 Open Access Article
Open Access ArticleTotal synthesis of kottamide E†
Thomas B.
Parsons
,
Neil
Spencer
,
Chi W.
Tsang
and
Richard S.
Grainger
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: r.s.grainger@bham.ac.uk; Fax: +44 (0)121 4144403; Tel: +44 (0)121 4144465
First published on 1st February 2013
Abstract
The first synthesis of kottamide E, a marine natural product containing a 5,6-dibromoindole linked via a (Z)-enamide to an unusual 1,2-dithiolane-containing amino acid, is reported.
Kottamide E (1), isolated as a minor component from the New Zealand ascidian Pycnoclavella kottae in 2003 by Appleton and Copp, is the only natural product reported to date containing the unusual 4-amino-1,2-dithiolane-4-carboxylic acid residue.1 Related mono- and dibrominated Z-enamide-containing indole alkaloids, kottamides A–D, isolated from the same source, have been shown to exhibit anti-inflammatory, antitumour and anti-metabolic activities.2 Given the paucity of the natural product and absence of biological data, we have investigated a total synthesis of kottamide E, which we report herein.
Retrosynthetically we envisaged that union of a protected (Z)-enamide 2 with an appropriate oxalic acid derivative and the amide 3 offered a viable approach to kottamide A (Scheme 1). The synthetically challenging, thermodynamically less stable (Z)-enamide 2 was planned to arise from reaction of a vinyl isocyanate with 2-trimethylsilylethanol, the isocyanate in turn produced from Curtius rearrangement of an acyl azide derived from α,β-unsaturated ester 4. We were particularly attracted to this general approach3,4 to stereodefined enamides given its successful utilization in complex natural product synthesis,5,6 including indolic enamides.7 The (Z)-double bond geometry within 4 would be set through an appropriate Horner–Wadsworth–Emmons reaction of 5,6-dibromoindole 5, which we have previously shown can be readily accessed in 3 steps from commercially available methyl indole-3-carboxylate via regioselective dibromination.8–10
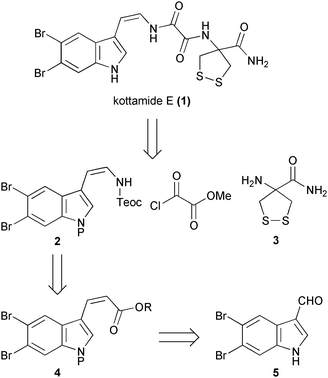 | ||
| Scheme 1 Retrosynthetic analysis of kottamide E. | ||
Although the Ando-modified Horner–Wadsworth–Emmons reaction11 of 5 proceeded with no selectivity, protection of the indole nitrogen gave rise to good to excellent levels of stereocontrol in favour of the expected (Z)-isomer 7, which could be readily separated from the corresponding (E)-isomer by column chromatography. Attempted ester hydrolysis of 7a–c was complicated by the faster removal of the indole protecting group and loss of integrity of the alkene double bond geometry. For example, in the case of the Boc-protected indole 7a, sodium hydroxide caused complete removal of the Boc group after 16 hours at room temperature, with no ester hydrolysis and a small amount of alkene isomerisation. Upon warming to 65 °C saponification was complete within 3 hours but the (Z)-alkene was completely converted to its (E)-isomer. Similarly the Teoc group of 7b and the tosyl group of 7c were removed within 30 and 5 minutes respectively at 0 °C. However, the SEM-protected indole 7d could be progressed via the carboxylic acid to the acyl azide 8 with complete retention of double bond geometry.
Heating 8 in the presence of 2-(trimethylsilyl)ethanol gave the unstable Teoc-protected enecarbamate 9. This was found to decompose rapidly, even when stored in the freezer under an atmosphere of argon. N-Acylation of 9via deprotonation with NaHMDS and quenching with methyl chlorooxoacetate gave methyl oxoacetate 10, which was not amenable to purification. Instead, treatment of the crude reaction mixture with TBAF afforded 11 in acceptable yield. Unfortunately the acylation step proved somewhat capricious and was not amenable to scale-up, which required recourse to multiple smaller scale reactions. The combined crude reaction mixtures were subjected to Teoc deprotection to afford 11, which is stable for several weeks at low temperature. Saponification of 11 gave acid 12 in quantitative yield.
The instability of 10 is consistent with the observations of Kitahara on Teoc-protected enamines appended to indoles, where N-acylated products could not be purified until the Teoc-group had been removed.7 We observed that 10 was highly sensitive to base and that acylation was somewhat reversible: if crude 10 was treated with triethylamine it rapidly reverted to Teoc-protected enamide 9. It is thought that this is the reason for the modest yield of the acylation-deprotection sequence: the basic nature of TBAF can cause competitive deacylation of intermediate 10 to return 9.‡
The novel amide 3 was prepared as its hydrochloride salt in 3 steps from the known12 protected amino acid 13 in 88% overall yield (Scheme 2). Coupling of 13 with carboxylic acid 3 proceeded smoothly using HBTU in DMF at 50 °C over 3 days to afford SEM-protected kottamide E 14 in 66% yield. Unfortunately, clean removal of the SEM group proved to be highly problematic. A wide variety of methods were tested without success.13 In each case reaction progress was followed by tlc, mass spectrometry and HPLC but we were unable to find conditions to deprotect kottamide E without concomitant fragmentation of the target. In most cases HPLC analysis of the reaction mixture indicated the presence of numerous by-products; mass spectrometric analysis suggested that various mono- and dibrominated fragmentation products were being formed, but sufficient material could not be isolated to confirm the identity of any such by-products.§
 | ||
| Scheme 2 Synthesis of SEM-protected kottamide E. Reagents and conditions: (i) for 6a: NaH, di-tert-butyl dicarbonate, THF, 95%; for 6b: NaH, 4-nitrophenyl 2-(trimethylsilyl)ethyl carbonate, THF, 95%; for 6c: NaH, TsCl, THF, 81%; for 6d: NaH, SEMCl, THF, 95%; (ii) EtO2CCH2P(O)(OPh)2, NaH, THF, −40 °C. For 7a: 81% + 13% (E)-isomer; for 7b: 81% + 13% (E)-isomer; for 7c: 80%; for 7d: 68% + 31% (E)-isomer 15; (iii) aq. NaOH, MeOH, THF, 65 °C then NaH, DPPA, THF, 91%; (iv) TMSCH2CH2OH, toluene, reflux, 77%; (v) NaHMDS, MeO2CC(O)Cl, THF, 0 °C; (vi) TBAF, THF, 0 °C, 66% over 2 steps; (vii) aq. NaOH, MeOH, THF, 100%; (viii) aq. NaOH, THF, MeOH, 99%; (ix) i-BuOC(O)Cl, Et3N, THF, 0 °C then NH3, MeOH, −20 °C, 89%; (x) SOCl2, MeOH, 50 °C, 100%; (xi) HBTU, DMF, Et3N, 50 °C, 66%. | ||
Given the difficulties encountered in removing the SEM protecting group from 14 to unveil kottamide E, an alternative approach was devised whereby the potentially sensitive natural product would be accessed directly upon formation of the amide bond. Considering the previously observed labile nature of the tosyl protecting group in 7c under saponification conditions, this new approach sought to exploit the expected removal of this group alongside methyl ester hydrolysis in the penultimate step (Scheme 3).
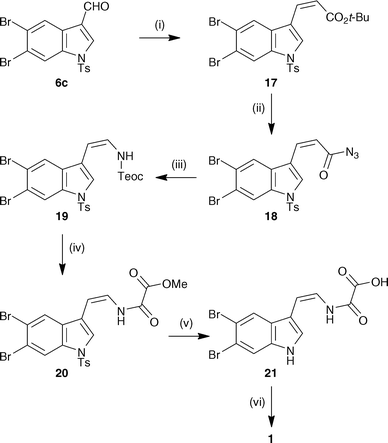 | ||
| Scheme 3 Total synthesis of kottamide E. Reagents and conditions: (i) t-BuO2CCH2P(O)(OPh)2, NaH, THF, −78 °C, 78% + 5% (E)-isomer; (ii) TFA, CH2Cl2 then NaH, DPPA, THF; (iii) TMSCH2CH2OH, toluene, reflux; (iv) NaHMDS, MeO2CC(O)Cl, THF then TBAF, THF, 32% over 5 steps; (v) aq. NaOH, MeOH, THF, 97%; (vi) 3·HCl, HBTU, DMF, Et3N, 38%. | ||
Towards this goal, tosyl-protected indole-3-carbaldehyde 6c underwent Ando-modified Horner–Wadsworth–Emmons reaction11 to afford (Z)-tert-butyl ester 17 as the major product, along with the separable (E)-isomer (Scheme 3). Hydrolysis of the ester under acidic conditions followed by reaction with DPPA gave acyl azide 18 as a 3.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of double bond isomers. Curtius rearrangement and trapping gave protected enamine 19 which was subjected to acylation and immediate deprotection to afford enamide 20. At this point the unwanted (E)-isomer could be removed by flash column chromatography. As expected, removal of the tosyl protecting group and concomitant saponification of the ester moiety of 20 gave carboxylic acid 21. This was subjected to the previously optimised conditions for amide bond formation with amine 3. Purification of the reaction mixture required semi-preparative and analytical HPLC to give kottamide E in modest yield and in approximately 85% purity, whose spectroscopic data were in agreement with those reported by Appleton and Cobb.1,14
:1 mixture of double bond isomers. Curtius rearrangement and trapping gave protected enamine 19 which was subjected to acylation and immediate deprotection to afford enamide 20. At this point the unwanted (E)-isomer could be removed by flash column chromatography. As expected, removal of the tosyl protecting group and concomitant saponification of the ester moiety of 20 gave carboxylic acid 21. This was subjected to the previously optimised conditions for amide bond formation with amine 3. Purification of the reaction mixture required semi-preparative and analytical HPLC to give kottamide E in modest yield and in approximately 85% purity, whose spectroscopic data were in agreement with those reported by Appleton and Cobb.1,14
In conclusion, the first total synthesis of kottamide E has been achieved in 8 steps starting from the known dibromoindole 5, itself prepared in three steps from commercially available methyl indole-3-carboxylate. The successful approach necessitated a change in protecting group strategy, but still suffered from a modest-yielding final step and recourse to purification by HPLC. This, along with the lack of tolerance towards a wide range of SEM deprotection conditions, suggests an inherent sensitivity of the natural product that means the choice of final steps in future syntheses of kottamide E should be of particular concern.
We thank EPSRC for funding. The NMR spectrometers used in this research were obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).
Notes and references
- D. R. Appleton and B. R. Copp, Tetrahedron Lett., 2003, 44, 8963 CrossRef CAS.
- D. R. Appleton, M. J. Page, G. Lambert, M. V. Berridge and B. R. Copp, J. Org. Chem., 2002, 67, 5402 CrossRef CAS.
- R. Brettle and A. J. Mosedale, J. Chem. Soc., Perkin Trans. 1, 1988, 2185 RSC.
- K. Kuramochi, H. Watanabe and T. Kitahara, Synlett, 2000, 397 CAS.
- A. B. Smith III and J. Zheng, Synlett, 2001, 1019 CrossRef; A. B. Smith III and J. Zheng, Tetrahedron, 2002, 58, 6455 CrossRef.
- J. T. Feutrill, M. J. Lilly and M. A. Rizzacasa, Org. Lett., 2002, 4, 525 CrossRef CAS; J. T. Feutrill, M. J. Lilly, J. M. White and M. A. Rizzacasa, Tetrahedron, 2008, 64, 4880 CrossRef.
- K. Kuramochi, Y. Osada and T. Kitahara, Tetrahedron, 2003, 59, 9447 CrossRef CAS.
- T. B. Parsons, C. Ghellamallah, L. Male, N. Spencer and R. S. Grainger, Org. Biomol. Chem., 2011, 9, 5021 CAS.
- For a similar approach to 3 see: E. M. Boyd and J. Sperry, Synlett, 2011, 826 CAS.
- For recent syntheses of 5,6-dibromoindole containing natural products, in addition to ref. 8 and 9, see: A. Mollica, A. Stefanucci, F. Feliciani, G. Lucente and F. Pinnen, Tetrahedron Lett., 2011, 52, 2583 CrossRef CAS; J. Sperry, Tetrahedron Lett., 2011, 52, 4042 CrossRef; J. Sperry, Tetrahedron Lett., 2011, 52, 4537 CrossRef.
- K. Ando, J. Org. Chem., 1997, 62, 1934 CrossRef CAS; K. Ando, J. Org. Chem., 1999, 64, 8406 CrossRef.
- E. Morera, M. Nalli, F. Pinnen, D. Rossi and G. Lucente, Bioorg. Med. Chem. Lett., 2000, 10, 1585 CrossRef CAS; E. Morera, G. Lucente, G. Ortar, M. Nalli, F. Mazza, E. Gavuzzo and S. Spisani, Bioorg. Med. Chem., 2002, 10, 147 CrossRef.
- See ESI† for a complete list of conditions with references.
- We have been unable to identify the impurities present in the aliphatic region of the NMR spectra of synthetic 1.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data for all new compounds. See DOI: 10.1039/c3cc39062d |
| ‡ In order to gain further insight into the stability issues, the minor (E)-enoate 15 from the Ando-modified Horner–Wadsworth–Emmons reaction was subjected to the same reaction sequence to give (E)-enamide 16 (see ESI† for details). Notably the acylation and Teoc-deprotection steps proceeded smoothly and could be conducted on larger scale than in the (Z)-series with no decrease in reaction yield. Presumably this is a reflection of the substantially lower steric crowding around the enamide in the (E)-series compared to the (Z)-series, where, particularly in the case of 10, a planar, conjugated arrangement is difficult to achieve. Similarly, reaction of (Z)-enamide 9 with TBAF was significantly faster than the corresponding (E)-isomer (1 hour at room temperature vs. ca. 50% consumption after 2 days), indicating orders of magnitude difference in reactivity between the two double bond isomers. |
| § The (E)-enamide 16 was also progressed to the same stage but the SEM group again could not be cleanly removed to afford the (E)-isomer of kottamide E. See ESI† for details. |

| This journal is © The Royal Society of Chemistry 2013 |