 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix isolation and spectroscopic properties of the methylsulfinyl radical CH3(O)S˙†
Hans Peter Reisenauer*a, Jarosław Romańskib, Grzegorz Mlostoń*b and Peter R. Schreinera
aInstitute of Organic Chemistry, Justus-Liebig University, Heinrich-Buff-Ring 58, D-35392 Giessen, Germany. E-mail: reisenauer@org.chemie.uni-giessen.de; Fax: +49-641-9934309; Tel: +49-641-9934380
bDepartment of Organic and Applied Chemistry, University of Lodz, Tamka 12, 91-493 Lodz, Poland. E-mail: gmloston@uni.lodz.pl; Fax: +48 42 6655162; Tel: +48 42 6355761
First published on 15th August 2013
Abstract
The atmospherically highly relevant methylsulfinyl radical CH3(O)S˙ was generated thermally under flash pyrolysis conditions and isolated in Ar matrices at 10 K; the allyl radical is a byproduct. CH3(O)S˙ and its D3- and 13C-isotopologues were characterized through the excellent agreement between experimental and computed IR and UV/Vis spectra.
Elemental sulfur and its compounds are essential for the earth's biogeochemical system. Volatile organosulfur compounds are of key importance to the aerosol budget at the marine atmospheric boundary.1 Sulfate aerosol particles present above the oceans that evolve from planktonic algae-derived dimethyl sulfide (1) emissions affect the earth's radiation balance either by direct backscattering of solar radiation or indirectly via the cloud albedo by forming cloud condensation nuclei.1 The oxidation of 1 is believed to be initiated by reaction with OH or NO3 radicals and eventually leads to the formation of sulfuric and methanesulfonic acids (Scheme 1). While the methanethiyl (2), and methylsulfinyl radicals (3) were postulated as intermediates, 3 is insufficiently spectroscopically characterized.2
 | ||
| Scheme 1 Postulated reaction cascade for dimethyl sulfide oxidation to sulfuric acid and methylsulfonic acid in the atmosphere.2 | ||
Sulfinyl radicals of type 3 have also been postulated to be products of scavenging of peroxide radicals by sulfenic acids causing the antioxidative effect of allium species widely used both in alternative and traditional medicine.3
Contrary to numerous theoretical reports,4 experimental spectroscopic studies for 3 have rarely been described. Whereas methods such as ESR,5 mass spectrometry,6 and photoionization7 were successfully applied, there are no reports on the use of IR, UV/Vis or microwave spectra for the characterization of 3. Only very recently a step scan FT-IR study on the oxidation of 2 with O2 tentatively assigned the most intense IR band.8 Methods for the generation of 3 comprise photochemical9 or multiphoton IR absorption,10 decomposition of DMSO, or the reaction of 1 with atomic oxygen.7,11 The focus of atmospheric chemistry studies was the determination of reaction rates for scavenging of 3 with O2, O3, and NO2.12 However, many uncertainties concerning the reaction pathways and involved intermediates still exist.13
In connection with earlier experiments on the matrix isolation of sulfur containing intermediates14 we re-examined the vacuum flash pyrolysis of allylmethyl sulfoxide (4a) in combination with matrix isolation techniques. In contrast to reported results,15 only traces of the expected sulfine 5 and propene were detected in the matrix (Scheme 2, Fig. 1, also see Experimental in the ESI†).
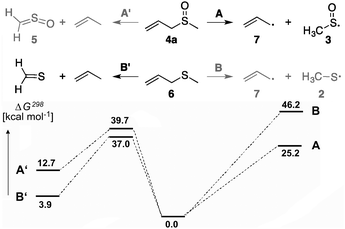 | ||
| Scheme 2 Homolytic bond cleavage vs. concerted propene elimination from allylmethyl sulfoxide (4a) and allylmethyl sulfide (6); free energies at 298 K in kcal mol−1 computed at the G4 level of theory. | ||
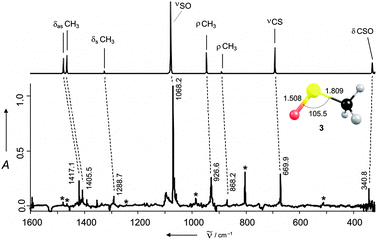 | ||
| Fig. 1 IR spectrum and structure (bond lengths in Å, angles in deg.) of 3. Top: computed at AE-CCSD(T)/cc-pVTZ (harmonic approx., no scaling); bottom: experimental (Ar matrix, 10 K) difference spectrum, obtained by subtracting the spectrum of the photolyzed matrix (254 nm, 15 h) from the non-irradiated pyrolysis products of precursor 4a (2 h at 660 °C), bands of 7 are marked with *. | ||
Since the thermal propene elimination from allyl sulfides is a standard method for the generation of thiocarbonyl compounds in the gas phase,16 the various reaction pathways in the pyrolysis of allylmethyl sulfide (6) and its S-oxide 4a deserve a comment. In both cases competitive processes (Scheme 2) are conceivable. On one hand, the concerted retro-ene reactions lead to propene and thioformaldehyde (for 6) or to thioformaldehyde S-oxide (5) (for 4a). On the other hand, homolytic cleavage of the C–S bonds in 4a and 6 yield the allyl radical (7) formed next to 3 or 2, respectively. The different behaviour of 4a and 6 under pyrolytic conditions can be explained by comparison of the ΔG‡ values for the concerted retro-ene reactions with the BDEs computed at the Gaussian-4 (G4) level of theory. While the ΔG‡ values of the retro-ene reactions are in both cases nearly equal, the energy for radical pair formation is more than 20 kcal mol−1 lower for 4a. Hence, only the radical pair forms in this case; this is also supported by the differences in BDEs for the breaking bonds.16b
Indeed, very intense IR absorption bands corresponding to 717 and 3 are evident (cf. Fig. S1 in ESI†). The unambiguous assignment of the latter follows directly from the comparison of the experimental data with the ab initio computed (AE-CCSD(T)/cc-pVTZ) IR spectrum (Fig. 1, Table S1 in ESI†). The most intense band (SO stretching vibration) appears at 1068.2 cm−1. This value is in good agreement with the reported gas phase value derived from the FT-IR step scan experiment 1071 cm−1.8 The less intense absorption band of the CS stretching vibration was found at 669.9 cm−1 along with the another band at 340.8 cm−1 for the CSO bending mode. Moreover, seven additional bands were attributed to CH stretching and bending vibrations. Only one out of the total number of twelve fundamental bands, namely the twisting vibration of the methyl group, was not observed in the experiment because its expected position (134 cm−1) was located below the range of the spectrometer used.
The geometry of 3 (cf.Fig. 1) bears no surprises. The location of the unpaired electron spin density, however, is less clear. The 2A′′ state (π-radical) is more favourable than the 2A′ state (σ-radical) by more than 40 kcal mol−1 at the UHF and UB3LYP levels of theory utilizing a large 6-311+G(3df, 3pd) basis set (cf. Fig. S5, ESI†). The spin distribution for the 2A′′ state is approximately even on the sulfur and oxygen atoms.2d
To support the proposed assignments, the spectra of the D3-and 13C-isotopologues 4b and 4c were recorded under the same experimental conditions. The intermediate allylmethyl sulfides 6b and 6c were prepared by treatment of sodium allylthiolate with CD3I or 13CH3I, respectively, and were subsequently oxidized in methanolic solution using H2O2 (Scheme 3, see also ESI†).18
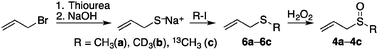 | ||
| Scheme 3 Synthesis of 4a and its labelled derivatives 4b and 4c. | ||
The observed isotopic band shifts of the two isotopologues 4b and 4c are in excellent agreement with the computed values. For example, the CS band is shifted by −13.9 cm−1, compared to the computed value of −14.1 cm−1, whereas the SO stretching showed practically no shift (exp. −0.3, computed 0.0 cm−1) upon 13C-labelling. Similarly, the experimental and computed IR band shifts of the D3-isotopologue 4b are in good agreement. Thus, in the case of the CS stretching vibration, which mixes with one of the CH bending modes, the experimental data show a shift of −55.7 cm−1 (calcd −60.2 cm−1). In accordance with the computations, the shift of the SO stretching vibration displays a small positive value (exp. +1.5, calcd +4.7 cm−1).
The matrix isolated products of pyrolysis were also characterized using UV/Vis spectroscopy (Fig. 2). As expected, the recorded spectra showed a weak absorption band of 7 with its characteristic vibrational fine structure between 409 nm and about 320 nm, which is well known from literature data.17a From the second, much stronger band of 7, (λmax = 213 nm) only a part of the absorption could be observed. Structure 3 shows two absorption bands that differ strongly in intensities. One is very weak displaying a pronounced vibrational fine structure starting at 635 nm and terminating at around 450 nm (λmax ≅ 530 nm). The second much more intense one starts near 320 nm (λmax at ca. 260 nm) and overlaps with the very strong band of 7. Both bands of 3 correlate well with the values of the electronic excitations computed using time-dependent density functional theory (TD-DFT) computations at TD-UB3LYP/6-311+G(3df,3pd) (Table 1).
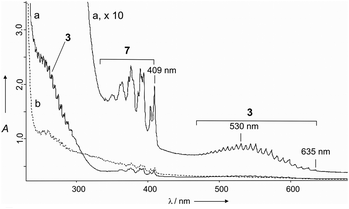 | ||
| Fig. 2 The UV/Vis absorption spectrum of the matrix isolated (Ar, 10 K) pyrolysis products of precursor 4a (2 h pyrolysis at 660 °C); upper trace (×10); (b) after 50 min irradiation with 248 nm light (KrF excimer laser). | ||
| State | E (eV) | λmax (nm) | Osc. str. | Trans. | Exptl. λmax (nm) |
|---|---|---|---|---|---|
| A, 2A′′ | 2.295 | 540 | 0.0005 | n → π* | ca. 530 |
| B, 2A′′ | 4.853 | 255 | 0.0001 | π → π* | n.o. |
| C, 2A′ | 4.954 | 250 | 0.0343 | π → π* | ca. 260 |
Our computations place the lowest energy transition from the electronic ground state to the first excited electronic state ((A)2A′′ ← (X)2A′′) at 540 nm with an extremely low oscillator strength of 0.0005. The second transition (B(2A′′) ← X(2A′′)), theoretically at 255 nm with an even lower intensity, is hidden under the very close-lying third absorption band (C(2A′) ← X(2A′′)) (250 nm) that is also two orders of magnitudes more intense. According to the orbitals involved in the corresponding electronic excitations (see ESI†), the weak absorption band in the visible range is an n → π* transition and the short wavelength intense band a π → π* transition.
The knowledge about the position of the absorption bands of 3 allows irradiation at specific wavelengths. While excitation with visible light did not induce photochemical transformations even after long irradiation times, photolysis of the matrix with selected wavelengths corresponding to the more intense UV-band of 3 (248 nm, 254 nm, and 290 nm) led to the simultaneous disappearance of both the IR as well as UV/Vis bands of 3. After photolysis a mixture of products such as CO, COS, H2O, CS, and thioformic acid was identified based on the IR absorption bands. Despite this rather unspecific decomposition, the differences between the spectra taken before and after irradiation allowed enhancement of the spectral features of 3, as shown in Fig. 1. Note that upon photolysis the photocylization of 7 leading to the cyclopropyl radical was largely suppressed.17d
In an extension of this study, we also tested dimethyl sulfoxide (8) as an alternative precursor for thermally generated 3 in the reaction H3C(SO)CH3 → 3 + ˙CH3. In accordance with the expected increase of the CS bond dissociation energy as compared to precursor 4a (computed at B3LYP/6-311+G(3df,3pd): 21 kcal mol−1), this pyrolysis had to be performed at a significantly higher temperature (800 °C).
The pyrolysate collected in the Ar matrix was analysed by means of IR spectroscopy. In this case, however, the characteristic bands of 3 and those of the methyl radical were observed along with the unconverted starting material, forming a major component of the collected pyrolysate. In addition, small amounts of 5, and methane were also observed. Hence, 8 is a poor precursor of 3.
In summary, radical 3 as well as its D3- and 13C-isotopologues were generated by high-vacuum flash pyrolysis and isolated in solid argon at 10 K starting either with allylmethyl sulfoxide (4a) or its isotopologues 4b and 4c, respectively, or with dimethyl sulfoxide (8). The identity of 3 was unambiguously confirmed based on the good agreement of measured and computed IR spectra at the AE-CCSD(T)/cc-pVTZ level of theory. The recorded UV/Vis spectrum showing a very weak band in the visible range (635–450 nm) due to an n → π* transition and a more intense π → π* absorption (λmax ≅ 260 nm) corresponds well with TD-DFT computations. As evident from the IR spectra, photolysis of 3 using UV light (λ < 300 nm) leads to an unspecific decomposition. The presented new method for the efficient generation of 3 opens the door to further studies on the structures of products formed in its reactions with atmospheric gases such as O2, O2 or NO2. These reactions have been postulated to be the main degradation pathways for 3 in the atmosphere and they deserve further in-depth investigation.
We dedicate this work to our dear colleague Bogusław Kryczka on the occasion of his 70th birthday. This work was supported by the DAAD Partnership of the University of Lodz and the Justus-Liebig University. G. M. thanks the National Science Center (Poland) for financial support (Project Maestro-3; Dec-2012/06/A/ST5/00219).
Notes and references
- (a) R. J. Charlson, J. E. Lovelock, M. O. Andreae and S. G. Warren, Nature, 1987, 326, 655 CrossRef CAS; (b) B. Albrecht, Science, 1989, 245, 1227 CAS; (c) R. J. Charlson, S. E. Schwarz, J. M. Hales, R. D. Cess, J. A. Coakley, Jr., J. E. Hansen and D. J. Hofmann, Science, 1992, 425, 423 Search PubMed; (d) M. O. Andreae, C. D. Jones and P. M. Cox, Nature, 2005, 435, 1187 CrossRef CAS PubMed; (e) I. Faloona, Atmos. Environ., 2009, 43, 2841 CrossRef CAS PubMed.
- (a) G. S. Tyndall and A. R. Ravishankara, Int. J. Chem. Kinet., 1991, 23, 483 CrossRef CAS; (b) S. B. Barone, A. A. Turnipseed and A. R. Ravishankara, Faraday Discuss., 1995, 100, 39 RSC; (c) I. Barnes, J. Hjorth and N. Mihalopoulos, Chem. Rev., 2006, 106, 940 CrossRef CAS PubMed; (d) D. D. Gregory and W. S. Jencks, J. Org. Chem., 1998, 63, 3859 CrossRef CAS.
- (a) E. Block, Angew. Chem., Int. Ed., 1992, 31, 1135 CrossRef; (b) V. Vaidya, K. U. Ingold and D. A. Pratt, Angew. Chem., Int. Ed., 2009, 48, 157 CrossRef CAS PubMed.
- (a) S. M. Resende and F. R. Ornellas, Chem. Phys. Lett., 2003, 367, 489 CrossRef CAS; (b) X. Li, L. Meng and S. Zheng, THEOCHEM, 2007, 847, 52 CrossRef CAS PubMed; (c) X.-Y. Li, L.-P. Meng and S.-J. Zheng, Chin. J. Chem., 2007, 25, 1480 CrossRef CAS; (d) R. Asatryan and J. W. Bozzelli, Phys. Chem. Chem. Phys., 2008, 10, 1769 RSC; (e) X. Li, L. Meng, Z. Sun and S. Zheng, THEOCHEM, 2008, 870, 53 CrossRef CAS PubMed; (f) A. Lesar, Int. J. Quantum Chem., 2012, 112, 1904 CrossRef CAS; (g) Z. Salta, A. M. Kosmas and A. Lesar, Comput. Theor. Chem., 2012, 1001, 67 CrossRef CAS PubMed.
- (a) G. K. Buerk and G. Schoffa, Int. J. Protein Res., 1969, 1, 113 CrossRef CAS; (b) T. Kawamura, P. J. Krusic and J. K. Kochi, Tetrahedron Lett., 1972, 4075 CrossRef CAS; (c) J. C. Machado, R. Debuyst, F. Dejehet and D. Apers, Radiochem. Radioanal. Lett., 1972, 9, 363 CAS; (d) K. Nishikida and F. Williams, J. Am. Chem. Soc., 1974, 96, 4781 CrossRef CAS; (e) B. C. Gilbert, C. M. Kirk, R. O. C. Norman and H. A. H. Laue, J. Chem. Soc., Perkin Trans. 2, 1977, 497 RSC; (f) D. J. Nelson, J. Phys. Chem., 1978, 82, 1400 CrossRef CAS; (g) S. G. Swarts, D. Becker, S. DeBolt and M. D. Sevilla, J. Phys. Chem., 1989, 93, 155 CrossRef CAS.
- F. Turecek, D. E. Drinkwater and F. W. McLafferty, J. Am. Chem. Soc., 1989, 111, 7696 CrossRef CAS.
- W.-C. Hung, M.-Y. Shen, Y.-P. Lee, N.-S. Wang and B.-M. Cheng, J. Chem. Phys., 1996, 105, 7402 CrossRef CAS; B.-M. Cheng, E. P. Chew, W.-C. Hung, J. Eberhard and Y.-P. Lee, J. Synchrotron Radiat., 1998, 5, 1041 CrossRef PubMed.
- L.-K. Chu and Y.-P. Lee, J. Chem. Phys., 2010, 133, 184303 CrossRef PubMed.
- (a) D. A. Blank, S. W. North, D. Stranges, A. G. Suits and Y. P. Lee, J. Chem. Phys., 1997, 106, 539 CrossRef CAS; (b) G. M. Thorson, C. M. Cheatum, M. J. Coffey and F. Fleming Crim, J. Chem. Phys., 1999, 110, 10843 CrossRef CAS; (c) J.-W. Ho, W.-K. Chen and P. Y. Cheng, J. Am. Chem. Soc., 2007, 129, 3784 CrossRef CAS PubMed.
- (a) H. Gross, Y. He, M. Quack, A. Schmid and G. Seyfang, Chem. Phys. Lett., 1993, 213, 121 CrossRef; (b) H. Gross, Y. He, M. Quack and G. Seyfang, Springer Proc. Phys., 1994, 74, 169 CrossRef CAS.
- D. L. Singleton, R. S. Irwin and R. J. Cvetanovic, Can. J. Chem., 1983, 61, 968 CrossRef CAS.
- (a) F. Domine, T. P. Murrells and C. J. Howard, J. Phys. Chem., 1990, 94, 5839 CrossRef CAS; (b) G. S. Tyndall and A. R. Ravishankara, J. Phys. Chem., 1989, 93, 2426 CrossRef CAS; (c) F. Domine, A. R. Ravishankara and C. J. Howard, J. Phys. Chem., 1992, 96, 2171 CrossRef CAS; (d) A. Kukui, V. Bossoutrot, G. Laverdet and G. Le Bras, J. Phys. Chem. A, 2000, 104, 935 CrossRef CAS; (e) D. Borissenko, A. Kukui, G. Laverdet and G. Le Bras, J. Phys. Chem., 2003, 107, 1155 CrossRef CAS.
- D. D. Lucas and R. G. Prinn, Atmos. Chem. Phys., 2005, 5, 1505 CrossRef CAS.
- (a) J. Romanski, H. P. Reisenauer, H. Petzold, W. Weigand, P. R. Schreiner and G. Mloston, Eur. J. Org. Chem., 2008, 2998 CrossRef CAS; (b) G. Mloston, J. Romanski, H. P. Reisenauer and G. Maier, Angew. Chem., Int. Ed., 2001, 113, 401 CrossRef; (c) H. P. Reisenauer, G. Mloston, J. Romanski and P. R. Schreiner, Eur. J. Org. Chem., 2011, 6269 CrossRef CAS; (d) H. P. Reisenauer, G. Mloston, J. Romanski and P. R. Schreiner, J. Am. Chem. Soc., 2010, 132, 7240 CrossRef PubMed; (e) H. P. Reisenauer, G. Mloston, J. Romanski and P. R. Schreiner, Eur. J. Org. Chem., 2012, 3480 Search PubMed . For review, see: ; (f) G. Mloston, J. Romanski, H. P. Reisenauer and P. R. Schreiner, Phosphorous, Sulfur, Silicon, 2011, 186, 1175 CrossRef CAS.
- D. E. Powers, C. A. Arrington, W. C. Harris, E. Block and V. F. Kalasinsky, J. Phys. Chem., 1979, 83, 1890 CrossRef CAS.
- (a) H. Bock, T. Hirabayashi and S. Mohmand, Chem. Ber., 1982, 115, 492 CrossRef CAS; (b) A. J. McGrath, G. E. Garrett, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2010, 132, 16759 CrossRef CAS PubMed.
- (a) G. Maier, H. P. Reisenauer, B. Rohde and K. Dehnicke, Chem. Ber., 1983, 116, 7323 CrossRef; (b) A. K. Mal'tsev, V. A. Korolov and O. M. Nefedov, Bull. Acad. Sci. USSR Div. Chem. Sci., 1982, 2131 Search PubMed; (c) A. K. Mal'tsev, V. A. Korolov and O. M. Nefedov, Bull. Acad. Sci. USSR Div. Chem. Sci., 1984, 510 Search PubMed; (d) K. Holtzhauer, C. Cometta-Morini and J. F. M. Oth, J. Phys. Org. Chem., 1990, 219 CrossRef CAS.
- B. Prabhuswamy and S. Y. Ambekar, Synth. Commun., 1999, 29, 3477 CrossRef CAS; M. L. Kline, N. Beutow, J. K. Kim and M. C. Caserio, J. Org. Chem., 1979, 44, 1904 CrossRef; D. E. O'Connor and W. I. Lyness, J. Am. Chem. Soc., 1964, 86, 3840 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Table, experimental, additional IR spectra and computational details. See DOI: 10.1039/c3cc45379k |
| This journal is © The Royal Society of Chemistry 2013 |