 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a phosphine imide bearing a hydrosilane moiety, and its water-driven reduction to a phosphine†‡
Naokazu
Kano
*,
Kazuhide
Yanaizumi
,
Xiangtai
Meng
,
Nizam
Havare
and
Takayuki
Kawashima§
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kano@chem.s.u-tokyo.ac.jp; Tel: +81 3 5841 4338
First published on 28th June 2013
Abstract
Organosilanes bearing a phosphine imide moiety were synthesized and crystallographically characterized. Reaction of the pentacoordinated hydrodiphenylsilyl derivative with water gave [2-(diphenylphosphino)phenyl]diphenylsilanol accompanied by both reduction of the phosphine imide moiety and hydrolytic oxidation of the Si–H moiety.
Hydrosilanes, R3Si–H, are useful for hydrosilylation of multiple bonds,1reduction of various functional groups,2 reductive aldol reactions,3 and preparation of a silanol, R3Si–OH, by hydrolysis.4 In some hydrosilanes, the silicon-bound hydrogen migrates intramolecularly through the 1,2-shift of the dyotropic rearrangement,5 1,3-shift,6 and 1,5-shift.7 As represented by the 1,3-hydride shift, the hypercoordinated hydrosilanes show hydride reducing reactivity without catalysis.8 Some neutral pentacoordinated hydrosilanes react easily with alcohols, carboxylic acids, and water in the absence of a catalyst and evolve hydrogen gas,8a,c,d whereas tetracoordinated hydrosilanes react with them quite slowly even under harsh reaction conditions.2 In these pentacoordinated hydrosilanes, the intramolecular coordination of a tertiary amino group with a suitable tether to the silicon atom is crucial for the pentacoordinated state of silicon and enhancement of the hydride reactivity. If a hydrosilyl group is located in the neighborhood of a phosphine imide, R3P
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR′, the intramolecular N⋯Si coordination, which should increase its hydride reactivity, is expected. This expectation is based on the reports in which some phosphine imides have been used as a chelating ligand for several transition metal complexes.9 As far as we know, amongst the few reported silicon compounds bearing a phosphine imide ligand, no such hydrosilane has been reported.10 We report here the synthesis and structure of a hydrosilane bearing a phosphine imide moiety and a unique conversion of its triarylphosphine imide moiety to the triarylphosphine moiety by hydride transfer from the hydrosilane moiety triggered by the reaction with water without any catalyst.
NR′, the intramolecular N⋯Si coordination, which should increase its hydride reactivity, is expected. This expectation is based on the reports in which some phosphine imides have been used as a chelating ligand for several transition metal complexes.9 As far as we know, amongst the few reported silicon compounds bearing a phosphine imide ligand, no such hydrosilane has been reported.10 We report here the synthesis and structure of a hydrosilane bearing a phosphine imide moiety and a unique conversion of its triarylphosphine imide moiety to the triarylphosphine moiety by hydride transfer from the hydrosilane moiety triggered by the reaction with water without any catalyst.
Phosphine imide 1 was lithiated with methyllithium in Et2O at room temperature,9,11,12 and then treated with chlorosilanes to give the corresponding silylated phosphine imides 2a–c in 56, 59 and 64% yields, respectively, from 1 (Scheme 1). Compounds 2a–c are thermally stable at room temperature in contrast to the phosphorus ylide analogue of 2a which was reported to undergo 1,4-silyl migration at 0 °C.13 Compounds 2a and 2b are not sensitive to water. However, in contrast, compound 2c is very sensitive to moisture. Treatment of 2c with water at room temperature gave [2-(diphenylphosphino)phenyl]diphenylsilanol (3) quantitatively, based on the monitoring of the reaction using 31P NMR spectroscopy and it was isolated in 87% yield (Scheme 2). The structure of 3 was determined using 1H, 13C, 29Si, and 31P NMR spectroscopy, IR spectroscopy and mass spectrometry. The 31P NMR spectrum (δP −10.1 ppm) established the existence of a triarylphosphine moiety in 3, instead of the corresponding triarylphosphine oxide moiety. Formation of the other product, tert-butylamine, which was derived from the imino moiety of 2c, was confirmed using GC-MS. Interestingly, the reaction of 2c with water resulted in the conversion from a hydrosilane and a triarylphosphine imide to a silanol and a set of a triarylphosphine and an amine, respectively. Therefore, this is not a usual hydrolysis of an iminophosphorane to give the corresponding phosphine oxide.
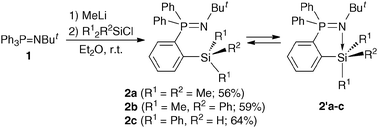 | ||
| Scheme 1 | ||
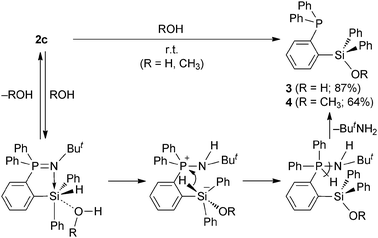 | ||
| Scheme 2 | ||
In order to reveal the causes of the reactivity of 2c, we analyzed the structure in detail. The 1H, 13C, 29Si, and 31P NMR spectroscopy results and elemental analysis of 2a–c were consistent with the silylated triarylphosphine imides. The 29Si NMRspectra of 2a and 2b in CDCl3 showed only small downfield shifts (2a: δSi −2.8 ppm; 2b: δSi −5.2 ppm) compared to trimethylphenylsilane (δSi −4.1 ppm)14 and dimethyldiphenylsilane (δSi −8.6 ppm),15 respectively. In contrast, 2c showed a significant upfield shift and an increased Si–H coupling constant (δSi −26.4 ppm (d, 1JSiH = 231 Hz)) compared to triphenylsilane (δSi −17.4 ppm (d, 1JSiH = 199 Hz)) in C6D6 at room temperature. In the VT-NMR experiment, lowering the temperature of a THF-d8 solution of 2c caused an increased upfield shift [δSi −25.6 ppm at 25 °C, δSi −29.7 ppm at −60 °C]. These results indicate the existence of a dynamic equilibrium between the chelated pentacoordinated and unchelated tetracoordinated silicon states in solution as shown in Scheme 1 and, especially at low temperature, a certain degree of contribution of the pentacoordination state represented by 2′c.16
To confirm the contribution of a pentacoordination state of silicon in 2c, the crystal structures of trimethylsilane 2a and hydrodiphenylsilane 2c were determined by X-ray crystallographic analysis (Fig. 1). The N1 atom is located on the backside of the Si1–C23 bond and directed to the silyl group in both 2a and 2c. The N1⋯Si1 interatomic distance in 2c (2.768(3) Å) is much shorter than the corresponding distance in 2a (3.353(1) Å) and the sum of the van der Waals radii (3.54 Å). The bond angle ∠C1–Si1–C23 in 2c (104.05(12)°) is somewhat narrower than that (107.08(6)°) in 2a. These structural features show that 2c is a capped tetrahedral structure around the pentacoordinated Si1 atom. The difference in the degree of the intramolecular N⋯Si coordination between 2a and 2c is ascribed to the difference in the electrophilicity of silicon. Considering that the hydrodiphenylsilane bearing 2-(phenylazo)phenyl group provides no N⋯Si interaction,17 the phosphine imide acts as a good coordinating ligand to achieve the pentacoordinated silicon state, reflecting the polarized P–N bond character.9b
![ORTEP drawings of 2a (left) and 2c (right) with the thermal ellipsoid plot (50% probability). Hydrogen atoms except for H1 in 2c are omitted for clarity. Selected bond lengths [Å] and bond angles [°] of 2a: Si1–C1 1.9043(12), Si1–C23 1.8796(14), Si1–C24 1.8617(13), Si1–C25 1.8693(13), Si1⋯N1 3.353(1), N1–P1 1.5479(10), C1–Si1–C23 107.08(6), C1–Si1–C24 110.22(6), C1–Si1–C25 114.01(5), C23–Si1–C24 106.51(6), C23–Si1–C25 105.50(6), C24–Si1–C25 112.94(6). 2c: Si1–C1 1.900(3), Si1–C23 1.905(3), Si1–C29 1.880(3), Si1–H1 1.43(2), Si1⋯N1 2.768(3), N1–P1 1.561(2), C1–Si1–C23 104.05(12), C1–Si1–C29 111.97(12), C23–Si1–C29 104.65(13), C1–Si1–H1 114.6(9), C23–Si1–H1 105.6(9), C29–Si1–H1 114.7(9).](/image/article/2013/CC/c3cc43955k/c3cc43955k-f1.gif) | ||
| Fig. 1 ORTEP drawings of 2a (left) and 2c (right) with the thermal ellipsoid plot (50% probability). Hydrogen atoms except for H1 in 2c are omitted for clarity. Selected bond lengths [Å] and bond angles [°] of 2a: Si1–C1 1.9043(12), Si1–C23 1.8796(14), Si1–C24 1.8617(13), Si1–C25 1.8693(13), Si1⋯N1 3.353(1), N1–P1 1.5479(10), C1–Si1–C23 107.08(6), C1–Si1–C24 110.22(6), C1–Si1–C25 114.01(5), C23–Si1–C24 106.51(6), C23–Si1–C25 105.50(6), C24–Si1–C25 112.94(6). 2c: Si1–C1 1.900(3), Si1–C23 1.905(3), Si1–C29 1.880(3), Si1–H1 1.43(2), Si1⋯N1 2.768(3), N1–P1 1.561(2), C1–Si1–C23 104.05(12), C1–Si1–C29 111.97(12), C23–Si1–C29 104.65(13), C1–Si1–H1 114.6(9), C23–Si1–H1 105.6(9), C29–Si1–H1 114.7(9). | ||
Based on the contribution of the pentacoordinated silicon in 2c, a mechanism of formation for 3 was proposed as shown in Scheme 2. Formation of 3 from 2c is initiated by reversible coordination of water to the silicon atom, whose Lewis acidity is enhanced by the contribution of the pentacoordinated state based on the intramolecular N⋯Si coordination in 2c,18 and proton transfer to the neighboring nitrogen forms a silicate intermediate. Hydride transfer from silicon to phosphorus generates an amino(hydro)phosphorane intermediate, and its facile reductive deamination gives 3 and tert-butylamine as the final products. In another possible reaction mechanism, the phosphine imide moiety, which may work as a base, is hydrolyzed to a phosphine oxide and tert-butylamine before the conversion of the hydrosilane moiety to silanol. We carried out three experiments to determine the reaction mechanism: (i) monitoring of the reaction solution using VT-NMRspectroscopy, (ii) a labeling experiment using D2O instead of H2O, and (iii) a reaction of 2c with methanol. In experiment (i), however, the formation of 3 from 2c was so fast that no intermediate could be observed using 1H and 31P NMR spectroscopy even at −20 °C. In experiment (ii), quantitative formation of 2-(Ph2P)C6H4Si(OD)Ph2 (3-ddd) and ButNHD in the reaction of 2c with D2O was confirmed using 1H and 31P NMR spectroscopy. The GC-MS measurement also supported the formation of ButNHD (m/z = 74, M+). Therefore, silicon-bound hydrogen in 2c was used for the quantitative formation of ButNH2 but not used for the formation of hydrogen gas in the reaction with water. The experimental result is consistent with the proposed reaction mechanism in Scheme 2. Combination of the characteristics of both pentacoordinated silicon and phosphorus should be a key for this reaction of 2c. In experiment (iii), the reaction of 2c with methanol gave methoxysilane 4 in 64% yield (Scheme 2). Therefore, even though OH− or MeO− may attack the phosphorus in any of the steps, the intermediate should give 2c back and finally give 3 or 4. Experiments (ii) and (iii) rule out other reaction mechanisms involving the hydrolysis of the phosphine imide moiety.
In the previous reports, phosphine imides were reduced with strong reductants such as lithium aluminum hydride and trichlorosilane, which react vigorously with water.19 In contrast, the reduction of the phosphine imide of 2c to phosphine was promoted by the reaction with water and it proceeded quickly at room temperature. Simultaneously, the hydrosilane moiety was converted to the silanol moiety. In association with this reactivity of 2c to give 3 and 4, dehydrogenative condensation of hydrosilanes with alcohols or amines without additional catalyst has been reported by combination of a triarylborane moiety in a single molecule, although condensation with water was not mentioned.20 In addition, some hypercoordinated silanes evolve hydrogen gas in the condensation with alcohols, carboxylic acids and water.8a,c,d Therefore, it is notable that the conversion of 2c to 3 and 4 proceeds at room temperature without any catalyst evolving hydrogen gas.
In summary, three organosilicon compounds2a–c bearing a phosphine imide moiety were synthesized. The hydrosilane moiety of the hydrodiphenylsilyl derivative 2c was hydrolyzed without any catalyst to the corresponding silanol accompanied by the reduction of the iminophosphorane moiety to triarylphosphine. Its high reactivity can be ascribed to the characteristics of both highly coordinated silicon and phosphorus in the intermediates. The unique structure of the product, which bears both the silanol and the triarylphosphine moiety at the ortho-position, is expected to be useful as a precursor of an O,P-chelating ligand for transition metal complexes.21
We thank Tosoh Finechem Corp. and Shin-etsu Chemical Co. Ltd. for gifts of alkyllithiums and silicon reagents, respectively. This work was partially supported by Grants-in-Aid for the Global COE Program for Chemistry Innovation and for Scientific Research (No. 22108508, 22685005 and 25109512) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Notes and references
- (a) B. Marciniec, J. Gulinski, W. Urbaniak and Z. W. Kornetka, in Comprehensive Handbook on Hydrosilylation, ed. B. Marciniec, Pergamon Press, Oxford, United Kingdom, 1992 Search PubMed; (b) B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluc, in Hydrosilylation. A Comprehensive Review on Recent Advances, ed. B. Marciniec, Springer, 2009 Search PubMed; (c) D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004 CrossRef CAS.
- M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000, ch. 8, pp. 171–188 Search PubMed.
- H. Nishiyama and T. Shiomi, Top. Curr. Chem., 2007, 279, 105 CrossRef CAS.
- P. D. Lickiss, Adv. Inorg. Chem., 1995, 42, 147 CrossRef CAS.
- J. Fassler and S. Bienz, Organometallics, 1994, 13, 4704 CrossRef.
- (a) R. J. P. Corriu, G. F. Lanneau and M. Perrot, Tetrahedron Lett., 1987, 28, 3941 CrossRef CAS; (b) M. Yamamura, N. Kano and T. Kawashima, Tetrahedron Lett., 2007, 48, 4033 CrossRef CAS; (c) E. Kertsnus-Banchik, I. Kalikhman, B. Gostevskii, Z. Deutsch, M. Botoshansky and D. Kost, Organometallics, 2008, 27, 5285 CrossRef CAS; (d) K. Lippe, D. Gerlach, E. Kroke and J. Wagler, Organometallics, 2009, 28, 621 CrossRef CAS; (e) M. Yamamura, N. Kano and T. Kawashima, Z. Anorg. Allg. Chem., 2009, 635, 1295 CrossRef CAS.
- (a) S. Anwar and A. P. Davis, J. Chem. Soc., Chem. Commun., 1986, 831 RSC; (b) S. W. McCombie, C. Ortiz, B. Cox and A. K. Ganguly, Synlett, 1993, 541 CrossRef CAS.
- (a) J. Boyer, C. Breliere, R. J. P. Corriu, A. Kpoton, M. Poirier and G. Royo, J. Organomet. Chem., 1986, 311, C39 CrossRef CAS; (b) R. J. P. Corriu, G. F. Lanneau and M. Perrot, Tetrahedron Lett., 1988, 29, 1271 CrossRef CAS; (c) P. Arya, R. J. P. Corriu, K. Gupta, G. F. Lanneau and Z. Yu, J. Organomet. Chem., 1990, 399, 11 CrossRef CAS; (d) R. J. P. Corriu, G. F. Lanneau, M. Perrot-Petta and V. D. Nehta, Tetrahedron Lett., 1990, 31, 2585 CrossRef CAS; (e) R. J. P. Corriu, G. F. Lanneau and Z. Yu, Tetrahedron, 1993, 49, 9019 CrossRef CAS; (f) Y. Domoto, A. Fukushima, Y. Kasuga, S. Sase, K. Goto and T. Kawashima, Org. Lett., 2010, 12, 2586 CrossRef CAS.
- (a) S. Wingerter, H. Gornitzka, G. Bertrand and D. Stalke, Eur. J. Inorg. Chem., 1999, 173 CrossRef CAS; (b) A. Steiner, S. Zacchini and P. I. Richards, Coord. Chem. Rev., 2002, 227, 193 CrossRef CAS; (c) J. Vicente, J.-A. Abad, R. Clemente, J. Lopéz-Serrano, M. C. Ramírez de Arellano, P. G. Jones and D. Bautista, Organometallics, 2003, 22, 4248 CrossRef CAS; (d) R. Bielsa, A. Larrea, R. Navarro, T. Soler and E. P. Urriolabeitia, Eur. J. Inorg. Chem., 2005, 1724 CrossRef CAS; (e) D. Aguilar, M. Contel, R. Navarro and E. P. Urriolabeitia, Organometallics, 2007, 26, 4604 CrossRef CAS; (f) R. Bielsa, R. Navarro, E. P. Urriolabeitia and A. Lledós, Inorg. Chem., 2007, 46, 10133 CrossRef CAS; (g) E. Martinez-Arripe, F. Jean-Baptiste-dit-Dominique, A. Auffrant, X.-F. LeGoff, J. Thuilliez and F. Nief, Organometallics, 2012, 31, 4854 CrossRef CAS.
- (a) W. Wolfberger, H.-H. Pickel and H. Schnzidbaur, Chem. Ber., 1971, 104, 1830 CrossRef; (b) A. Sivaramakrishna, I. Kalikhman, E. Kertsnus, A. A. Korlyukov and D. Kost, Organometallics, 2006, 25, 3665 CrossRef CAS; (c) Y. Xiong, S. Yao, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 10074 CrossRef CAS.
- (a) P. Wei, K. T. K. Chan and D. W. Stephan, Dalton Trans., 2003, 3804 RSC; (b) K. T. K. Chan, L. P. Spencer, J. D. Masuda, J. S. J. McCahill, P. Wei and D. W. Stephan, Organometallics, 2004, 23, 381 CrossRef CAS; (c) L. Boubekeur, L. Ricard, N. Mézailles, M. Demange, A. Auffrant and P. Le Floch, Organometallics, 2006, 25, 3091 CrossRef CAS.
- N. Kano, K. Yanaizumi, X. Meng and T. Kawashima, Heteroat. Chem., 2012, 23, 429 CrossRef CAS.
- A. Kawachi, T. Yoshioka and Y. Yamamoto, Organometallics, 2006, 25, 2390 CrossRef CAS.
- C. Breliére, F. Carré, R. J. P. Corriu, M. Poirier and G. Royo, Organometallics, 1986, 5, 388 CrossRef.
- R. H. Cragg and R. D. Lane, J. Organomet. Chem., 1984, 277, 199 CrossRef CAS.
- (a) B. J. Helmer, R. West, R. J. P. Corriu, M. Poirier, G. Royo and A. De Saxce, J. Organomet. Chem., 1983, 251, 295 CrossRef CAS; (b) N. Auner, R. Probst, F. Hahn and E. Herdtweck, J. Organomet. Chem., 1993, 459, 25 CrossRef CAS.
- (a) N. Kano, F. Komatsu and T. Kawashima, Chem. Lett., 2001, 338 CrossRef CAS; (b) N. Kano, F. Komatsu, M. Yamamura and T. Kawashima, J. Am. Chem. Soc., 2006, 128, 7097 CrossRef CAS.
- M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000, chap. 4, pp. 97–114 Search PubMed.
- (a) L. D. Quin, G. Keglevich and K. C. Caster, Phosphorus, Sulfur Relat. Elem., 1987, 31, 133 CrossRef CAS; (b) J. Barluenga, F. López and F. Palacios, Tetrahedron Lett., 1987, 28, 2875 CrossRef CAS; (c) J. Barluenga, F. López and F. Palacios, Synthesis, 1989, 298 CrossRef CAS.
- (a) A. Kawachi, M. Zaima, A. Tani and Y. Yamamoto, Chem. Lett., 2007, 362 CrossRef CAS; (b) L. Könczöl, A. Kawachi and D. Szieberth, Organometallics, 2012, 31, 120 CrossRef.
- (a) P. W. N. M. van Leeuwen, C. F. Roobeek and A. G. Orpen, Organometallics, 1990, 9, 2179 CrossRef CAS; (b) M. A. Garralda, R. Hernández, L. Ibarlucea, E. Pinilla, M. R. Torres and M. Zarandona, Organometallics, 2007, 26, 1031 CrossRef CAS; (c) N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 9590 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Renji Okazaki on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and spectral data. CCDC 867751 (2a) and 867750 (2c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc43955k |
| § Present address: Graduate School of Science and Technology, Gunma University, 1-5-1 Tenjin-cho, Kiryu, Gunma 376-8515, Japan. |
| This journal is © The Royal Society of Chemistry 2013 |