 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemically crosslinked isoreticular metal–organic frameworks†
Corinne A.
Allen
,
Jake A.
Boissonnault
,
Jordi
Cirera
,
Ryan
Gulland
,
Francesco
Paesani
and
Seth M.
Cohen
*
Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA, USA 92093. E-mail: scohen@ucsd.edu
First published on 6th March 2013
Abstract
Herein we report the synthesis of canonical isoreticular metal–organic frameworks (IRMOFs) containing interligand crosslinks. Chemically crosslinking two molecules of 2-amino-1,4-benzene dicarboxylic acid (NH2-BDC) gives ligands that readily form IRMOF-1 analogs, producing crosslinked MOFs that may be designed to have novel properties.
Hybrid organic–inorganic porous materials have been of increasing interest for numerous large-scale applications such as CO2 sequestration,1 H2 storage,2–4 separations,5–7 catalysis,8 biomedicine, and many others.9–12 Due to their large surface areas and tunable pore features, metal–organic frameworks (MOFs) have become the hybrid material of choice as next generation porous, crystalline solids.13 Tuning the chemical and physical properties of MOFs, such as rendering them more stable to moisture, can be achieved by covalent modification of the ligands.14 Ligand modification can be achieved both presynthetically (prior to MOF synthesis) or postsynthetically by executing chemical reactions on the intact, crystalline MOF.15 Herein, it is shown that a presynthetic approach allows for incorporation of a chemically crosslinked ligand that results in unusual guest entrapment when compared to the parent IRMOF-3 (IRMOF = isoreticular metal–organic framework) material.
One method to alter the properties of materials, such as polymers and protein crystals, is through chemical crosslinking.16–18 While crosslinking is well established in many fields of chemistry and materials science,19–22 applications of crosslinking to MOFs has been somewhat limited.23–30 Zaworotko and co-workers have described one form of crosslinking, by preparing MOFs using discrete metal–organic polyhedral (MOPs) that have been connected by a linker. However, the strategy of connecting MOPs together to obtain MOFs is conceptually distinct from what is described here. In Zaworotko's approach crosslinking refers to the connection of supramolecular polyhedral or secondary building units (SBUs) that dictate the overall topology of the framework. In these studies, the pore size and shape of the parent framework is not altered, as the MOP substructure predefines these spaces. The work here focuses on covalent linkages between the ‘rod’ (ligand) portions of a MOF of known structure (i.e. IRMOF), which has isolated SBUs, but does not contain MOP-like subunits. In this manner, the pores of the MOF are directly affected by the presence of the chemical crosslink (vide infra). The only example of direct chemical crosslinking of a MOF-like material was reported over a decade ago, when Lee and coworkers explored the stability and characteristics of a chemically crosslinked coordination solid based on Ag(I) ions and tritopic nitrile ligands.31 Using a postsynthetic modification (PSM) approach, Lee's seminal study clearly demonstrated that ligands within an intact coordination solid could be directly crosslinked resulting in enhanced framework stability.
In light of the latter example, initial attempts to produce crosslinked MOFs employed a PSM strategy. Specifically, crystals of IRMOF-3, containing free aniline groups, were treated with diacylchlorides in an attempt to link pairs of NH2-BDC ligands via amide bonds. It was reasoned that such an approach should be effective based on a number of studies showing that the amine groups in IRMOF-3 can be readily acylated by anhydrides.32 However, all attempts to treat IRMOF-3 with these reagents resulted in framework degradation, likely due to the HCl byproduct generated from the diacylchlorides during the PSM.
To overcome this limitation, a series of pre-crosslinked NH2-BDC ligands were prepared according to Scheme 1. These ligands contain between 5–9 atoms (3–7 methylene groups and two carbonyl groups) between the amide moieties. All of the ligands were straightforward to prepare, in good yields, on a multigram scale (see ESI†).
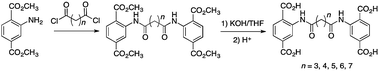 | ||
| Scheme 1 Synthesis of crosslinked, tetracarboxylate ligands (L3–7). | ||
With these crosslinked, tetracarboxylate ligands in hand, the direct solvothermal synthesis of a series of IRMOFs was pursued. It was somewhat surprising to find that all of the ligands (L3–7) produced the desired IRMOF analogues (termed IRMOF-3-AMnXL, AM = amide, n = number of methylene units, XL = crosslinked) under standard solvothermal synthesis conditions (Scheme 2). IRMOF-3-AM3XL (78%) and IRMOF-3-AM4XL (46%) formed as colorless, block crystals after 18 h. Longer linkers required longer reaction times (48 h) to obtain optimal yields, producing IRMOF-3-AM5XL (75%), IRMOF-3-AM6XL (72%), and IRMOF-3-AM7XL (52%) as small, cubic crystals, with the IRMOF-3-AM7XL producing a truncated cube morphology (Fig. S1–S5, ESI†).
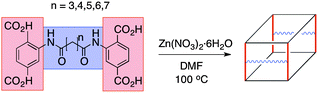 | ||
| Scheme 2 Cartoon representation of the synthesis of crosslinked IRMOFs. Other linker orientations are possible (Fig. S8, ESI†). | ||
All IRMOFs were characterized using single-crystal (XRD) and powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA), 1H NMR, and gas sorption studies. Single-crystals XRD structures of the materials unambiguously confirm the formation of the desired IRMOF topology (Fig. S6, Tables S1 and S2, ESI†); however, electron density for the linker groups could not be assigned in any of the structures. The PXRD patterns of all IRMOF-AMnXL samples matched the simulated IRMOF-3 powder patterns (Fig. 1).
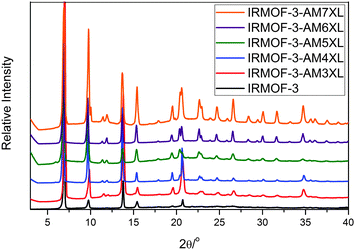 | ||
| Fig. 1 Experimental PXRD patterns for IRMOFs (as synthesized) compared to the simulated pattern for IRMOF-3. | ||
The relative thermal stabilities of the crosslinked IRMOFs are on par with IRMOF-3, with framework degradation occurring at ∼400 °C (Fig. S1–S5, ESI†). This is in contrast with the earlier work by Lee and co-workers, who noted that their fragile coordination network became more thermally stable upon crosslinking.31 Although no enhancement in thermal stability was observed for the IRMOF-3-AMnXL solids, presumably due to the thermal lability of simple alkyl chains, a measurable difference in thermal activation was found. For activation at 105 °C, the TGA analysis did reveal trapped, residual guests in IRMOF-3-AM4XL.
Standard activation for IRMOF samples involves three days of solvent exchange with CHCl3 followed by heating at 105 °C under vacuum. This procedure results in full activation of IRMOF-3 as evidenced by TGA, 1H NMR, and gas sorption analysis, all of which indicate that the material is essentially devoid of residual solvent. This standard thermal treatment at 105 °C can fully activate IRMOF-3, as well as all of the IRMOF-3-AMnXLs, except for n = 4. Activation of IRMOF-3-AM4XL at 105 °C resulted in a material that showed a significant weight loss (12% mass loss) at ∼140 °C and 1H NMR analysis indicated the presence of DMF (Fig. S2, ESI†). The residual DMF originates from the solvothermal synthesis process despite extensive washing of the crystals with CHCl3. When activated at 150 °C, IRMOF-3-AM4XL showed no substantial mass loss at temperatures below 350 °C and no residual DMF in the 1H NMR (Fig. S2, ESI†), suggesting complete desolvation.
Solvent entrapment was further evidence by Brunauer–Emmett–Teller (BET) surface area measurements (Table S3, ESI†). After activation at 105 °C under vacuum, IRMOF-3-AMnXL (where n = 3, 5–7) samples show surface areas (∼1575–2000 m2 g−1), reflective of the IRMOF structure (Fig. S7, ESI†). The values obtained were independent of the activation temperature (105 °C versus 150 °C) and consistent with those obtained for similarly acylated IRMOF-3 structures.33 IRMOF-3-AM4XL behaved differently, depending on the activation temperature. When activated at 105 °C, IRMOF-3-AM4XL gave a BET surface area of 1618 m2 g−1 even after 48 h of heating. However, when activated at 150 °C, the BET surface area measurably and reproducibly increased by ∼10% (1774 m2 g−1, Table S3, ESI,†Fig. 2), again indicative of better activation and complete removal of DMF. For all the crosslinked MOFs, the presence of the linkers occluding the MOF pores was observed as a monotonic decrease of the average pore size of the IRMOF-3-AMnXL materials with increasing tether length (Horvath–Kawazoe model, Table S3, ESI†).
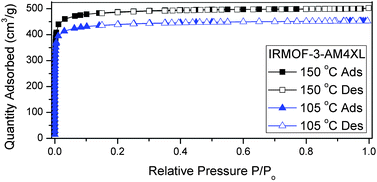 | ||
| Fig. 2 N2 isotherms of IRMOF-3-AM4XL after activation at both 105 °C (blue triangles) and 150 °C (black squares). Filled and open symbols represent the absorption and desorption curves, respectively. | ||
Several observations and experiments were performed to confirm these ligands were truly crosslinking the framework and to exclude the possibility that only one half of the ligand was participating in MOF formation. That is, it is possible that only one BDC component was incorporated into the MOF lattice, with the tether and second BDC group simply behaving as a dangling substituent within the MOF pores. Combustion analysis data obtained (Table S4, ESI†) clearly indicate that the carbon content of the MOF is consistent with both BDC groups participating in lattice formation. If the tether and one of the BDC units was acting simply as a substituent the carbon content would be nearly double that measured by combustion analysis (Table S5, ESI†). In addition, the gas sorption data supports the crosslinked formulation. The BET values obtained clearly indicate that both BDC groups are used structurally in the MOF. If the tether and one BDC group was dangling within the MOF pores, the BET values would be much lower than those obtained, as evidence by several previous studies on IRMOFs containing such bulky substituents.33
The bulk analytical methods used to characterize these new IRMOFs (e.g. TGA, BET) do not reveal the conformation of the different linkers across the pore of each IRMOF. As described above, XRD data failed to provide assignable electron density for the linkers (Fig. S6, ESI†). The linkers, depending on length, can be arranged is several possible ways across the pore of the MOF (Fig. S8, ESI†). Molecular dynamics calculations on the crosslinked MOFs were performed (ESI†), placing the different tethers within the distance constraints defined by the crystal structure. These calculations revealed that IRMOF-3-AM3XL and IRMOF-3-AM4XL can only connect adjacent ligands (‘isomer 1’, Fig. S8, ESI†) and are not sufficiently long to bisect the pore of the IRMOF (‘isomer 2’, Fig. S8, ESI†). However, IRMOF-3-AM5XL, -AM6XL, and -AM7XL can access multiple crosslinking conformations (Table S6, ESI†). Experimental and computational studies are ongoing to try to elucidate the conformation of these linkers within the MOFs.
It is somewhat remarkable that a single carbon difference in the tether length (IRMOF-3-AM4XL) can elicit a clearly measurable difference in the encapsulation properties of MOF. However, previous studies on alkyl-decorated DMOFs (DMOF = dabco MOF) have also shown similarly subtle differences. When varying the length of the alkyl substituent on these series of DMOFs ‘breathing’ behaviour was observed only for DMOF-1-AM3, while other chain lengths did not induce such structural plasticity into the MOF.34 The data obtained here on IRMOF-3-AM4XL show that even very small changes in ligand substituents can result in measurable differences in physical behaviour. We anticipate that further exploration of linker lengths, structure, and composition will lead to more unanticipated MOF properties.
In conclusion, we have obtained a unique crosslinked variant of the prototypical IRMOF system. This was achieved by direct solvothermal synthesis using a tethered, tetracarboxylate BDC ligand. Despite the use of a crosslinked ligand, the topology of the IRMOF lattice remained unaltered in these systems. Simple diacyl chlorides with alkyl chains of varying lengths were used to synthesize these ligands. In general, the properties of these MOFs are similar to the parent IRMOF-3; however, a notable difference in guest entrapment was observed with IRMOF-3-AM4XL. The robust inclusion of DMF in this MOF suggests that the physical properties of the MOF can be substantially altered by even very small changes in tether length. This observation, combined with the ability to incorporate additional chemical functionality into the tethering group, opens up a new subclass of MOFs with even more highly tailored chemical and physical properties solely within the pores.
We thank Dr Min Kim for helpful discussions, Dr Y. Su (U.C.S.D.) for performing mass spectrometry experiments, and Dr C. Moore, Prof. A. Rheingold (U.C.S.D.), and Phuong V. Dau for assistance with single-crystal X-ray crystallography. This work was supported by a grant from Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award No. DE-FG02-08ER46519. C.A.A. was supported by a National Science Foundation Graduate Research Fellowship (C.A.A.) under Award No. DGE1144086. J.C. was supported by the The Hellman Fellowship Program (UCSD).
Notes and references
- L. Wen, P. Cheng and W. B. Lin, Chem. Sci., 2012, 3, 2288–2292 RSC.
- S. Hong, M. Oh, M. Park, J. W. Yoon, J. S. Chang and M. S. Lah, Chem. Commun., 2009, 5397–5399 RSC.
- Y. Yan, X. Lin, S. H. Yang, A. J. Blake, A. Dailly, N. R. Champness, P. Hubberstey and M. Schroder, Chem. Commun., 2009, 1025–1027 RSC.
- Y. Yan, I. Telepeni, S. H. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schroder, J. Am. Chem. Soc., 2010, 132, 4092–4094 CrossRef CAS.
- W. G. Lu, J. P. Sculley, D. Q. Yuan, R. Krishna, Z. W. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS.
- X. S. Wang, S. Q. Ma, P. M. Forster, D. Q. Yuan, J. Eckert, J. J. Lopez, B. J. Murphy, J. B. Parise and H. C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7263–7266 CrossRef CAS.
- B. S. Zheng, J. F. Bai, J. G. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS.
- X. M. Lin, T. T. Li, L. F. Chen, L. Zhang and C. Y. Su, Dalton Trans., 2012, 41, 10422–10429 RSC.
- G. A. Carriedo, A. Presa, M. L. Valenzuela and M. Ventalon, J. Organomet. Chem., 2009, 694, 249–252 CrossRef CAS.
- L. R. Macgillivray, G. S. Papaefstathiou, T. Friscic, T. D. Hamilton, D. K. Bucar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS.
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817–7818 CrossRef CAS.
- D. Zhao, D. Q. Yuan, D. F. Sun and H. C. Zhou, J. Am. Chem. Soc., 2009, 131, 9186–9188 CrossRef CAS.
- S. Basu, M. Maes, A. Cano-Odena, L. Alaerts, D. E. De Vos and I. F. J. Vankelecom, J. Membr. Sci., 2009, 344, 190–198 CrossRef CAS.
- J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560–4561 CrossRef CAS.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS.
- C. J. Lusty, J. Appl. Crystallogr., 1999, 32, 106–112 CrossRef CAS.
- Y. Wang and O. Annunziata, Langmuir, 2008, 24, 2799–2807 CrossRef CAS.
- L. Ma, C. Y. Gao, Z. W. Mao, J. C. Shen, X. Q. Hu and C. M. Han, J. Biomater. Sci., Polym. Ed., 2003, 14, 861–874 CrossRef CAS.
- Z. T. Xu, S. Lee, Y. H. Kiang, A. B. Mallik, N. Tsomaia and K. T. Mueller, Adv. Mater., 2001, 13, 637–641 CrossRef CAS.
- E. A. Appel, J. Del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- M. A. B. Meador, E. F. Fabrizio, F. Ilhan, A. Dass, G. H. Zhang, P. Vassilaras, J. C. Johnston and N. Leventis, Chem. Mater., 2005, 17, 1085–1098 CrossRef CAS.
- J. J. Perry, V. C. Kravtsov, G. J. McManus and M. J. Zaworotko, J. Am. Chem. Soc., 2007, 129, 10076–10077 CrossRef CAS.
- S. Chavan, J. G. Vitillo, M. J. Uddin, F. Bonino, C. Lamberti, E. Groppo, K. P. Lillerud and S. Bordiga, Chem. Mater., 2010, 22, 4602–4611 CrossRef CAS.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS.
- D. K. Kumar, A. Das and P. Dastidar, Cryst. Growth Des., 2006, 6, 1903–1909 CAS.
- C. Q. Li, W. G. Qiu, W. Shi, H. B. Song, G. M. Bai, H. He, J. Li and M. J. Zaworotko, CrystEngComm, 2012, 14, 1929–1932 RSC.
- L. Q. Ma, C. D. Wu, M. M. Wanderley and W. B. Lin, Angew. Chem., Int. Ed., 2010, 49, 8244–8248 CrossRef CAS.
- X. Roy, J. K. H. Hui, M. Rabnawaz, G. J. Liu and M. J. MacLachlan, Angew. Chem., Int. Ed., 2011, 50, 1597–1602 CrossRef CAS.
- S. T. Zheng, F. Zuo, T. Wu, B. Irfanoglu, C. S. Chou, R. A. Nieto, P. Y. Feng and X. H. Bu, Angew. Chem., Int. Ed., 2011, 50, 1849–1852 CrossRef CAS.
- Y. Zou, M. Park, S. Hong and M. S. Lah, Chem. Commun., 2008, 2340–2342 RSC.
- Y. H. Kiang, G. B. Gardner, S. Lee and Z. Xu, J. Am. Chem. Soc., 2000, 122, 6871–6883 CrossRef CAS.
- S. J. Garibay, Z. Q. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 7341–7349 CrossRef CAS.
- K. K. Tanabe, Z. Q. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef CAS.
- Z. Q. Wang and S. M. Cohen, J. Am. Chem. Soc., 2009, 131, 16675–16677 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and synthetic details, Fig. S1–S8, and Tables S1–S3. CCDC 906534–906538. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc40635k |
| This journal is © The Royal Society of Chemistry 2013 |