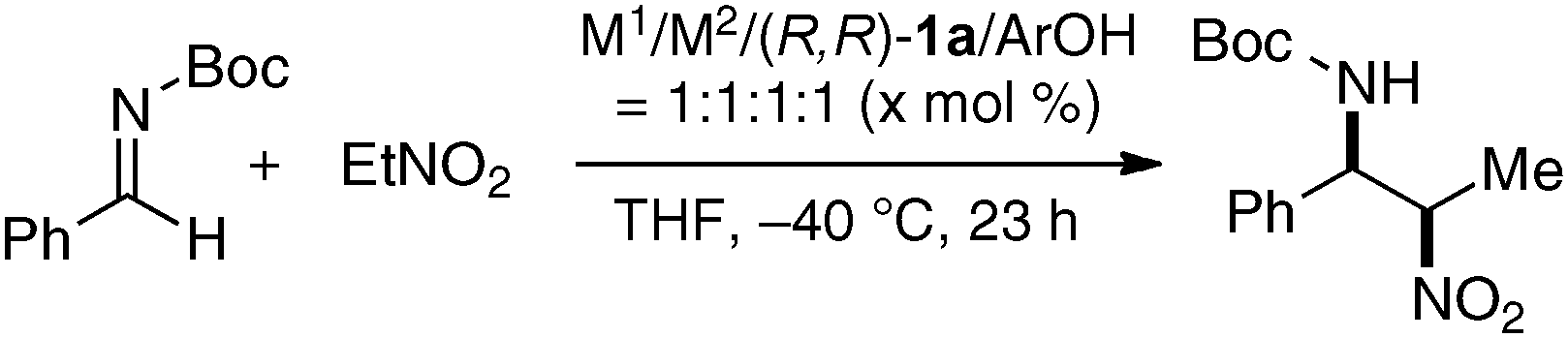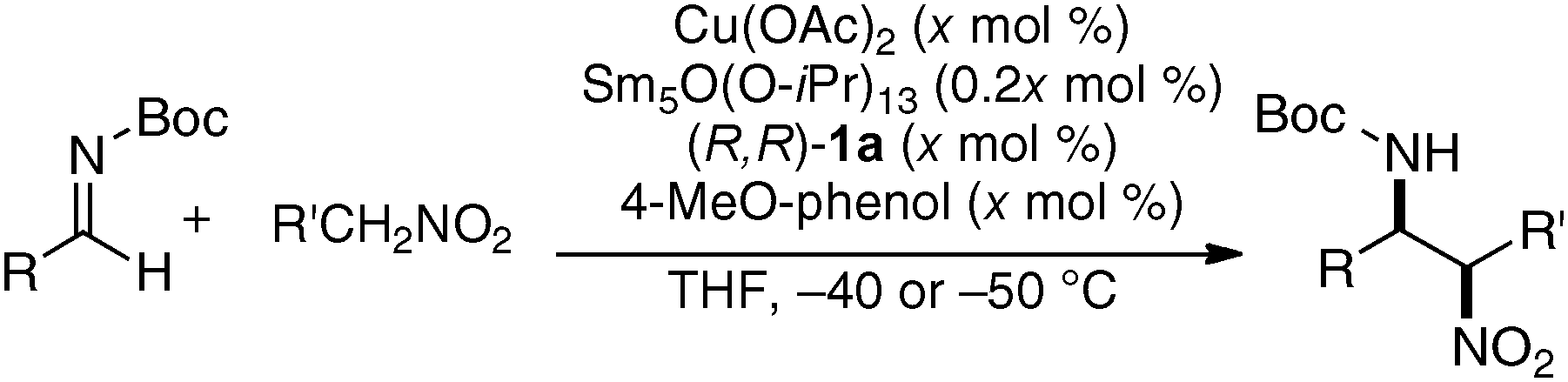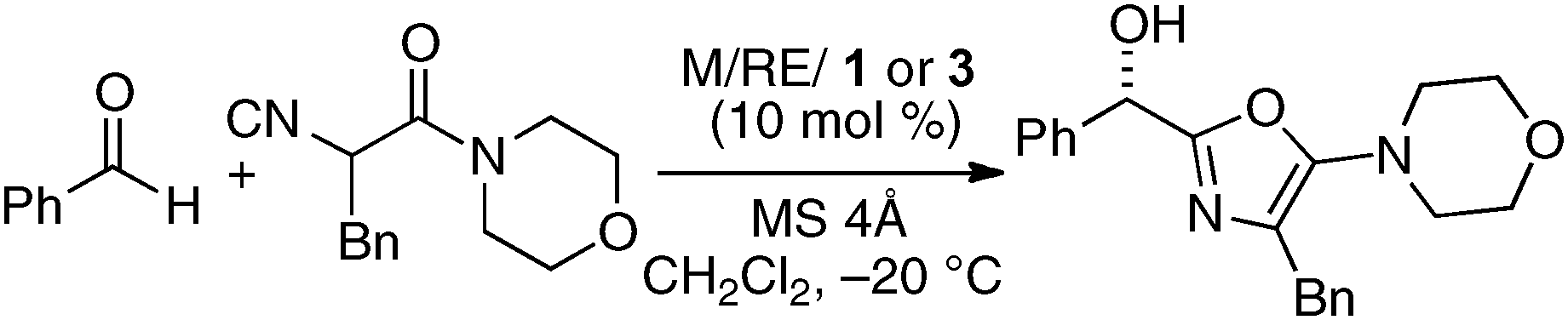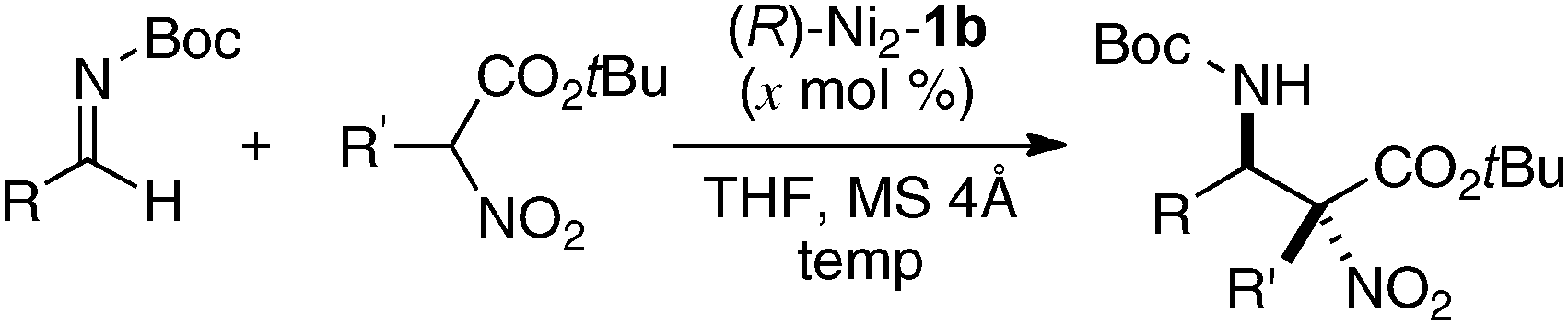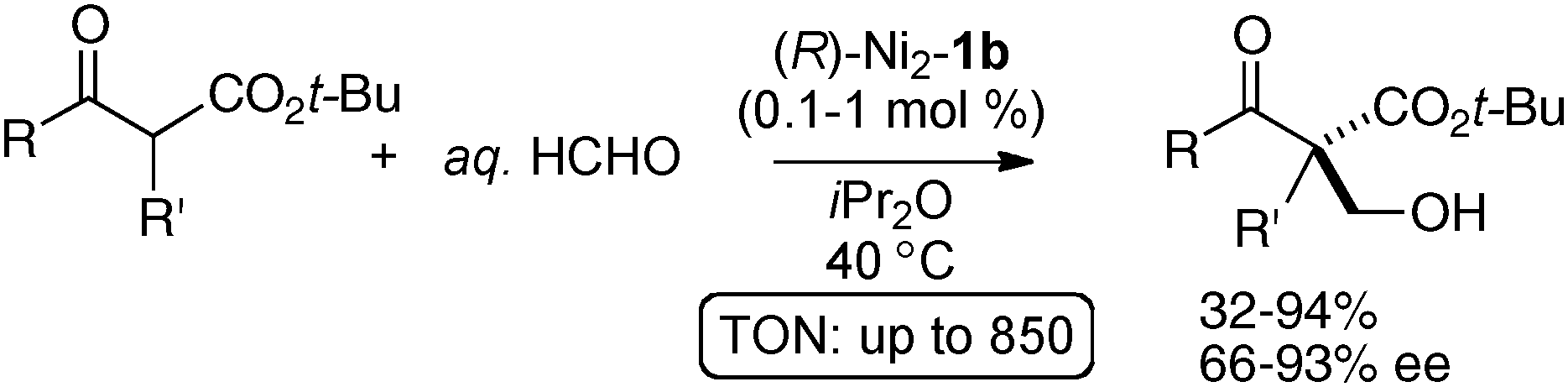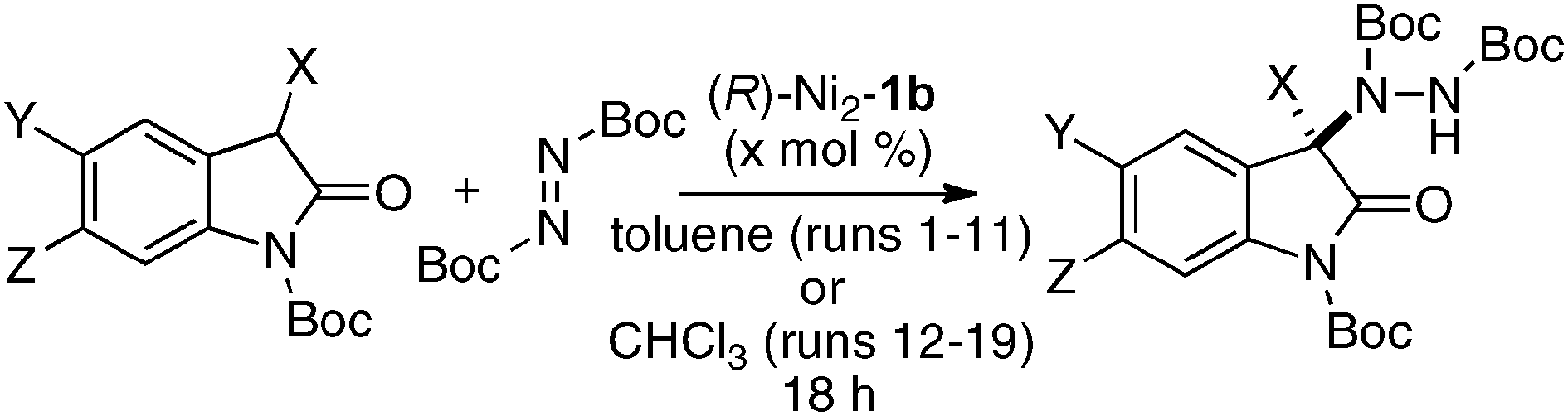DOI:
10.1039/C3CC47587E
(Feature Article)
Chem. Commun., 2014,
50, 1044-1057
Recent advances in cooperative bimetallic asymmetric catalysis: dinuclear Schiff base complexes
Received
3rd October 2013
, Accepted 4th November 2013
First published on 5th November 2013
Abstract
Cooperative catalysis has proven to be a powerful strategy for realizing high reactivity and selectivity in asymmetric transformations. A variety of cooperative asymmetric catalysts have been developed over the last two decades. In this feature article, recent advances from our research on cooperative asymmetric catalysis, focusing on dinuclear Schiff base catalysis, are described. Design of dinuclear Schiff base catalysts and their applications in several asymmetric C–C and C–N bond-forming reactions under simple proton transfer conditions with perfect atom-economy are discussed in detail.

Shigeki Matsunaga
| Shigeki Matsunaga is an associate professor at the University of Tokyo. He was born in 1975, and received his PhD from the University of Tokyo under the direction of professor M. Shibasaki. He started his academic career in 2001 as an assistant professor in professor Shibasaki's lab. He was promoted to a senior lecturer in 2008, and to his current position in 2011. He is the recipient of the Chemical Society of Japan Award for Young Chemists (2006), Mitsui Chemicals Catalysis Science Award of Encouragement (2009), Merck-Banyu Lectureship Award 2010, and others. His research interests are in cooperative asymmetric catalysis, C–H bond functionalization, and synthesis of biologically active compounds. |

Masakatsu Shibasaki
| Masakatsu Shibasaki received his PhD from the University of Tokyo in 1974 under the direction of the late professor Shun-ichi Yamada before conducting postdoctoral studies with Professor E. J. Corey at Harvard University. In 1977, he returned to Japan and joined Teikyo University as an associate professor. In 1983, he moved to Sagami Chemical Research Center as a group leader, and in 1986 took up a professorship at Hokkaido University, before returning to the University of Tokyo as a professor in 1991. Currently, he is a director of the Institute of Microbial Chemistry (Tokyo). His research interests include asymmetric catalysis and medicinal chemistry of biologically significant compounds. |
Introduction
Asymmetric catalysis has established its firm position as an efficient method for constructing a wide range of enantiomerically enriched compounds. Intensive efforts have been devoted to this field over decades, leading to the development of numerous excellent asymmetric catalysts.1 Many organic transformations can now be rendered into catalytic and enantioselective processes. The aim of synthetic methodology development has gradually shifted towards environmentally benign organic synthesis, and not only high yield and high stereoselectivity of organic transformations, but also atom-economy2 and step-economy3 of the processes are currently considered to be important factors. Conventional studies on asymmetric catalysis realized high yield as well as high regio- and stereoselectivity, but often with the help of stoichiometric activating reagents. It is highly desirable to minimize the waste and by-products to achieve high overall efficiency in organic synthesis and match the criteria of green chemistry.
Intermolecular direct catalytic asymmetric C–C bond-forming reactions are highly attractive because of their atom-economical processes.4 In the direct C–C bond-forming reaction, an active carbon nucleophile, such as an enolate, is catalytically generated in situ via deprotonation of the pronucleophile (Nu–H in Fig. 1) with a Brønsted basic catalyst. Addition of the carbon nucleophile to an electrophile followed by protonation provides the product. In principle, the direct catalytic reaction proceeds with 100% atom economy under simple proton transfer conditions, thus providing useful building blocks without any waste derived from stoichiometric reagents. To induce high reactivity and stereoselectivity in the direct catalytic asymmetric C–C bond-forming reactions, the concurrent activation of pronucleophiles and electrophiles is highly desirable to control the stereochemical outcome of reactions, possibly through both Brønsted basic as well as Lewis acidic and/or Brønsted acidic sites of catalysts. Acid and base cooperative functions are often utilized in enzymes to promote enantioselective reactions under remarkably mild conditions via a simple proton transfer process. For example, the postulated transition state model of class II aldolase, a Zn-dependent metalloenzyme, clearly indicates the power of acid and base cooperative functions to activate both nucleophiles and electrophiles.5 As shown in Fig. 2, the glutamate-73 residue functions as a Brønsted base to deprotonate a pronucleophile, dihydroxyacetone phosphate, which is activated by the coordination to Lewis acidic Zn2+. On the other hand, the position of the aldehyde is controlled by the Brønsted acidic tyrosine-113′ residue. With the appropriate control of two reactants in close proximity, high reactivity, stereoselectivity as well as high atom-economy were achieved in enzymatic aldol reactions under mild reaction conditions.
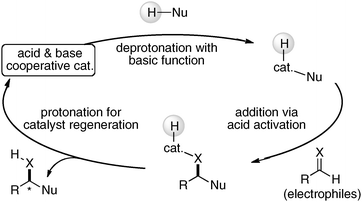 |
| | Fig. 1 Direct catalytic asymmetric reactions via a simple proton transfer process. | |
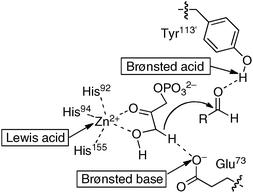 |
| | Fig. 2 Postulated acid and base cooperative mechanism of class II aldolase. | |
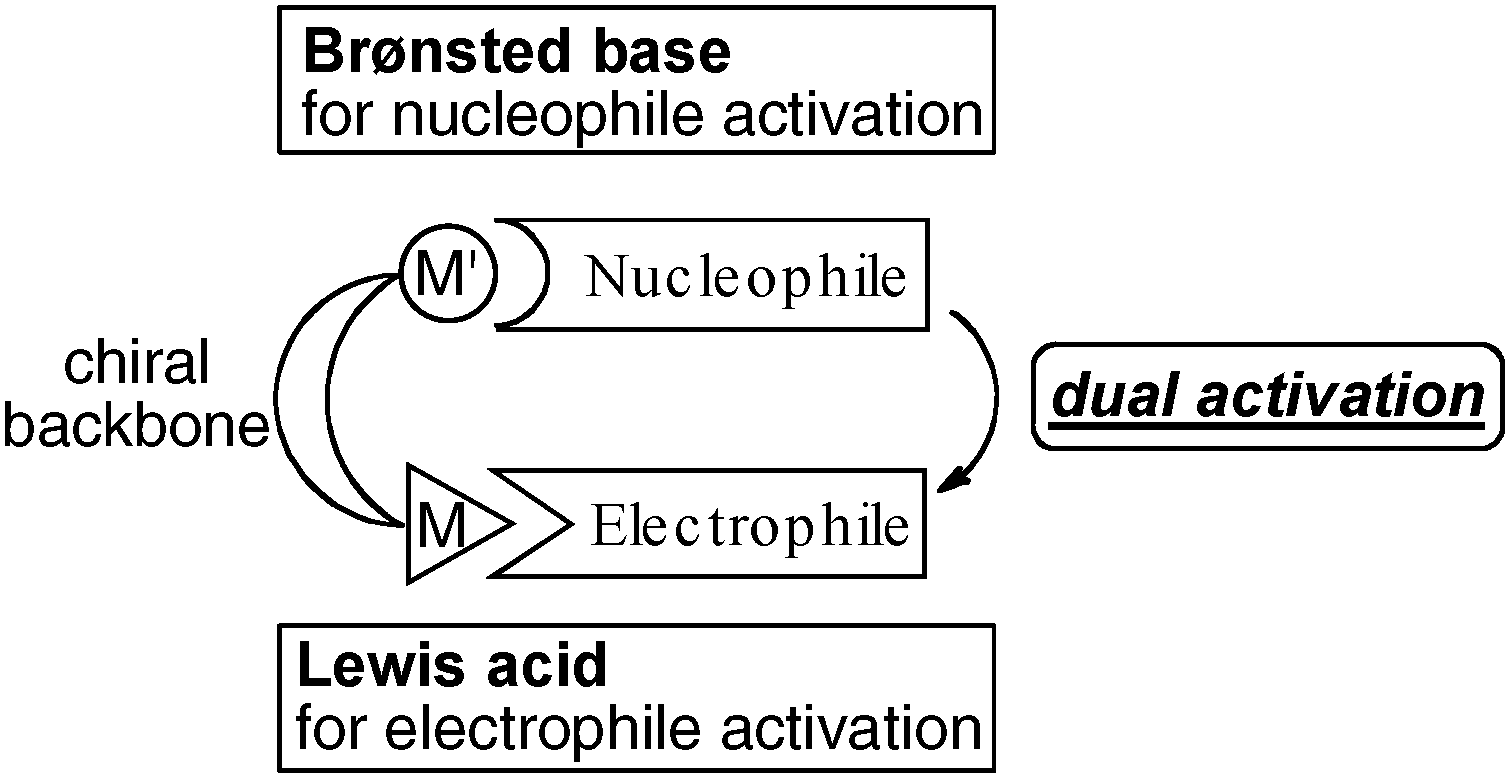
Inspired by the intriguing cooperative mechanism of enzymes, a variety of artificial cooperative asymmetric metal- and organo-catalysts have been developed over the last two decades.6 Our group has intensively worked in this field and developed various bimetallic Lewis acid and Brønsted base cooperative asymmetric catalysts. The concept of bimetallic cooperative asymmetric catalysis is shown in Fig. 3. One metal center together with its counter ion acts as a Brønsted base to activate pronucleophiles, while the other Lewis acidic metal center activates electrophiles. Both reaction partners are simultaneously activated by fine-tuned bimetallic catalysts with an appropriate chiral environment. In this account, we highlight our own research on the development of a new family of bimetallic cooperative asymmetric catalysts, dinuclear Schiff base catalysts, and their applications in asymmetric C–C and C–N bond-forming reactions since 2007.
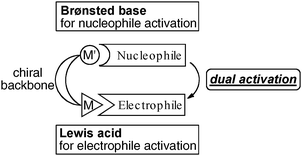 |
| | Fig. 3 Dual activation with bimetallic cooperative asymmetric catalysts. | |
Heterodinuclear Schiff base catalysis
Design of dinucleating Schiff bases
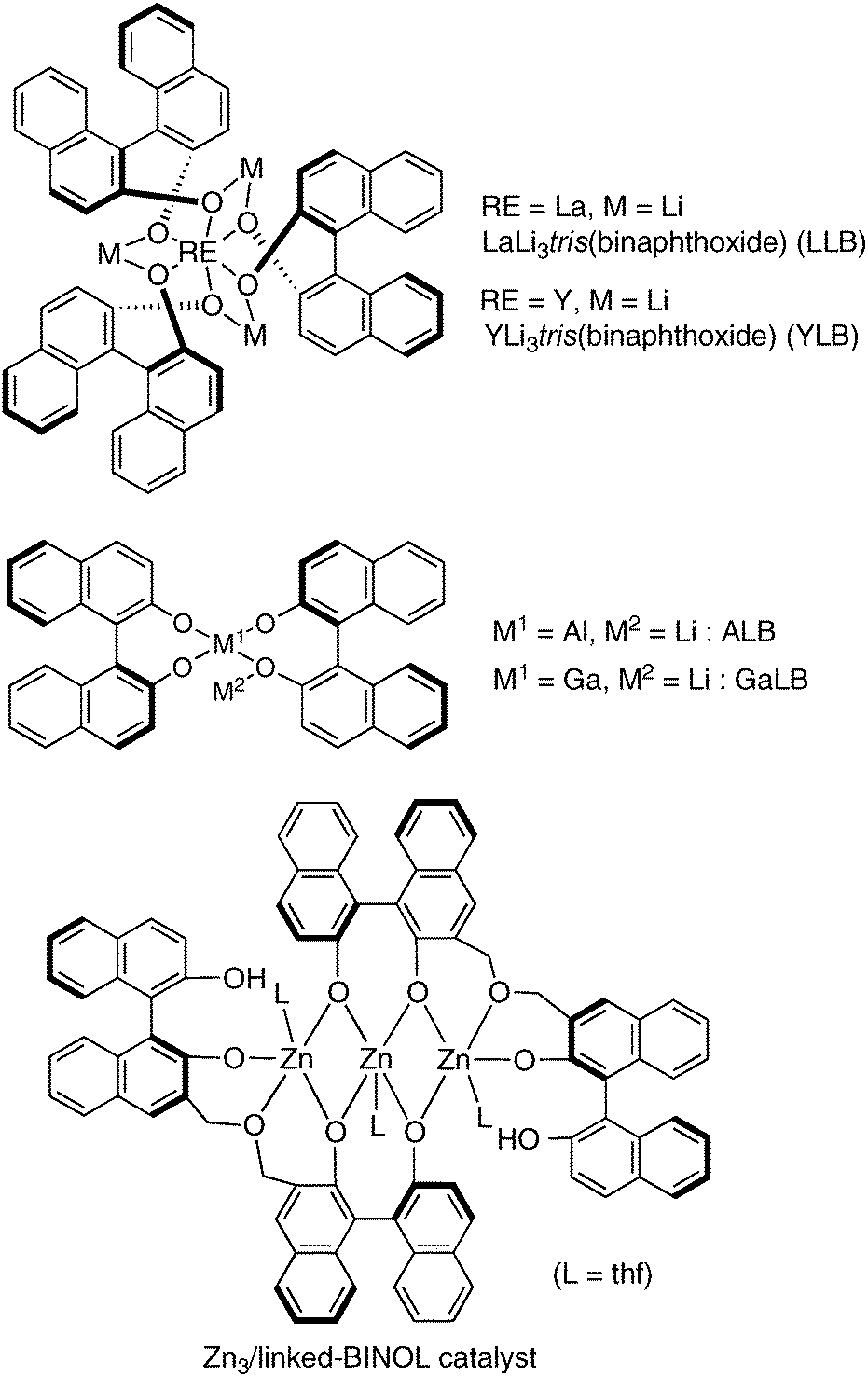
When designing cooperative asymmetric catalysts, the construction of a suitable chiral environment for each targeted reaction is important for achieving efficient dual activation of nucleophiles and electrophiles through the cooperation of two metal centers. Optimization of enantioselectivity and reactivity requires a strategy for constructing a readily tunable chiral environment. Various chiral scaffolds have been utilized by our group to construct multimetallic self-assembled complexes for realizing efficient cooperative dual activation. At the early stage of our work, we mainly utilized BINOL-based ligands, and various multimetallic Lewis acid and Brønsted base cooperative asymmetric catalysts, such as heterobimetallic rare earth metal/alkali metal/binaphtholate = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3 complexes,7 heterobimetallic group 13 metal/alkali metal/binaphtholate = 1:1:2 complexes,8 a trinuclear Zn/linked-BINOL = 3:2 complex,9 and others, have been developed (Fig. 4). Those BINOL-based multimetallic catalysts were powerful and promoted a variety of asymmetric reactions. The use of BINOL-based ligands, however, restricted the applicable metal sources to relatively oxophilic ones, such as alkali metals, rare earth metals, group 13 metals, zinc, and others. Transition metals, like Mn, Co, Ni, and Cu, are often not suitable for BINOL-based chiral ligands. In order to broaden the scope of bimetallic cooperative asymmetric catalysis, the development of new multidentate chiral ligands is highly desirable. We believe that new metal combinations will lead to novel catalytic activity and selectivity in cooperative catalysis.
:3:3 complexes,7 heterobimetallic group 13 metal/alkali metal/binaphtholate = 1:1:2 complexes,8 a trinuclear Zn/linked-BINOL = 3:2 complex,9 and others, have been developed (Fig. 4). Those BINOL-based multimetallic catalysts were powerful and promoted a variety of asymmetric reactions. The use of BINOL-based ligands, however, restricted the applicable metal sources to relatively oxophilic ones, such as alkali metals, rare earth metals, group 13 metals, zinc, and others. Transition metals, like Mn, Co, Ni, and Cu, are often not suitable for BINOL-based chiral ligands. In order to broaden the scope of bimetallic cooperative asymmetric catalysis, the development of new multidentate chiral ligands is highly desirable. We believe that new metal combinations will lead to novel catalytic activity and selectivity in cooperative catalysis.
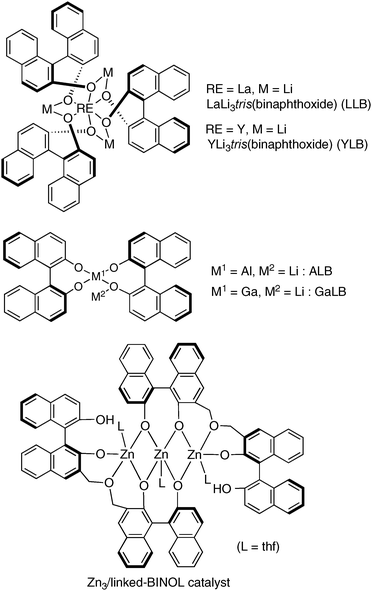 |
| | Fig. 4 Early examples of BINOL-based Lewis acid and Brønsted base multimetallic cooperative asymmetric catalysts. | |
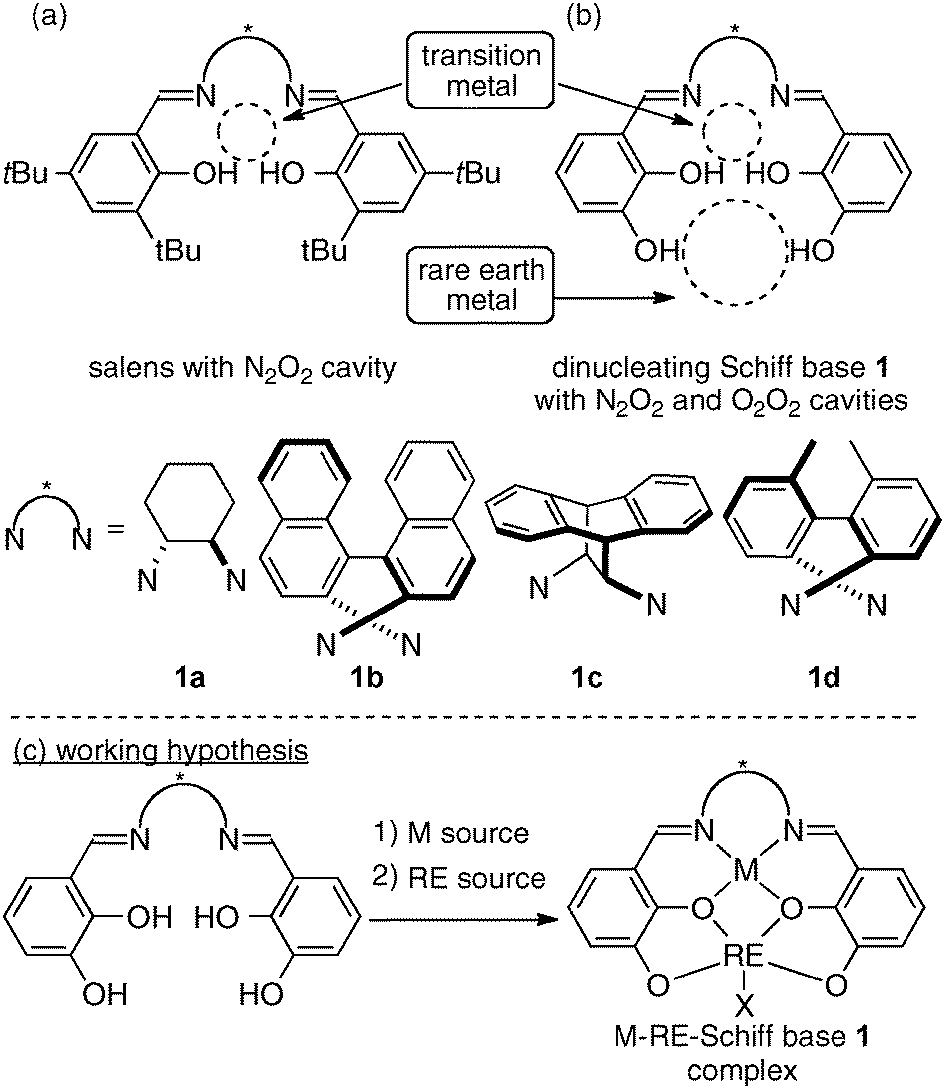
Chiral Schiff base ligands, such as salens, are one of the privileged ligand classes in asymmetric catalysis.1c,10,11 Over the last two decades, various chiral transition metal–salen complexes have been widely utilized for a broad range of catalytic asymmetric reactions (Fig. 5a). As to the cooperative catalysis field, Jacobsen and coworkers have developed an elegant intermolecular bimolecular cooperative system using two metal–salen complexes.12 Attempts to strengthen the catalytic cooperativity of two metal–salen complexes by linking two metal–salen units have also been reported.13 Furthermore, several groups have expanded the potential of Schiff base ligands for heterobimetallic transition metal/alkali metal catalysts and transition metal/group 13 metal combined catalysts.14 The advantages of heterobimetallic Schiff base complexes in comparison with mononuclear Schiff complexes for intermolecular bimolecular cooperative systems, however, had been rather limited at the time we started our project.
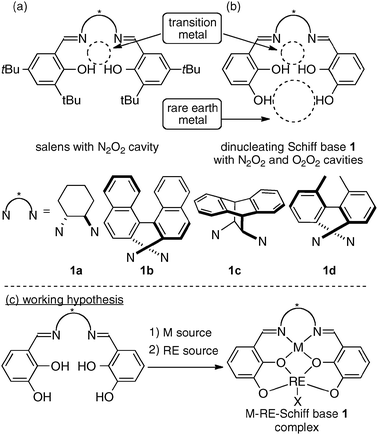 |
| | Fig. 5 (a) Salens for mononuclear complexes; (b) our design of dinucleating Schiff base 1 with two distinct cavities; (c) working hypothesis to form heterodinuclear cooperative Schiff base complexes with transition metal/rare earth metal combination. | |
When developing bimetallic asymmetric catalysts for intramolecular cooperative catalysis, the design of a suitable multidentate ligand is important for appropriately controlling the positions of two metals in the complex. The positions of the two metals substantially affect the reactivity and stereoselectivity of the bimetallic complexes. At the initial stage of our study, we tried several dinucleating Schiff base ligands, and new dinucleating Schiff bases 1 with additional phenolic hydroxyl units, in comparison with standard salens, gave promising results (Fig. 5b). Based on precedents in the field of coordination chemistry,15 we hypothesized that the Schiff bases 1 would selectively incorporate a transition metal into the N2O2 inner cavity and an oxophilic rare earth metal with a much larger ionic radius into the O2O2 outer cavity (Fig. 5c).
Heterobimetallic transition metal/Brønsted basic rare earth metal/Schiff base catalysts
As a model reaction to evaluate bimetallic Schiff base complexes, we selected aza-Henry reaction, which is a powerful C–C bond-forming reaction to provide chiral 1,2-diamines.16 Effects of metal combination on the aza-Henry reaction are summarized in Table 1, runs 1–8. As expected, the selection of a suitable metal combination for the targeted reaction was critical for achieving good stereoselectivity. A heterobimetallic complex, prepared from Cu(OAc)2, Sm(O-iPr)3, and dinucleating Schiff base 1a was the best, giving product in >20:1 syn-selectivity and 80% ee. Other metal combinations resulted in much lower selectivity and/or stereoselectivity. Either Cu or Sm alone resulted in poor reactivity and selectivity (runs 9–10), suggesting that both Cu and Sm are essential for good performance.17 Further optimization studies revealed that an achiral phenol source improved the enantioselectivity. The use of oxo-samarium alkoxide, Sm5O(O-iPr)13, which has a well-ordered structure, gave better enantioselectivity when reducing the catalyst loading to 5 mol%.
Table 1 Effects of metal combination and achiral phenol in catalytic asymmetric aza-Henry reaction
Selected examples of the substrate scope of the Cu/Sm/1a-catalyzed aza-Henry reaction are shown in Table 2. Not only aryl and heteroaryl N-Boc-imines (runs 1–9 and 13–15), but also readily isomerizable aliphatic N-Boc-imines were applicable (runs 10–12). The Cu/Sm/1a-catalyst was also applicable to β-silyloxy-substituted aliphatic imine, and the utility of the present reaction was demonstrated in a concise catalytic asymmetric synthesis of nemonapride, which is used clinically as an antipsychotic agent (Scheme 1).18
Table 2 Cu/Sm/1a-catalyzed asymmetric aza-Henry reaction
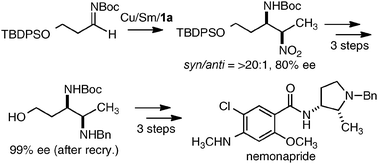 |
| | Scheme 1 Catalytic asymmetric synthesis of nemonapride. | |
To obtain insight into the structure of the active species and the role of the achiral phenol source, a mixture of the Cu:Sm:ligand 1a = 1:1:1 system was analyzed by ESI-MS with and without added achiral phenol. In the absence of any achiral phenol, no peak corresponding to the monomeric Cu:Sm:ligand 1a = 1:1:1 complex was detected. Instead, several peaks corresponding to oligomeric species, such as the μ-oxo-trimer [Cu3Sm3O(ligand 1a)3]+, a hexamer, a heptamer, an octamer, and a decamer, were observed. In the presence of an achiral phenol, the peak due to the μ-oxo-trimer [Cu3Sm3O(ligand 1a)3]+ (m/z = 1710) was similarly observed. Peaks for higher m/z for higher order oligomers, however, became much weaker, suggesting that such higher order oligomers partially dissociate into the μ-oxo-μ-aryloxy trimer in the presence of an achiral phenol (Fig. 6). On the basis of these ESI-MS observations as well as the positive non-linear-effects observed in this system, we speculated that the μ-oxo-μ-aryloxy trimer complex is the most enantioselective active species. Based on the initial kinetic studies and kinetic isotope effect studies, the catalytic cycle shown in Fig. 7 was postulated for this reaction. For this catalyst system, the results shown in Table 1 demonstrate that both Cu and Sm are essential. We assume that the cooperative dual activation of nitroalkanes and imines with Cu and Sm-aryloxide is important to achieve this syn-selective catalytic asymmetric aza-Henry reaction. The Sm-aryloxide moiety in the catalyst would act as a Brønsted base to generate Sm-nitronate, while Cu(II) would act as a Lewis acid to control the position of the N-Boc-imine. Among the possible transition states, the sterically less hindered TS-1 is more favorable than TS-2. Thus, the stereoselective C–C bond-formation via TS-1 followed by protonation by the phenolic proton affords the syn-product and regenerates the catalyst.
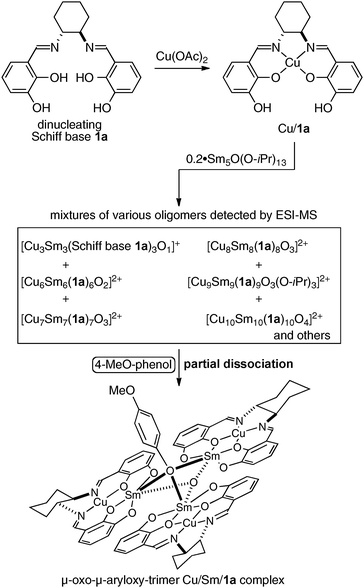 |
| | Fig. 6 Postulated role of achiral phenol additive and the postulated structure of the μ-oxo-μ-aryloxy-trimer Cu/Sm/1a active species. | |
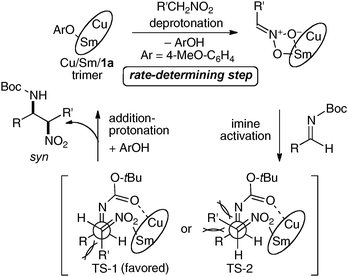 |
| | Fig. 7 Postulated catalytic cycle of aza-Henry reaction under heterobimetallic cooperative catalysis. | |
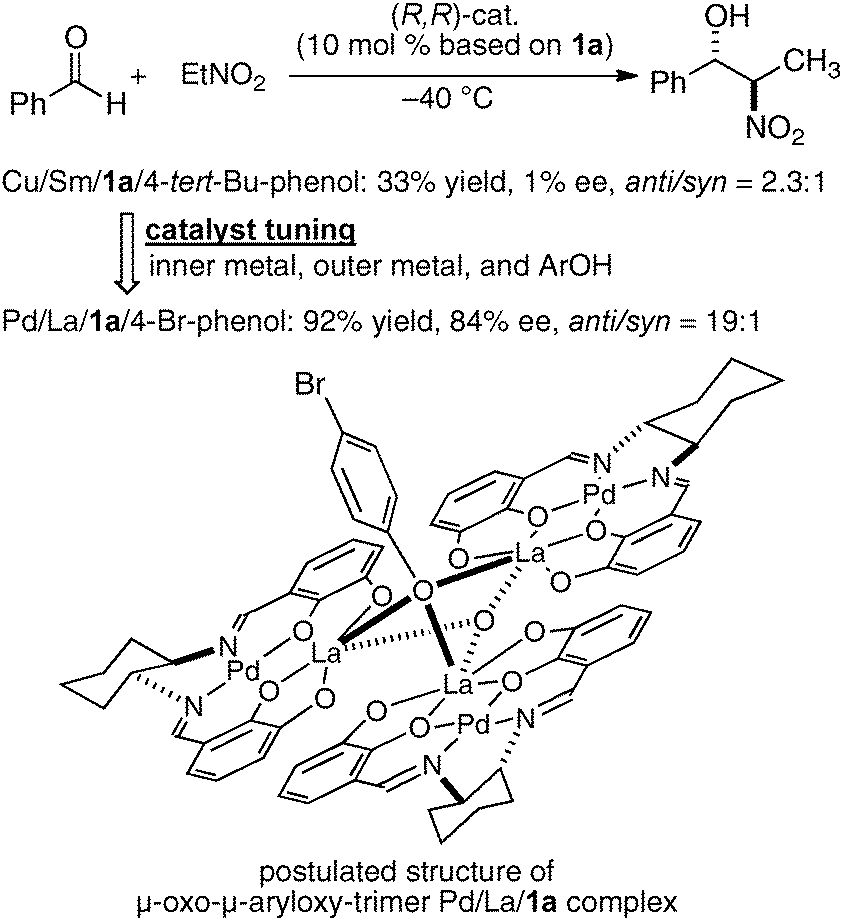
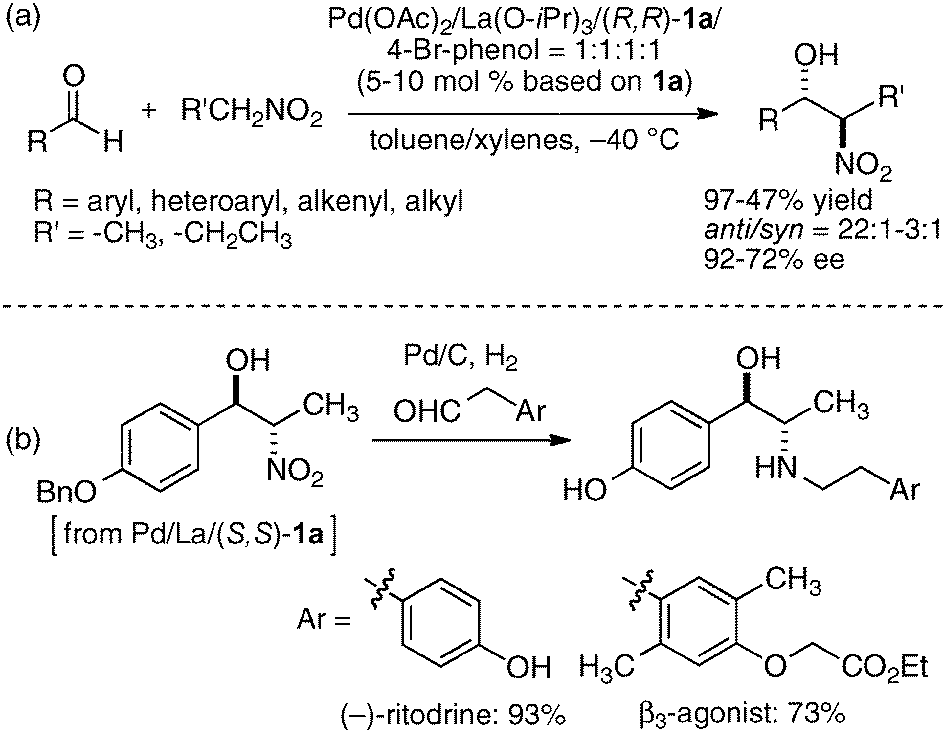
In the bimetallic system, the choice of transition metal and rare earth metal combination drastically affected the enantioselectivity of aza-Henry reaction, as observed in Table 1. We assumed that a variety of chiral environments could be readily constructed using the same ligand 1a just by changing each component of the heterobimetallic complex, transition metal, rare earth metal, and an achiral phenol additive. As a proof of concept, we applied the dinucleating Schiff base 1a to the anti-selective asymmetric Henry reaction.19,20 As shown in Scheme 2, the Cu–Sm–Schiff base complex gave poor results in the reaction with aldehyde, giving the product in only 33% yield, anti/syn ratio = 2.3:1, and 1% ee. Screening of the transition metal, rare earth metal, phenol additive, and other reaction conditions finally revealed that Pd/La/Schiff base 1a with the 4-bromophenol additive was optimum, giving the product in 92% yield, anti/syn ratio = 19:1, and 84% ee. The results support our initial assumption that the dinucleating Schiff base is suitable for constructing diverse chiral environments which enable optimization of targeted asymmetric reactions by appropriate selection of each catalyst component. With the optimized Pd–La–1a system, the anti-selective catalytic asymmetric Henry reaction of various aldehydes proceeded with up to 92% ee and 22:1 anti-selectivity (Scheme 3a).21 The reaction was also applied to the short syntheses of some β-adrenoceptor agonists (Scheme 3b).
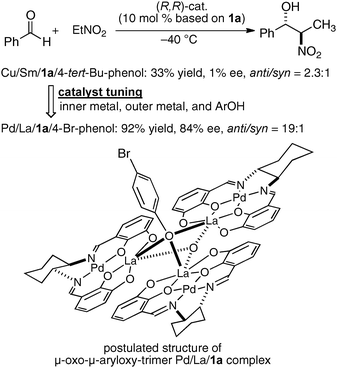 |
| | Scheme 2 Catalyst tuning for anti-selective Henry reaction and a postulated structure of the Pd–La–1a complex. | |
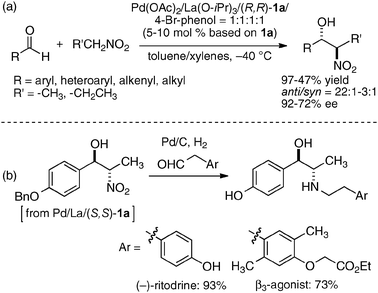 |
| | Scheme 3 (a) Pd/La/1a-catalyzed anti-selective catalytic asymmetric Henry reaction; (b) catalytic asymmetric synthesis of β-adrenoceptor agonists. | |
The heterobimetallic system with transition metal and Brønsted basic rare earth metal alkoxide was further expanded to include the use of a reduced-type dinucleating amino-phenol ligand 2.22 A catalyst prepared from Ni(OAc)2, La(O-iPr)3, and ligand 2 in a ratio of 1:1:1 was the best for catalytic asymmetric decarboxylative 1,4-addition of a malonic acid half-thioester. The addition of achiral phosphine oxide was effective in improving the reactivity of the catalyst, possibly by enhancing the Brønsted basicity of the lanthanum aryloxide moiety. Decarboxylated 1,4-adducts were obtained in 40–99% yield and 66–92% ee (Scheme 4).23 The products of this reaction are useful synthetic intermediates for γ-amino acids,24 and the Ni/La/ligand 2 catalyst was applied to the catalytic asymmetric synthesis of (S)-rolipram, an anti-deppresant agent.
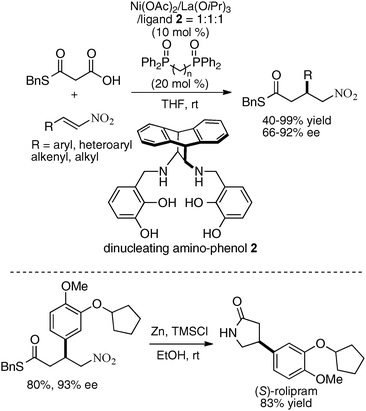 |
| | Scheme 4 Heterobimetallic Ni–La system for decarboxylative 1,4-addition of malonic acid half-thioester. | |
Heterobimetallic catalysts containing cationic rare earth metal as a Lewis acid unit
In the previous section, heterobimetallic systems including Brønsted basic rare earth metal aryloxide moieties were described. As to rare earth metal sources, cationic rare earth metal sources have also been widely utilized as Lewis acids.25 If a cationic rare earth metal can be incorporated into the outer cavity of dinucleating Schiff bases, the resulting heterobimetallic system should be also useful as different types of cooperative asymmetric catalysts.
To incorporate a cationic rare earth metal into the outer O2O2 cavity of a dinucleating Schiff base, we utilized Schiff base 3 derived from o-vanillin (Fig. 8). We selected α-additions of α-isocyanoacetamides to aldehydes as a model reaction because a salen–Al catalyst gave only modest enantioselectivity and reactivity for the transformation.26,27 Our working hypothesis for bimetallic Schiff base catalysis is shown in Fig. 9. We assumed that the bimetallic Schiff base complex would not only activate the aldehyde with one metal, but might also interact with the α-isocyanoacetamide through the other metal to effectively control the orientation of the two substrates in the enantio-discriminating step (Fig. 9, Lewis acid/Lewis acid cooperative catalysis). After screening suitable metal combinations for Schiff base 3, Ga(O-iPr)3/Yb(OTf)3 was found to afford promising results. The chiral diamine backbone affected both the reactivity and enantioselectivity (Table 3, runs 1–3). Schiff base 3a gave unsatisfactory results, but 3b containing a binaphthyl diamine unit gave 78% ee (run 2). The best reactivity and selectivity were achieved using Schiff base 3c with an anthracene-derived diamine unit (run 3, 67% yield, 94% ee). After optimization of the Ga/Yb ratio (1:0.95), the product was obtained in >95% yield and 96% ee after 24 h (run 4). To confirm the utility of the Ga(O-iPr)3/Yb(OTf)3 combination, several control experiments were performed with the best ligand 3c (runs 5–12). Neither Ga–3c alone nor Yb–3c alone efficiently promoted the reaction (runs 5–6). For Ga(O-iPr)3 and other rare earth metal triflates, the reactivity decreased in correlation with the Lewis acidity of the rare earth metal (Yb > Gd > Nd > La),28 while good to excellent enantioselectivity was maintained in runs 7–9 (89–96% ee). We investigated Et2AlCl, Al(O-iPr)3, and In(O-iPr)3 as other sources of group 13 metals for the inner cavity (runs 10–12), but the results were less satisfactory than that of entry 4. In entry 13, Schiff base 1c (Fig. 5) was utilized instead of Schiff base 3c, but no reaction occurred. The results described in runs 5–13 indicated that the Ga(O-iPr)3/Yb(OTf)3 combination as well as Schiff base 3c derived from o-vanillin were essential for obtaining high reactivity and enantioselectivity in the present reaction. The optimized Lewis acid–Lewis acid cooperative system was applicable to a broad range of aryl, heteroaryl, alkenyl, and alkyl aldehydes, giving products in 88–98% ee (Scheme 5).29
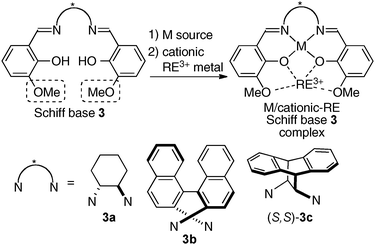 |
| | Fig. 8 Design of Schiff bases 3 for the preparation of heterobimetallic complexes incorporating cationic rare earth metals (RE3+). | |
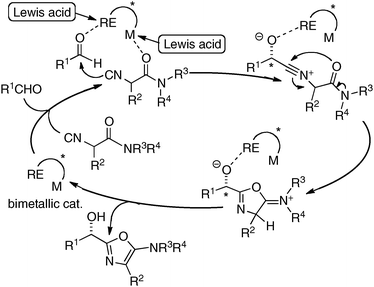 |
| | Fig. 9 Working hypothesis for Lewis acid/Lewis acid cooperative catalysis with a heterobimetallic complex including a cationic rare earth metal. | |
Table 3 Effects of metal combination and Schiff bases for α-addition of isocyanoacetamide to benzaldehyde
|
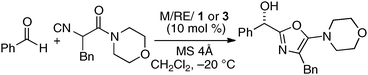
|
| Run |
M source |
RE source (x mol%) |
Ligand |
Time (h) |
Yield (%) |
ee (%) |
| 1 |
Ga(O-iPr)3 |
Yb(OTf)3 (10) |
3a
|
74 |
55 |
29 |
| 2 |
Ga(O-iPr)3 |
Yb(OTf)3 (10) |
3b
|
71 |
27 |
78 |
| 3 |
Ga(O-iPr)3 |
Yb(OTf)3 (10) |
3c
|
74 |
67 |
94 |
| 4 |
Ga(O-iPr)3 |
Yb(OTf)3 (9.5) |
3c
|
24 |
>95 |
96 |
| 5 |
Ga(O-iPr)3 |
None |
3c
|
24 |
Trace |
— |
| 6 |
None |
Yb(OTf)3 (10) |
3c
|
24 |
Trace |
— |
| 7 |
Ga(O-iPr)3 |
Gd(OTf)3 (9.5) |
3c
|
24 |
62 |
96 |
| 8 |
Ga(O-iPr)3 |
Nd(OTf)3 (9.5) |
3c
|
24 |
33 |
95 |
| 9 |
Ga(O-iPr)3 |
La(OTf)3 (9.5) |
3c
|
24 |
11 |
89 |
| 10 |
Et2AlCl |
Yb(OTf)3 (9.5) |
3c
|
48 |
68 |
37 |
| 11 |
Al(O-iPr)3 |
Yb(OTf)3 (9.5) |
3c
|
24 |
>95 |
79 |
| 12 |
In(O-iPr)3 |
Yb(OTf)3 (9.5) |
3c
|
24 |
55 |
80 |
| 13 |
Ga(O-iPr)3 |
Yb(OTf)3 (9.5) |
1c
|
24 |
Trace |
— |
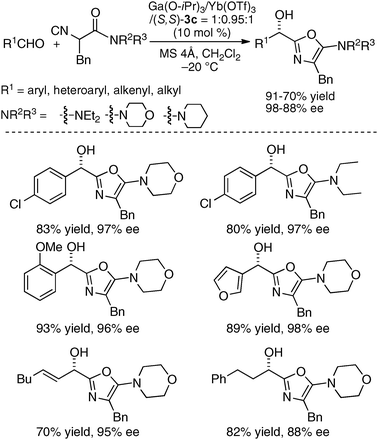 |
| | Scheme 5 Catalytic asymmetric α-addition of isocyanides to aldehydes under heterobimetallic Lewis acid/Lewis acid cooperative catalysis. | |
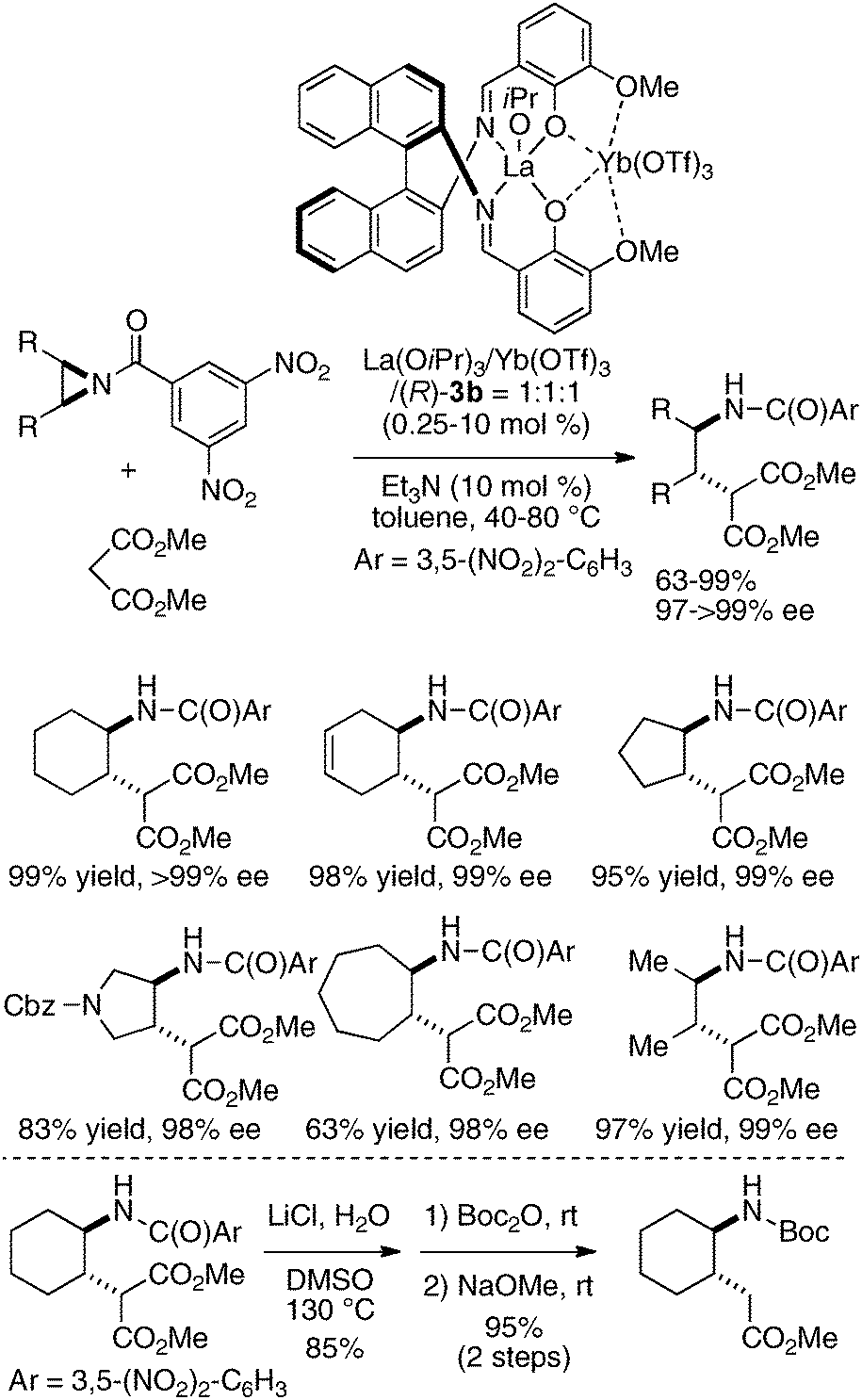
The utility of the dinucleating Schiff bases 3 derived from o-vanillin30 was further expanded to a heterobimetallic catalyst composed of two distinct rare earth metals, a cationic Lewis acidic rare earth metal and a Brønsted basic rare earth metal alkoxide. Lewis acidic Yb(OTf)3 and Brønsted basic La(O-iPr)3 were successfully placed onto the same dinucleating ligand 3b, and the heterobimetallic La/Yb/3b = 1:1:1 catalyst promoted the asymmetric ring-opening reaction of meso aziridines31 with malonates. The desymmetrized ring-opening adducts were obtained in 63–99% yield and 98–99% ee from cyclic and acyclic meso aziridines (Scheme 6).32 The utility of the products was demonstrated through transformation of the ring-opened products into cyclic γ-amino acids, which are useful building blocks in foldamer research.33
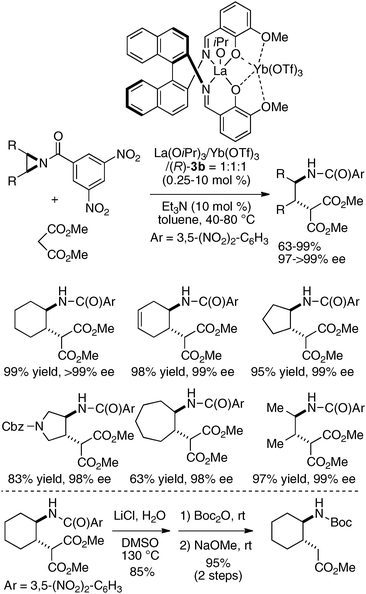 |
| | Scheme 6 Catalytic asymmetric ring opening of meso aziridines promoted by a heterodinuclear rare earth metal Schiff base 3c catalyst and its application to cyclic γ-amino acid synthesis. | |
Homobimetallic first-row transition metal catalysts
In the heterodinuclear Schiff base catalysts described in previous sections, it is important to establish the method of obtaining uniform catalytically active species by selectively placing two different metals into two distinct cavities. If different heterodinuclear complexes exist in equilibrium, that may lead to low enantioselectivity. To avoid difficulty in the preparation of dinuclear Schiff base complexes, the possible metal combination was limited to some extent, that is, large and oxophilic rare earth metals were used as the second metal to be selectively incorporated into the outer O2O2 cavity. In order to further expand the diversity of dinuclear Schiff base catalysis, we planned to develop dinuclear Schiff base cooperative catalysts without using rare earth metals.
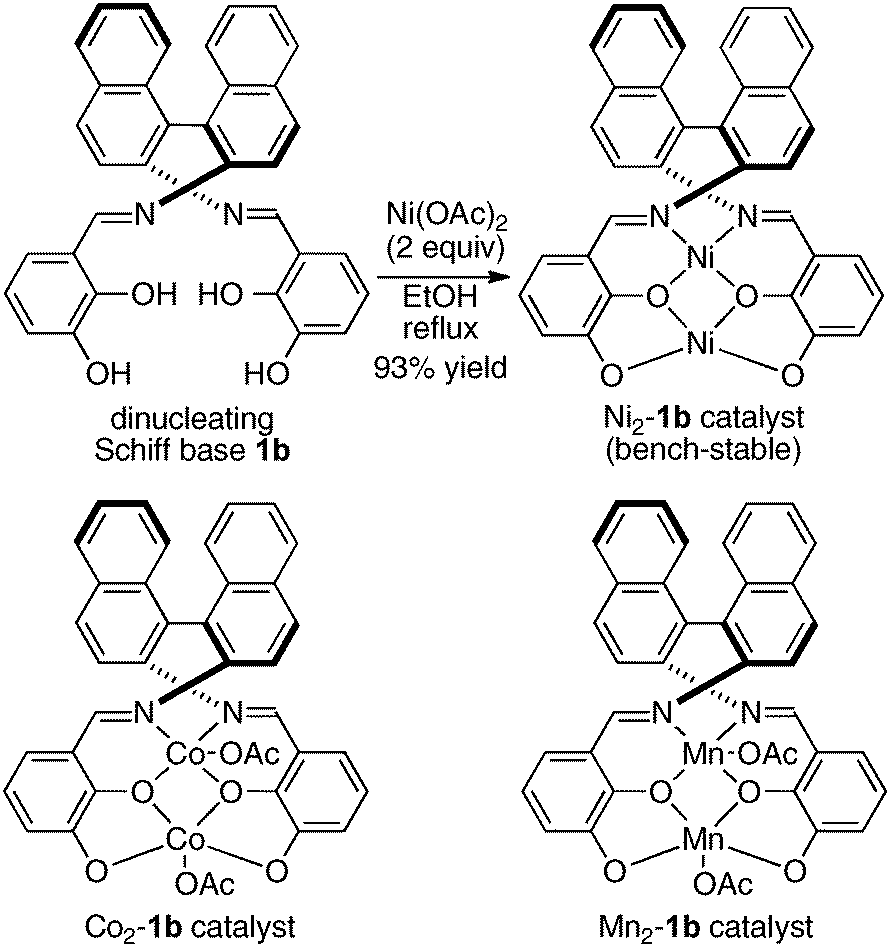
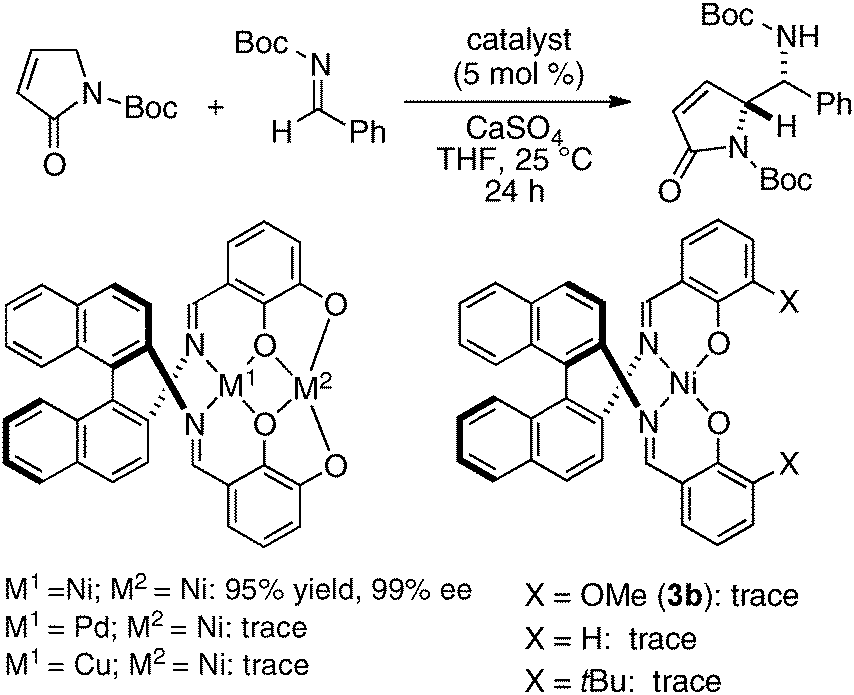
To avoid difficulty in the two metals-one-ligand complexation process, we decided to use the two same metals in the design of dinuclear cooperative catalysts. For creating unique catalytic intramolecular cooperative activity of homodinuclear catalysts different from mononuclear metal–salen intermolecular cooperative systems, it would be desirable to place the two metals into two distinct cavities because two metals in a coordinatively different environment should have different functions.34 While addressing this issue, dinucleating Schiff base 1b derived from 1,1′-binaphthyl-2,2′-diamine was found to be the best. Because the size of the O2O2 outer cavity in 1b was smaller than that in related Schiff bases derived from other diamines, various first-row transition metals were successfully incorporated into the O2O2 outer cavity. As shown in Scheme 7, the reaction of the dinucleating Schiff base 1b with two equivalents of Ni(OAc)2·4H2O gave a homodinuclear Ni2–1b catalyst in 93% yield as a bench-stable solid.35 Homodinuclear Co2–1b catalyst36 and Mn2–1b catalyst37 were also synthesized starting from corresponding metal acetates under dry air-atmosphere without difficulty.
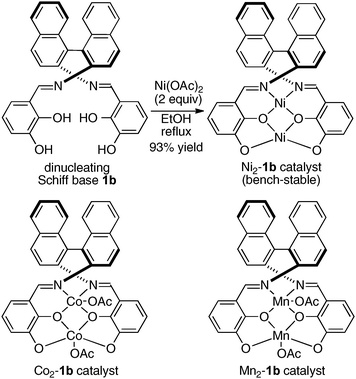 |
| | Scheme 7 Preparation of bench-stable homodinuclear transition metal catalysts from dinucleating Schiff base 1b derived from 1,1′-binaphthyl-2,2′-diamine. | |
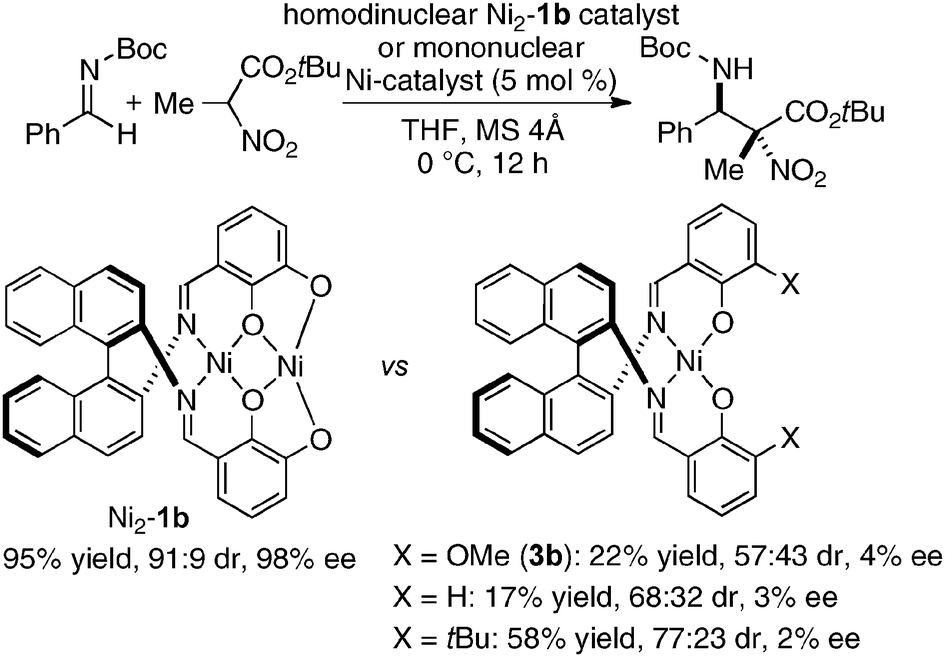
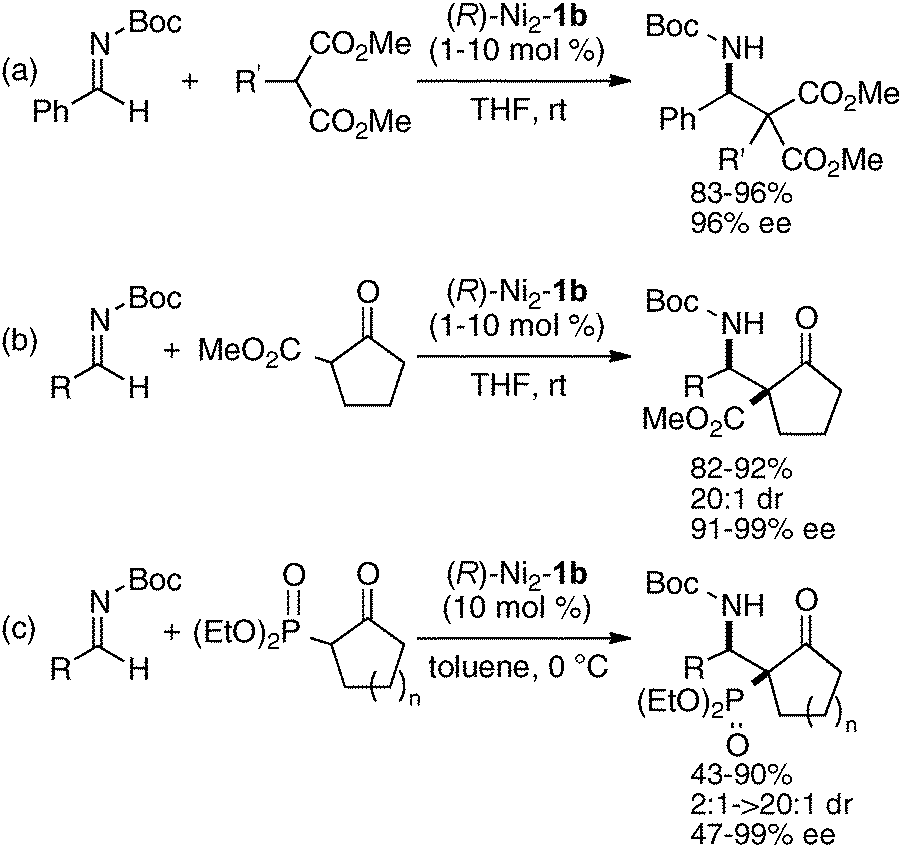
Homodinuclear transition metal–1b complexes were utilized for the catalytic asymmetric carbon–carbon bond-forming reactions with pronucleophiles bearing a relatively acidic proton. As summarized in Table 4, the homodinuclear Ni2–1b catalyst promoted direct Mannich-type reaction38 of N-Boc imines with α-substituted nitroacetates. Not only aryl and heteroaryl imines, but also isomerizable alkyl imines were applicable, and products were obtained in 91 → 99% ee and 86:14 → 97:3 diastereoselectivity.35 Products were readily converted into α,β-diamino acids with an α-tetrasubstituted stereocenter. As shown in run 13, catalyst loading was successfully reduced to 1 mol%, while maintaining high enantioselectivity. The homodinuclear Ni2–1b catalyst showed unique catalytic activity and stereoselectivity in comparison with related mononuclear Ni–Schiff base complexes as summarized in Scheme 8. Mononuclear Ni–Schiff base complexes gave only poor reactivity, diastereoselectivity, and enantioselectivity. Thus, we believe that the cooperative function of the two Ni centers would be important. The postulated cooperative mechanism of the dinuclear Ni-catalysis is shown in Fig. 10. One of the Ni–O bonds in the outer O2O2 cavity works as a Brønsted base to deprotonate the nitroacetate and generate a nickel-nitronate in situ. The other nickel in the inner N2O2 cavity functions as a Lewis acid to control the position of the imines. The carbon–carbon bond-formation, followed by protonation, affords the Mannich product and regenerates the Ni2–1b catalyst.
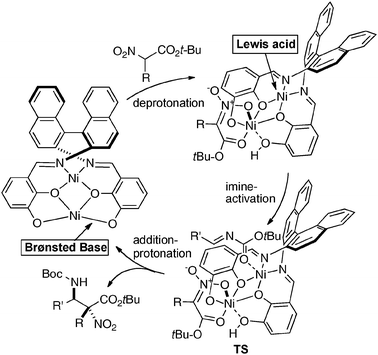 |
| | Fig. 10 Postulated Lewis acid–Brønsted base cooperative mechanism of homodinuclear Ni2–1b catalyst. | |
Table 4 Ni2–1b-catalyzed asymmetric direct Mannich-type reaction
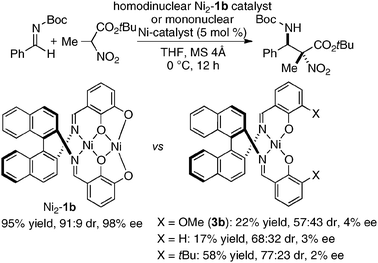 |
| | Scheme 8 Catalytic activity difference between homodinuclear Ni2–1b catalyst and mononuclear Ni–Schiff base catalysts. | |
The same Ni2–1b catalyst was also suitable for the direct catalytic asymmetric Mannich-type reaction of malonates,35 β-keto esters,35 and β-keto phosphonates,39 as summarized in Scheme 9. The scope of the Ni2–1b catalysis was further expanded to direct catalytic asymmetric vinylogous Mannich-type reaction.40 The reaction of N-Boc imines with an α,β-unsaturated γ-butyrolactam proceeded with good to high syn-selectivity and enantioselectivity at 25 °C (99% ee, Scheme 10).41 In the vinylogous Mannich-type reaction, the scope of imines was, however, limited to non-isomerizable aryl and heteroaryl N-Boc imines. When using isomerizable alkyl N-Boc imines, undesirable isomerization of imines to enamides via α-deprotonation of alkyl N-Boc imines competed with desired vinylogous Ni-enolate formation from the α,β-unsaturated γ-butyrolactam, thereby resulting in poor yield of Mannich products. The negative control experiments of the vinylogous Mannich-type reaction using heterodinuclear Pd or Cu–Ni–1b complexes and mononuclear Ni–Schiff base complexes resulted in trace, if any, products (Scheme 11). Thus, the importance of the cooperative functions of two Ni-centers in distinct cavities was confirmed again. We believe that the vinylogous Mannich-type reaction of α,β-unsaturated γ-butyrolactam was promoted in a similar manner as discussed in Fig. 10 for the Mannich-type reaction of nitroacetates.
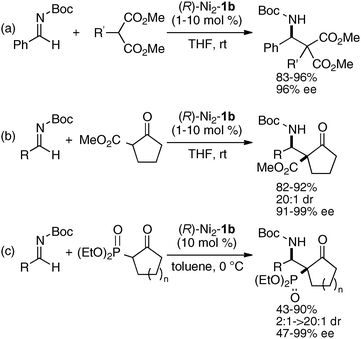 |
| | Scheme 9 Direct catalytic asymmetric Mannich-type reactions of (a) malonates, (b) β-keto esters, and (c) β-keto phosphonates under dinuclear Ni-catalysis. | |
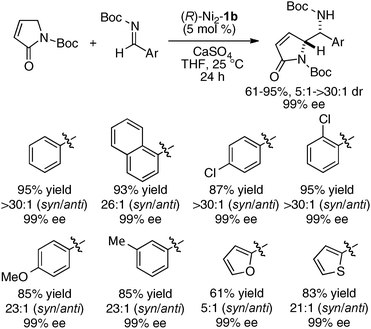 |
| | Scheme 10 Direct catalytic asymmetric vinylogous Mannich-type reaction of α,β-unsaturated γ-butyrolactam. | |
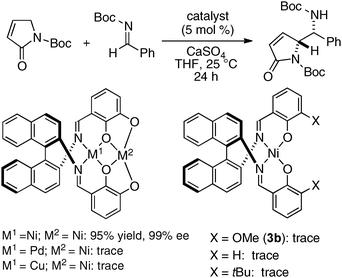 |
| | Scheme 11 Negative control experiments using heterodinuclear 1b complexes and mononuclear Ni-Schiff base complexes. | |
When using 1,2-dicarbonyl compounds as a pronucleophile, a slight modification of the chiral environment of the dinuclear Ni-catalyst was effective. The best selectivity was achieved with the Schiff base 1d derived from biphenyldiamine, and the Ni2–1d catalyst gave products in 91–95% ee and 10:1 → 50:1 dr at 0 °C (Scheme 12).42 The Mannich adducts in Scheme 12 were useful precursors for the synthesis of fully substituted azetidine carboxylic acid derivatives as unnatural α-amino acids. Stereoselective reduction of Mannich adducts with K-Selectride, followed by intramolecular Mitsunobu cyclization, gave azetidine-2-amides.
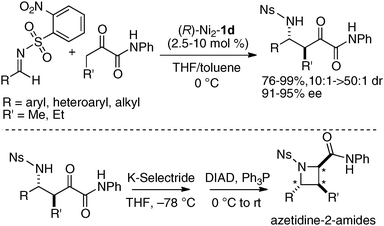 |
| | Scheme 12 Biphenyldiamine-based homodinuclear Ni2–1d-catalyzed asymmetric Mannich-type reaction of 1,2-dicarbonyl compounds, and its application to azetidine-2-amide synthesis. | |
The homodinuclear Ni2–1b catalyst was also applicable to electrophiles other than imines. For example, the aldol-type reaction also proceeded under simple proton transfer conditions. As shown in Scheme 13, direct catalytic asymmetric aldol reaction of 3-isothiocyanato oxindoles43 proceeded smoothly to give spiro-oxindoles in 80–99% ee and 4:1–10:1 dr from aliphatic aldehydes.44,45 It is noteworthy that the reaction proceeded smoothly even when using as little as 0.1 mol% of Ni2–1b catalyst (TON = 850). The Ni2–1b catalyst gave, however, poor selectivity in the case of aromatic aldehydes. For the aromatic aldehydes, a Sr(O-iPr)2/3b was used alternatively.46 Because the Ni2–1b catalyst was bench-stable and tolerant towards water, aqueous formaldehyde was utilized as an electrophile in catalytic asymmetric hydroxymethylation of β-keto esters, giving products in 66–94% ee. A high catalyst turnover number, up to 940, was also observed in the hydroxymethylation (Scheme 14).47
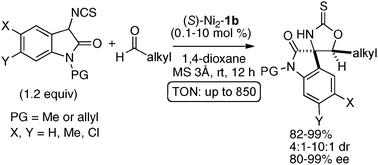 |
| | Scheme 13 Catalytic asymmetric aldol-type reaction of 3-isothiocyanato oxindoles to aldehydes. | |
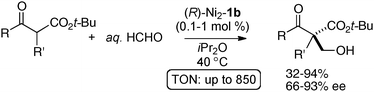 |
| | Scheme 14 Catalytic asymmetric aldol reaction of β-keto esters with aqueous formaldehyde. | |
For the catalytic asymmetric Michael reaction to nitroalkenes,48 the selection of a suitable metal source was important in the control of catalyst sites-substrates arrangement. Ni2–1b smoothly promoted asymmetric vinylogous Michael reaction with the α,β-unsaturated γ-butyrolactam, giving products in high enantio- and diastereoselectivity (Scheme 15a).41,49 When using 1,2-dicarbonyl compounds as donors, Ni2–1d gave much better stereoselectivity than Ni2–1b (Scheme 15b).50 On the other hand, homodinuclear Ni-catalysts gave poor to moderate enantio- and diastereoselectivity for other donors, such as β-keto esters and N-Boc oxindoles. After optimization studies, Co2–1b was found to be the best for asymmetric Michael reaction with β-keto esters (Scheme 15c),36,51 while Mn2–1b was required in the reaction with N-Boc oxindoles as donors (Scheme 15d).37 The related enantio- and diastereoselective double Michael reaction of N-Boc bisoxindole was applied for a concise catalytic enantioselective total synthesis of hexahydropyrrolo-indole alkaloids, such as (+)-chimonanthine, (+)-folicanthine, and (−)-calycanthine (Scheme 16).52–54 In the double Michael reaction of N-Boc bis-oxindole, Mn(O-4-F-benzoate)2 was utilized instead of Mn(OAc)2 as a metal source in the first Michael reaction. For the second Michael reaction, the chiral Mn–1b catalyst gave the double Michael product in excellent enantioselectivity via chiral amplification (99% ee), but only in moderate yield (44%) possibly due to the steric hindrance of vicinal quaternary carbon stereocenters;55 thus, the diastereoselectivity in the second Michael reaction was controlled by a catalytic amount of less hindered achiral Mg(OAc)2, giving the double Michael adduct in 69% yield (in two steps), 95% ee, and >20:1 dr. Reduction of the nitro groups in the double Michael adduct (87% yield), followed by reductive cyclization gave a known key intermediate in 61% yield. Enantioselective total synthesis of (+)-chimonanthine was thus achieved in overall 25% yield (5 steps from N-Boc bisoxindole). (+)-Folicanthine and (−)-calycanthine were also synthesized from (+)-chimonanthine using the reported procedure.52
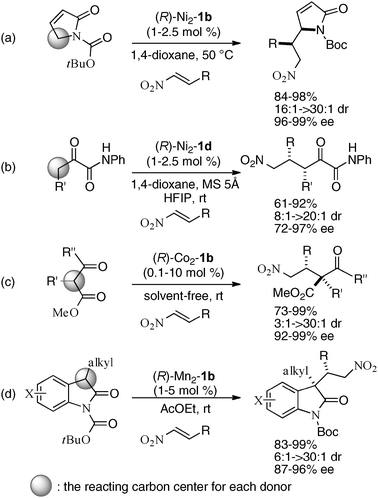 |
| | Scheme 15 Catalytic asymmetric Michael reactions of various bidentate donors to nitroalkenes under homodinuclear Schiff base catalysis. | |
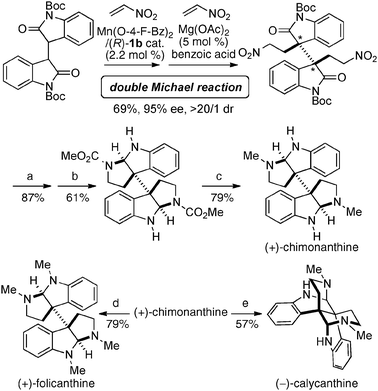 |
| | Scheme 16 Concise catalytic asymmetric total synthesis of (+)-chimonanthine, (+)-folicanthine, and (−)-calycanthine via double Michael reaction; reagents and conditions: (a) NiCl2, NaBH4, dimethyl dicarbonate, MeOH, 87% yield; (b) LiEt3BH, toluene, −78 °C to −40 °C, 1 h; 4 M HCl in AcOEt, rt, 5 h; TFA, rt, 15 h, 61% yield; (c) sodium bis(2-methoxyethoxy) aluminum hydride, toluene, reflux, 4.5 h, 79% yield; (d) aq. HCHO, NaBH(OAc)3, MeCN, rt, 0.5 h, 79% yield; (e) AcOH, H2O, 95 °C, 48 h, 57% yield. | |
Asymmetric Michael reactions with other acceptors were also achieved under homodinuclear Schiff base catalysis, such as alkynones (Scheme 17)36 and vinylidenebisphosphonates (Scheme 18).56 In the catalytic asymmetric Michael addition to alkynones, Co2–1b catalyst gave the best results under solvent-free conditions. Because the E/Z ratio was moderate after the Co2–1b-catalyzed addition/protonation process, the crude reaction mixture was directly treated with a catalytic amount of Ph2MeP to afford the thermodynamically more favorable E-isomer predominantly (>30:1). In the catalytic asymmetric 1,4-addition of nitroacetates to vinylidenebisphosphonates, Ni2–1b catalyst gave the best enantioselectivity. Products in Scheme 18 can be key synthetic intermediates for biologically important bisphosphonates with an α-amino acid unit.
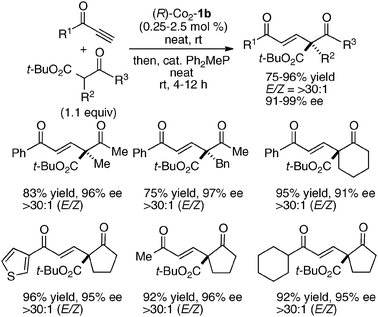 |
| | Scheme 17 Asymmetric Michael addition of β-keto esters to alkynones under homodinuclear Co2–1b catalysis. | |
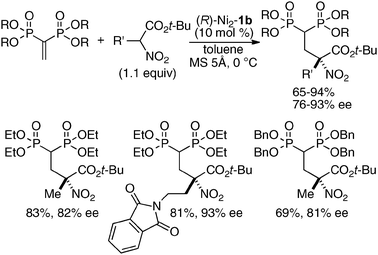 |
| | Scheme 18 Asymmetric Michael addition of nitroacetates to vinylidenebisphosphonates under homodinuclear Ni2–1b catalysis. | |
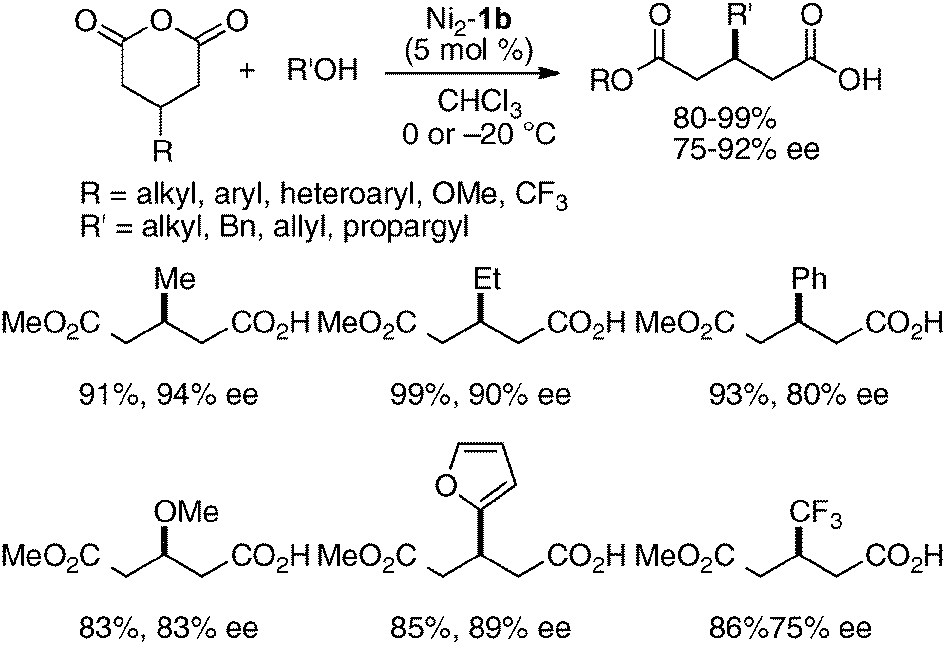
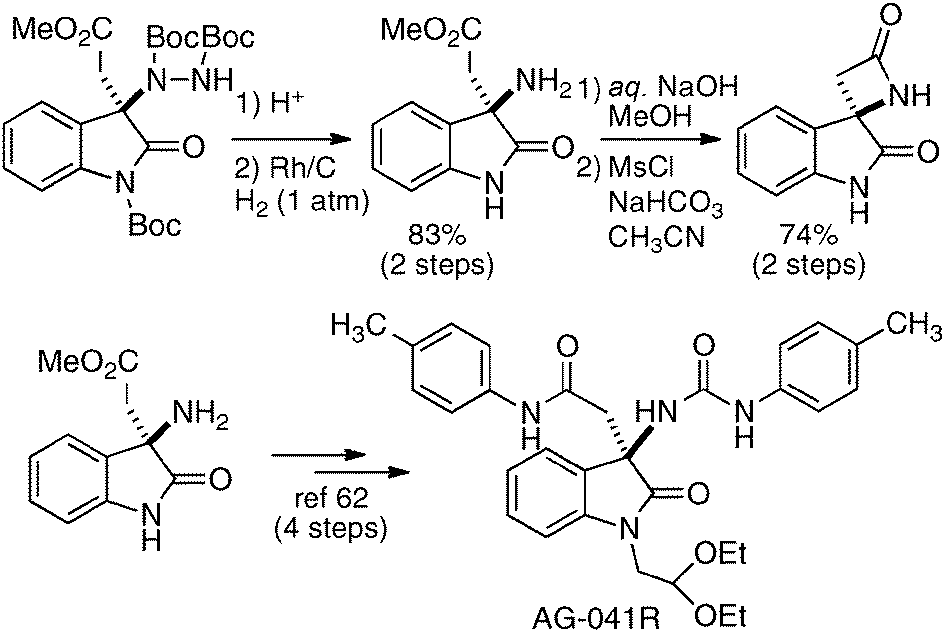
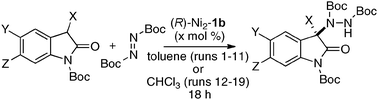
The utility of the homodinuclear Ni2–1b catalyst was further expanded to reactions other than C–C bond-forming reactions. Catalytic desymmetrization of 3-methylglutaric anhydride and its derivatives with alcohols gave hemi-esters in 80–99% yield and 75–92% ee (Scheme 19).57 The product of the desymmetrization reaction was utilized as a key building block in the synthetic studies of caprazamycin B.58 Ni2–1b also promoted asymmetric C–N bond-forming reaction.59 As shown in Table 5, 1 mol% of Ni2–1b was sufficient to promote asymmetric amination of 3-alkyl-substituted N-Boc oxindoles,60 giving products in high yield with 87–99% ee (runs 1–11). On the other hand, 3-aryl-substituted N-Boc oxindoles required much higher catalyst loading (10 mol%) to induce moderate to good enantioselectivity (runs 12–19), because the background racemic reaction competed in the case of 3-aryl-substituted donors.61 Amination adducts were readily converted into 3-amino-oxindoles via Boc-removal under acidic conditions, followed by Rh–C catalyzed N–N bond cleavage. A suitably functionalized amination product was, thus, successfully converted into an optically active oxindole bearing a spiro-β-lactam unit and a known intermediate for AG-041R synthesis (Scheme 20).62
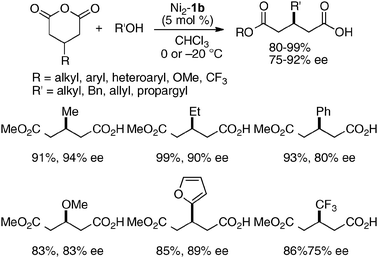 |
| | Scheme 19 Ni2–1b-catalyzed desymmetrization of 3-substituted-glutaric anhydrides. | |
Table 5 Ni2–1b-catalyzed asymmetric amination of oxindoles
|
|
| Run |
X |
Y |
Z |
x
|
Temp (°C) |
Yield (%) |
ee (%) |
| 1 |
Me |
H |
H |
1 |
50 |
99 |
99 |
| 2 |
Allyl |
H |
H |
1 |
50 |
99 |
97 |
| 3 |
(E)-Cinnamyl |
H |
H |
1 |
50 |
86 |
91 |
| 4 |
Bn |
H |
H |
1 |
50 |
93 |
99 |
| 5 |
Me |
MeO |
H |
1 |
50 |
91 |
94 |
| 6 |
Me |
F |
H |
1 |
50 |
95 |
96 |
| 7 |
Allyl |
F |
H |
1 |
50 |
90 |
98 |
| 8 |
Allyl |
Cl |
H |
1 |
50 |
93 |
95 |
| 9 |
Bn |
H |
Cl |
1 |
50 |
98 |
99 |
| 10 |
–CH2CO2Me |
H |
H |
1 |
50 |
98 |
96 |
| 11 |
–CH2CN |
H |
H |
1 |
50 |
89 |
87 |
| 12 |
Ph |
H |
H |
10 |
30 |
94 |
90 |
| 13 |
4-F–C6H4 |
H |
H |
10 |
30 |
91 |
82 |
| 14 |
3-MeO–C6H4 |
H |
H |
10 |
30 |
93 |
86 |
| 15 |
2-MeO–C6H4 |
H |
H |
10 |
30 |
75 |
98 |
| 16 |
Ph |
Me |
H |
10 |
30 |
72 |
74 |
| 17 |
Ph |
MeO |
H |
10 |
30 |
86 |
82 |
| 18 |
4-F–C6H4 |
MeO |
H |
10 |
30 |
72 |
73 |
| 19 |
3-MeO–C6H4 |
MeO |
H |
10 |
30 |
78 |
66 |
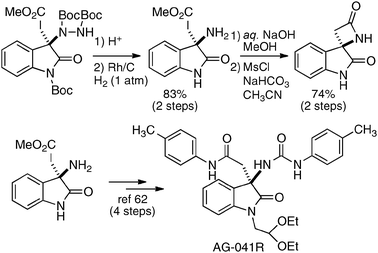 |
| | Scheme 20 Transformation of the amination adduct. | |
Conclusions
In summary, a new family of bimetallic cooperative asymmetric catalysts based on dinucleating Schiff bases has been developed. Dinucleating Schiff bases enabled the new metal combinations in bimetallic catalysis, which were impossible in our early studies with BINOL-based chiral ligands. The new either hetero- or homo-bimetallic combinations, such as transition metal/rare earth metal, group 13 metal/rare earth metal, Lewis acidic rare earth metal/Brønsted basic rare earth metal, and transition metal/transition metal led to much improved catalytic activity and selectivity. Readily tunable chiral environments were successfully achieved in the bimetallic Schiff base system, and its utility was demonstrated in a variety of catalytic asymmetric C–C and C–N bond-forming reactions under simple proton transfer conditions. High selectivity and catalytic activity have been realized through unique intramolecular cooperative functions of two metal centers in distinct cavities. Further applications of these bimetallic Schiff base cooperative catalysis are actively ongoing in our group.
Acknowledgements
We express deep gratitude to a highly-talented group of co-workers whose names appear in the references. We specially thank Dr V. Gnanadesikan, Dr S. Handa, and Dr Z. Chen for their pioneering contributions at the initial stage of the dinuclear Schiff base catalysis projects. Financial support was in part provided by ACT-C from JST, a Grant-in-Aid for Young Scientist (A) from JSPS, the Naito Foundation, Takeda Science Foundation, and the Inoue Science Foundation.
Notes and references
-
(a)
P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books: Sausalito, California, 2009 Search PubMed;
(b)
E. J. Corey and L. Kürti, Enantioselective Chemical Synthesis, Direct Book Publishing, 2010 Search PubMed;
(c)
Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, 2011 Search PubMed.
- B. M. Trost, Science, 1991, 254, 1471 CAS.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197 CrossRef CAS PubMed.
- N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760 CrossRef CAS PubMed.
-
Enzyme Catalysis in Organic Synthesis, ed. P. T. Anastas and R. H. Crabtree, Wiley-VCH, Weinheim, 2009 Search PubMed.
- Reviews:
(a) M. Sawamura and Y. Ito, Chem. Rev., 1992, 92, 857 CrossRef CAS;
(b) J.-A. Ma and D. Cahard, Angew. Chem., Int. Ed., 2004, 43, 4566 CrossRef CAS PubMed;
(c) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS PubMed;
(d) M. Kanai, N. Kato, E. Ichikawa and M. Shibasaki, Synlett, 2005, 1491 CrossRef CAS;
(e) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed;
(f) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed;
(g) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117 CrossRef CAS PubMed;
(h) J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931 RSC.
- Reviews:
(a) M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed. Engl., 1997, 36, 1236 CrossRef;
(b) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187 CrossRef CAS PubMed ; For recent applications, see also: ;
(c) T. Sone, G. Lu, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 1677 CrossRef CAS PubMed;
(d) T. Sone, A. Yamaguchi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 10078 CrossRef CAS PubMed.
- Review:
(a)
T. Ohshima and M. Shibasaki, in Multimetallic Catalysts in Organic Synthesis, ed. M. Shibasaki and Y. Yamamoto, Wiley-VCH, 2004, ch. 3, p. 77 Search PubMed; For recent applications, see:
(b) T. Ohshima, Y. Xu, R. Takita, S. Shimizu, D. Zhong and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 14546 CrossRef CAS PubMed;
(c) H. Mihara, Y. Sohtome, S. Matsunaga and M. Shibasaki, Chem.–Asian. J., 2008, 3, 359 CrossRef CAS PubMed.
- Reviews on linked-BINOL catalysts:
(a) S. Matsunaga and M. Shibasaki, Bull. Chem. Soc. Jpn., 2008, 81, 60 CrossRef CAS;
(b) M. Shibasaki and S. Matsunaga, Chem. Soc. Rev., 2006, 35, 269 RSC;
(c) S. Matsunaga, T. Ohshima and M. Shibasaki, Adv. Synth. Catal., 2002, 344, 3 CrossRef CAS ; For a recent application of linked-BINOL, see: ;
(d) T. Yukawa, B. Seelig, Y. Xu, H. Morimoto, S. Matsunaga, A. Berkessel and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 11988 CrossRef CAS PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed.
- Reviews:
(a) T. Katsuki, Chem. Soc. Rev., 2004, 33, 437 RSC;
(b) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC;
(c) S. J. Wezenberg and A. W. Kleij, Angew. Chem., Int. Ed., 2008, 47, 2354 CrossRef CAS PubMed;
(d) C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987 CrossRef CAS PubMed;
(e) E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563 CrossRef CAS PubMed.
- Review: E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421 CrossRef CAS PubMed.
- For general reviews on bimetallic Schiff base catalysts:
(a) R. M. Haak, S. J. Wezenberg and A. W. Kleij, Chem. Commun., 2010, 46, 2713 RSC;
(b) S. Matsunaga and M. Shibasaki, Synthesis, 2013, 421 CrossRef CAS.
- For selected early examples of heterobimetallic Schiff base intramolecular cooperative asymmetric catalysts, see:
(a) E. F. DiMauro and M. C. Kozlowski, Organometallics, 2002, 21, 1454 CrossRef CAS;
(b) V. Annamalai, E. F. DiMauro, P. J. Carroll and M. C. Kozlowski, J. Org. Chem., 2003, 68, 1973 CrossRef CAS PubMed;
(c) M. Yang, C. Zhu, F. Yuan, Y. Huang and Y. Pan, Org. Lett., 2005, 7, 1927 CrossRef CAS PubMed;
(d) J. Gao, F. R. Woolley and R. A. Zingaro, Org. Biomol. Chem., 2005, 3, 2126 RSC;
(e) W. Li, S. S. Thakur, S.-W. Chen, C.-K. Shin, R. B. Kawthekar and G.-J. Kim, Tetrahedron Lett., 2006, 47, 3453 CrossRef CAS;
(f) S.-W. Chen, S. S. Thakur, W. Li, C.-K. Shin, R. B. Kawthekar and G.-J. Kim, J. Mol. Catal. A: Chem., 2006, 259, 116 CrossRef CAS;
(g) R. B. Kawthekar, C.-H. Ahn and G.-J. Kim, Catal. Lett., 2007, 115, 62 CrossRef CAS;
(h) R. B. Kawthekar and G.-J. Kim, Synth. Commun., 2008, 38, 1236 CrossRef CAS;
(i) J. Sun, F. Yuan, M. Yang, Y. Pan and C. Zhu, Tetrahedron Lett., 2009, 50, 548 CrossRef CAS ; see also reviews in ref. 13.
- Review:
(a) M. Sakamoto, K. Manseki and H. Okawa, Coord. Chem. Rev., 2001, 219–221, 379 CrossRef CAS ; For selected examples, see also: ;
(b) R. Koner, G.-H. Lee, Y. Wang, H. H. Wei and S. Mohanta, Eur. J. Inorg. Chem., 2005, 1500 CrossRef CAS;
(c) R. Koner, H. H. Lin, H. H. Wei and S. Mohanta, Inorg. Chem., 2005, 44, 3524 CrossRef CAS PubMed;
(d) L. Salmon, P. Thuéry, E. Rivière and M. Ephritikhine, Inorg. Chem., 2006, 45, 83 CrossRef CAS PubMed;
(e) G. Novitchi, S. Shova, A. Caneschi, J.-P. Costes, M. Gdaniec and N. Stanica, Dalton Trans., 2004, 1194 RSC;
(f) R. Miyamoto, T. Sugai and S. Sudoh, Polyhedron, 2002, 21, 2127 CrossRef CAS;
(g) J.-P. Costes, F. Dahan and A. Dupuis, Inorg. Chem., 2000, 39, 165 CrossRef CAS PubMed;
(h) J.-P. Costes, F. Dahan, A. Dupuis and J.-P. Laurent, Chem.–Eur. J., 1998, 4, 1616 CrossRef CAS;
(i) A. Aguiari, E. Bullita, U. Casellato, P. Guerriero, S. Tamburini and P. A. Vigato, Inorg. Chim. Acta, 1994, 219, 135 CrossRef CAS.
- Reviews on catalytic asymmetric aza-Henry reactions:
(a) B. Westermann, Angew. Chem., Int. Ed., 2003, 42, 151 CrossRef CAS PubMed;
(b) E. Marqués-López, P. Merino, T. Tejero and R. P. Herrera, Eur. J. Org. Chem., 2009, 2401 CrossRef.
- S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 4900 CrossRef CAS PubMed.
- S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 4925 CrossRef CAS PubMed.
- Reviews on catalytic asymmetric Henry reactions:
(a) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561 CrossRef CAS;
(b) J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315 CrossRef CAS;
(c) C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442 CrossRef CAS PubMed.
- For selected leading examples of anti-selective direct catalytic asymmetric Henry reactions:
(a) T. Ogawa, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2013, 52, 6196 CrossRef CAS PubMed;
(b) D. Uraguchi, S. Nakamura and T. Ooi, Angew. Chem., Int. Ed., 2010, 49, 7562 CrossRef CAS PubMed;
(c) T. Nitabaru, A. Nojiri, M. Kobayashi, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 13860 CrossRef CAS PubMed;
(d) D. Uraguchi, S. Sakaki and T. Ooi, J. Am. Chem. Soc., 2007, 129, 12392 CrossRef CAS PubMed.
-
(a) S. Handa, K. Nagawa, Y. Sohtome, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2008, 47, 3230 CrossRef CAS PubMed;
(b) Y. Sohtome, Y. Kato, S. Handa, N. Aoyama, K. Nagawa, S. Matsunaga and M. Shibasaki, Org. Lett., 2008, 10, 2231 CrossRef CAS PubMed.
- For leading reports on the utility of salan and salalen ligands in asymmetric catalysis, see:
(a) H. Egami, T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 5886 CrossRef CAS PubMed;
(b) K. Suyama, Y. Sakai, K. Matsumoto, B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2010, 49, 797 CrossRef CAS PubMed;
(c) K. Matsumoto, T. Oguma and T. Katsuki, Angew. Chem., Int. Ed., 2009, 48, 7432 CrossRef CAS PubMed;
(d) B. Saito, H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 1978 CrossRef CAS PubMed;
(e) B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2005, 44, 4600 CrossRef CAS PubMed , and references therein.
- M. Furutachi, S. Mouri, S. Matsunaga and M. Shibasaki, Chem.–Asian. J., 2010, 5, 2351 CrossRef CAS PubMed.
- A review on stereoselective synthesis of γ-amino acids: M. Ordóñez and C. Cativiela, Tetrahedron: Asymmetry, 2007, 18, 3 CrossRef.
- Review on rare earth metal triflates in organic synthesis: S. Kobayashi, M. Sugiura, H. Kitagawa and W. W. L. Lam, Chem. Rev., 2002, 102, 2227 CrossRef CAS PubMed.
-
(a) S.-X. Wang, M.-X. Wang, D.-X. Wang and J. Zhu, Org. Lett., 2007, 9, 3615 CrossRef CAS PubMed;
(b) S.-X. Wang, M.-X. Wang, D.-X. Wang and J. Zhu, Eur. J. Org. Chem., 2007, 4076 CrossRef CAS.
- A review on the utility of isocyanide in organic synthesis: A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed.
- Evaluation of the relative Lewis acidity of rare earth metals: H. Tsuruta, K. Yamaguchi and T. Imamoto, Chem. Commun., 1999, 1703 RSC.
- H. Mihara, Y. Xu, N. E. Shepherd, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 8384 CrossRef CAS PubMed.
- For representative other applications of Schiff base 3b and related ligands in asymmetric catalysis:
(a) B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Angew. Chem., Int. Ed., 2009, 48, 1126 CrossRef CAS PubMed;
(b) T. Yoshino, H. Morimoto, G. Lu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 17068 CrossRef PubMed;
(c) G. Lu, T. Yoshino, H. Morimoto, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4382 CrossRef CAS PubMed;
(d) T. Yoshino and S. Matsunaga, Chem. Rec., 2011, 11, 260 CrossRef PubMed;
(e) Y. Suzuki, M. Kanai and S. Matsunaga, Chem.–Eur. J., 2012, 18, 7654 CrossRef CAS PubMed.
- A review on desymmetrization of meso epoxides and aziridines: C. Schneider, Angew. Chem., Int. Ed., 2009, 48, 2082 CrossRef CAS PubMed.
- Y. Xu, L. Lin, M. Kanai, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2011, 133, 5791 CrossRef CAS PubMed.
- Reviews on the foldamer research:
(a) C. M. Goodman, S. Choi, S. Shandler and W. F. Degrado, Nat. Chem. Biol., 2007, 3, 252 CrossRef CAS PubMed;
(b) D. Seebach, M. Brenner, M. Rueping and B. Jaun, Chem.–Eur. J., 2002, 8, 573 CrossRef CAS PubMed;
(c) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- For selected examples of homodinuclear cooperative asymmetric Schiff base catalysts where two metals were placed into two coordinatively same cavities, see:
(a) Q.-X. Guo, Z.-J. Wu, Z.-B. Luo, Q.-Z. Liu, J.-L. Ye, S.-W. Luo, L.-F. Cun and L.-Z. Gong, J. Am. Chem. Soc., 2007, 129, 13927 CrossRef CAS PubMed;
(b) C. Mazet and E. N. Jacobsen, Angew. Chem., Int. Ed., 2008, 47, 1762 CrossRef CAS PubMed;
(c) W. Hirahata, R. M. Thomas, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2008, 130, 17658 CrossRef CAS PubMed;
(d) S. Takizawa, T. Katayama, H. Somei, Y. Asano, T. Yoshida, C. Kameyama, D. Rajesh, K. Onitsuka, T. Suzuki, M. Mikami, H. Yamataka, D. Jayaprakash and H. Sasai, Tetrahedron, 2008, 64, 3361 CrossRef CAS;
(e) S. Takizawa, T. Katayama, C. Kameyama, K. Onitsuka, T. Suzuki, T. Yanagida, T. Kawai and H. Sasai, Chem. Commun., 2008, 1810 RSC;
(f) B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Angew. Chem., Int. Ed., 2009, 48, 1126 CrossRef CAS PubMed;
(g) Y. N. Belokon', D. Chusov, A. S. Peregudov, L. V. Yashkina, G. I. Timofeeva, V. I. Maleev, M. North and H. B. Kagan, Adv. Synth. Catal., 2009, 351, 3157 CrossRef;
(h) R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520 CrossRef CAS PubMed.
- Z. Chen, H. Morimoto, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 2170 CrossRef CAS PubMed.
- Z. Chen, M. Furutachi, Y. Kato, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 2218 CrossRef CAS PubMed.
- Y. Kato, M. Furutachi, Z. Chen, H. Mitsunuma, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 9168 CrossRef CAS PubMed.
- A review on direct Mannich-type reactions, see
(a) M. M. B. Marques, Angew. Chem., Int. Ed., 2006, 45, 348 CrossRef CAS PubMed ; A review on Mannich-type reactions for α,β-diamino acid synthesis: ;
(b) R. Gómez Arrayás and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940 RSC ; A general review on catalytic asymmetric synthesis of β-amino acids: ;
(c) B. Weiner, W. Szymanski, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656 RSC.
- Z. Chen, K. Yakura, S. Matsunaga and M. Shibasaki, Org. Lett., 2008, 10, 3239 CrossRef CAS PubMed.
- Reviews on asymmetric vinylogous C–C bond forming reactions and their utility:
(a) G. Casiraghi, F. Zanardi, L. Battistini and G. Rassu, Synlett, 2009, 1525 CrossRef CAS;
(b) G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929 CrossRef CAS PubMed.
- N. E. Shepherd, H. Tanabe, Y. Xu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 3666 CrossRef CAS PubMed.
- Y. Xu, G. Lu, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 3353 CrossRef CAS PubMed.
- For the utility of 3-isothiocyanato oxindoles in catalytic asymmetric spirooxindole synthesis, see:
(a) W.-B. Chen, Z.-J. Wu, J. Hu, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2011, 13, 2472 CrossRef CAS PubMed;
(b) S. Kato, T. Yoshino, M. Shibasaki, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2012, 51, 7007 CrossRef CAS PubMed;
(c) Y.-M. Cao, F.-F. Shen, F.-T. Zhang and R. Wang, Chem.–Eur. J., 2013, 19, 1184 CrossRef CAS PubMed;
(d) H. Wu, L.-L. Zhang, Z.-Q. Tian, Y.-D. Huang and Y.-M. Wang, Chem.–Eur. J., 2013, 19, 1747 CrossRef CAS PubMed;
(e) Q. Chen, J. Liang, S. Wang, D. Wang and R. Wang, Chem. Commun., 2013, 49, 1657 RSC;
(f) X.-L. Liu, W.-Y. Han, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2013, 15, 1246 CrossRef CAS PubMed.
- A general review on enantioselective synthesis of tetrasubstituted oxindoles: F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS.
- S. Kato, M. Kanai and S. Matsunaga, Chem.–Asian. J., 2013, 8, 1768 CrossRef CAS PubMed.
- S. Kato, M. Kanai and S. Matsunaga, Heterocycles DOI:10.3987/COM-13-S(S)60.
- S. Mouri, Z. Chen, S. Matsunaga and M. Shibasaki, Chem. Commun., 2009, 5138 RSC.
- A review on asymmetric 1,4-addition to nitroalkenes:
(a) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS;
(b) General reviews on asymmetric 1,4-addition to electron-deficient alkenes with metal catalysis: J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 1279 CrossRef CAS;
(c) With organocatalysis: S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS.
- H. Tanabe, Y. Xu, B. Sun, S. Matsunaga and M. Shibasaki, Heterocycles, 2012, 86, 611 CrossRef CAS.
- Y. Xu, S. Matsunaga and M. Shibasaki, Org. Lett., 2010, 12, 3246 CrossRef CAS PubMed.
- M. Furutachi, Z. Chen, S. Matsunaga and M. Shibasaki, Molecules, 2010, 15, 532 CrossRef CAS PubMed.
- For leading studies on enantioselective total synthesis:
(a) L. E. Overman, D. V. Paone and B. A. Stearns, J. Am. Chem. Soc., 1999, 121, 7702 CrossRef CAS;
(b) L. E. Overman, J. F. Larrow, B. A. Stearns and J. M. Vance, Angew. Chem., Int. Ed., 2000, 39, 213 CrossRef CAS;
(c) M. Movassaghi and M. A. Schmidt, Angew. Chem., Int. Ed., 2007, 46, 3725 CrossRef CAS PubMed ; See also a review;
(d) M. A. Schmidt and M. Movassaghi, Synlett, 2008, 313 CAS.
-
(a) H. Mitsunuma, M. Shibasaki, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2012, 51, 5217 CrossRef CAS PubMed;
(b) H. Mitsunuma and S. Matsunaga, Chem. Commun., 2011, 47, 469 RSC.
- Other catalytic asymmetric approaches, see:
(a) C. Guo, J. Song, J. Z. Huang, P. H. Chen, S. W. Luo and L. Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 1046 CrossRef CAS PubMed;
(b) R. R. Liu and J. L. Zhang, Org. Lett., 2013, 15, 2266 CrossRef CAS PubMed;
(c) B. M. Trost and M. Osipov, Angew. Chem., Int. Ed., 2013, 52, 9176 CrossRef CAS PubMed.
- General reviews on quaternary carbon construction:
(a) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS PubMed;
(b) B. M. Trost and C. H. Jiang, Synthesis, 2006, 369 CrossRef CAS.
- Y. Kato, Z. Chen, S. Matsunaga and M. Shibasaki, Synlett, 2009, 1635 CAS.
- P. Gopinaph, T. Watanabe and M. Shibasaki, Org. Lett., 2012, 14, 1358 CrossRef PubMed.
- P. Gopinaph, T. Watanabe and M. Shibasaki, J. Org. Chem., 2012, 77, 9260 CrossRef PubMed.
- For selected examples of catalytic asymmetric amination of oxindoles,
(a) L. Cheng, L. Liu, D. Wang and Y.-J. Chen, Org. Lett., 2009, 11, 3874 CrossRef CAS PubMed;
(b) Z.-Q. Qian, F. Zhou, T.-P. Du, B.-L. Wang, M. Ding, X.-L. Zhao and J. Zhou, Chem. Commun., 2009, 6753 RSC;
(c) T. Bui, M. Borregan and C. F. Barbas III, J. Org. Chem., 2009, 74, 4537 CrossRef PubMed;
(d) T. Bui, G. Hernández-Torres, C. Milite and C. F. Barbas III, Org. Lett., 2010, 12, 5696 CrossRef CAS PubMed;
(e) T. Zhang, L. Cheng, L. Liu, D. Wang and Y.-J. Chen, Tetrahedron: Asymmetry, 2010, 21, 2800 CrossRef CAS;
(f) Z. Yang, Z. Wang, S. Bai, K. Shen, D. Chen, X. Liu, L. Lin and X. Feng, Chem.–Eur. J., 2010, 16, 6632 CrossRef CAS PubMed;
(g) K. Shen, X. Liu, G. Wang, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4684 CrossRef CAS PubMed.
- S. Mouri, Z. Chen, H. Mitsunuma, M. Furutachi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 1255 CrossRef CAS PubMed.
- S. Mouri, Z. Chen, S. Matsunaga and M. Shibasaki, Heterocycles, 2012, 84, 879 CrossRef CAS.
-
(a) S. Sato, M. Shibuya, N. Kanoh and Y. Iwabuchi, Chem. Commun., 2009, 6264 RSC;
(b) S. Sato, M. Shibuya, N. Kanoh and Y. Iwabuchi, J. Org. Chem., 2009, 74, 7522 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a