., 2006, 348, 1899–1907 CrossRef CAS; (d) N. Debono, M. Iglesias and F. Sánchez, Adv. Synth. Catal., 2007, 349, 2470–2476 CrossRef CAS.
- T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS PubMed.
- Review on the topic of trace metal impurities in catalysis: I. Thome, A. Nijs and C. Bolm, Chem. Soc. Rev., 2012, 41, 979–987 RSC.
- (a) A. Corma, R. Juárez, M. Boronat, F. Sánchez, M. Iglesias and H. García, Chem. Commun., 2010, 46, 1446–1448 RSC; (b) M. Boronat, D. Combita, P. Concepción, A. Corma, H. García, R. Juárez, S. Laursen and J. D. López-Castro, J. Phys. Chem. C, 2012, 116, 24855–24867 CrossRef CAS.
- M. Stratakis and H. García, Chem. Rev., 2012, 102, 4469–4506 CrossRef PubMed.
- J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060–2061 CrossRef CAS PubMed.
- G. Li, D.-E. Jiang, C. Liu, C. Yu and R. Jin, J. Catal., 2013, 306, 177–183 CrossRef CAS PubMed.
- (a) G. Kyriakou, S. K. Beaumont, S. M. Humphrey, C. Antonetti and R. M. Lambert, ChemCatChem, 2010, 2, 1444–1449 CrossRef CAS; (b) V. K. Kanuru, G. Kyriakou, S. K. Beaumont, A. C. Papageorgiou, D. J. Watson and R. M. Lambert, J. Am. Chem. Soc., 2010, 132, 8081–8086 CrossRef CAS PubMed; (c) S. K. Beaumont, G. Kyriakou and R. M. Lambert, J. Am. Chem. Soc., 2010, 132, 12246–12248 CrossRef CAS PubMed.
- M. García-Mota, N. Cabello, F. Maseras, A. M. Echavarren, J. Pérez-Ramírez and N. López, ChemPhysChem, 2008, 11, 1624–1629 CrossRef PubMed.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
- (a) Y. Shi, K. E. Roth, S. D. Ramgren and S. A. Sand, J. Am. Chem. Soc., 2009, 131, 18022–18023 CrossRef CAS PubMed; (b) K. E. Roth and S. A. Blum, Organometallics, 2011, 30, 4811–4813 CrossRef CAS; (c) J. J. Hirner, Y. Shi and S. A. Blum, Acc. Chem. Res., 2011, 44, 603–613 CrossRef CAS PubMed; (d) Ni-catalysed coupling of organogold complexes: J. Hirner and S. A. Blum, Organometallics, 2011, 30, 1299–1302 CrossRef CAS.
- (a) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243–8246 CrossRef CAS PubMed; (b) A. S. K. Hashmi, R. Döpp, C. Lothschütz, M. Rudolph, D. Riedel and F. Rominger, Adv. Synth. Catal., 2010, 352, 1307–1314 CrossRef CAS; (c) A. S. K. Hashmi, M. Ghanbari, M. Rudolph and F. Rominger, Chem.–Eur. J., 2012, 18, 8113–8119 CrossRef CAS PubMed; (d) M. M. Hansmann, M. Pernpointner, R. Döpp and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 15290–15303 CrossRef CAS PubMed.
- (a) M. H. Pérez-Temprano, J. A. Casares and P. Espinet, Chem.–Eur. J., 2012, 18, 1864–1884 CrossRef PubMed; (b) J. delPozo, J. A. Casares and P. Espinet, Chem. Commun., 2013, 49, 7246–7248 RSC; (c) J. delPozo, D. Carrasco, M. H. Pérez-Temprano, M. García-Melchor, R. Álvarez, J. A. Casares and P. Espinet, Angew. Chem., Int. Ed., 2013, 52, 2189–2193 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. D. B. Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133–147 CrossRef CAS; (b) See also: P. Nösel, T. Lauterbach, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 8634–8641 CrossRef PubMed.
- (a) A. Tamaki and J. K. Kochi, J. Organomet. Chem., 1972, 40, C81–C84 CrossRef CAS; (b) A. Tamaki and J. K. Kochi, J. Chem. Soc., Dalton Trans., 1973, 2620–2626 RSC; (c) A. Johnson and R. J. Puddephatt, Inorg. Nucl. Chem. Lett., 1973, 9, 1175–1177 CrossRef CAS; (d) A. Tamaki and J. K. Kochi, J. Organomet. Chem., 1974, 64, 411–425 CrossRef CAS; (e) A. Johnson and R. J. Puddephatt, J. Organomet. Chem., 1975, 85, 115–121 CrossRef CAS.
- M. T. Johnson, J. M. J. van Rensburg, M. Axelsson, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2011, 2, 2373–2377 RSC.
- A radical mechanism was found in the reaction between [AuMePR3] and CF3I: A. Johnson and R. J. Puddephatt, J. Chem. Soc., Dalton Trans., 1976, 1360–1363 RSC.
- R. E. Bachman, S. A. Bodolosky-Bettis, C. J. Pyle and M. A. Gray, J. Am. Chem. Soc., 2008, 130, 14303–14310 CrossRef CAS PubMed.
- P. Gualco, S. Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2011, 50, 8320–8324 CrossRef CAS PubMed.
- P. S. D. Robinson, G. N. Khairallah, G. da Silva, H. Lioe and R. A. J. O'Hair, Angew. Chem., Int. Ed., 2012, 51, 3812–3817 CrossRef CAS PubMed.
- CCDC 891204 (2a), CCDC 891201 (2b), CCDC 891203 (4a), CCDC 964933 (4b), and CCDC 891202 (5).
- M. T. Reetz, H. Guo, J.-A. Ma, R. Goddard and R. J. Mynott, J. Am. Chem. Soc., 2009, 131, 4136–4142 CrossRef CAS PubMed.
- D. Morales-Morales, R. Redón, Y. Zheng and J. R. Dilwort, Inorg. Chim. Acta, 2002, 39–44 CrossRef CAS.
- See ESI† for additional details.
- M. Besora, C. Gourlaouen, B. Yates and F. Maseras, Dalton Trans., 2011, 40, 11089–11094 RSC.
- (a) S. Komiya, T. A. Albright, R. Hoffmann and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 7255–7265 CrossRef CAS; (b) Reductive elimination of MeI from LAuI2Me: V. J. Scott, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 4090–4096 CrossRef CAS.
- M. Besora, A. A. C. Braga, G. Ujaque, F. Maseras and A. Lledós, Theor. Chem. Acc., 2011, 128, 639–646 CrossRef CAS.
- Although the majority of Au(I) complexes are two-coordinated, linear 14-electron species, three- and four-coordinated Au(I) complexes are also known: (a) M. C. Gimeno and A. Laguna, Chem. Rev., 1997, 97, 511–522 CrossRef CAS PubMed; (b) V. R. Bojan, E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, R. C. Puelles and C. Silvestru, Inorg. Chem., 2010, 49, 5530–5541 CrossRef CAS PubMed , and references therein.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 891201–891204 and 964933. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc48914k |
| This journal is © The Royal Society of Chemistry 2014 |