 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic properties and reactivity of a mononuclear oxomanganese(IV) complex†
Domenick F.
Leto
,
Rena
Ingram
,
Victor W.
Day
and
Timothy A.
Jackson
*
Department of Chemistry, The University of Kansas, 1251 Wescoe Hall Drive, Lawrence, KS, USA. E-mail: taj@ku.edu; Fax: +1-785-864-5396; Tel: +1-785-864-3968
First published on 26th April 2013
Abstract
A non-porphyrinic, mononuclear oxomanganese(IV) complex was generated at room temperature and characterized by spectroscopic methods. The MnIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O adduct is capable of activating C–H bonds by a H-atom transfer mechanism and is more reactive in this regard than most MnIVO species.
O adduct is capable of activating C–H bonds by a H-atom transfer mechanism and is more reactive in this regard than most MnIVO species.
High-valent oxomanganese adducts are suggested as active oxidants for synthetic and biological manganese catalysts, including those involved in textile and paper bleaching with H2O2 and oxygen evolution from water.1 Oxomanganese(V) adducts with S = 0 and 1 spin states have been reported, and these invariably feature strongly electron-donating, anionic ligands.2 Oxomanganese(IV) complexes with neutral, non-porphyrinic ligands are comparatively less common.3 Detailed studies of substrate oxidation exist for a limited number of complexes.3a–c,4 In addition, few MnIV
O complexes have been characterized by Mn K-edge X-ray absorption spectroscopy (XAS),3b,5 a technique featuring prominently in the study of Mn enzymes1b and biominerals.6 In this report, we describe the spectroscopic properties and oxidative reactivity of an oxomanganese(IV) complex supported by the neutral, pentadentate N4py ligand (N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine). This MnIVO compound is more reactive than the majority of MnIVO complexes for C–H bond oxidation.
The manganese(II) complex [MnII(N4py)]2+ (1) was generated as the triflate salt. The X-ray diffraction (XRD) structure exhibits a distorted octahedral MnII center with pentadentate N4py and monodentate triflate ligands (Fig. 1A). The Mn–ligand bond lengths are 2.1 to 2.3 Å. The Mn K-edge XAS spectrum of a frozen aqueous sample of 1(OTf)2 displays a pre-edge feature at 6540.6 eV and an edge at 6547.3 eV. The EXAFS data are best fit with 1 O at 2.09 Å, 5 N at 2.26 Å, and 3 C at 3.00 Å, in excellent agreement with the XRD structure (cf. Tables S4 and S6; ESI†).
![(A) XRD structure of [MnII(N4py)(OTf)]+ (1(OTf), left) and DFT (BP/TZVP) structure of [MnIV(O)(N4py)]2+ (2, right). Hydrogen atoms are omitted for clarity. (B) 298 K electronic absorption spectra of 1 (black solid trace) and 2 (green dashed trace); both 2 mM in CF3CH2OH. (C) Fourier transform of the Mn K-edge EXAFS data and EXAFS spectrum (inset), experimental data (⋯) and fits (–), for 2 in CF3CH2OH.](/image/article/2013/CC/c3cc00244f/c3cc00244f-f1.gif) | ||
| Fig. 1 (A) XRD structure of [MnII(N4py)(OTf)]+ (1(OTf), left) and DFT (BP/TZVP) structure of [MnIV(O)(N4py)]2+ (2, right). Hydrogen atoms are omitted for clarity. (B) 298 K electronic absorption spectra of 1 (black solid trace) and 2 (green dashed trace); both 2 mM in CF3CH2OH. (C) Fourier transform of the Mn K-edge EXAFS data and EXAFS spectrum (inset), experimental data (⋯) and fits (–), for 2 in CF3CH2OH. | ||
The addition of excess PhIO (2.5 equiv.) to 1 in CF3CH2OH at 298 K led to the formation of a green species (2), with a broad electronic absorption band at 950 nm and weaker features at 600 and 450 nm (Fig. 1B). At 298 K, the formation of 2 finished in ∼10 minutes, and 2 showed a half-life of 30 minutes. The absorption features of 2 are very similar to those of other non-porphyrinic MnIVO complexes in tetragonal, six-coordinate environments, which display broad near-infrared bands from ∼1040–825 nm and weaker features at higher energies.3a,b,7 The perpendicular mode X-band EPR spectrum of 2 is typical of a mononuclear, S = 3/2 MnIV ion (Fig. S4; ESI†).3a,b,d,8 Hyperfine coupling for the geff = 5.76 feature is ∼76 G, in good agreement with that observed for other MnIVO complexes.3d High-resolution electrospray-ionization mass spectral (ESI-MS) data of 2 reveal a major ion peak at m/z 219.0502, consistent with [MnIV(O)(N4py)]2+ (m/z calc. 219.0563). When 2 is spiked with 10 μL H218O (97% 18O-enriched), a new molecular ion peak is observed at m/z 220.0537, indicating incorporation of 18O from H218O ([MnIV(18O)(N4py)]2+m/z calc. 220.0585). These data together support the formulation of 2 as [MnIV(O)(N4py)]2+.
As we have been unable to grow crystals of 2, its molecular structure was investigated by Mn K-edge XAS. The edge energy of 2 (6550.8 eV) is blue-shifted over 3 eV relative to that of 1, as expected for the higher oxidation state of Mn (Fig. 2). The edge energy of 2 is within 1 eV of those reported for MnIVO complexes supported by salen and porphyrin ligands (6549.9–6551.2 eV; Table S3; ESI†).5 The pre-edge peak of 2 at 6541.9 eV is significantly more intense than that of 1 (Fig. 2, inset), consistent with a large deviation from centrosymmetry.
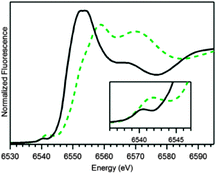 | ||
| Fig. 2 20 K Mn K-edge XAS near-edge region of 1(OTf)2 (black solid trace) and 2 (green dashed trace) in H2O and CF3CH2OH, respectively. | ||
The Fourier transform (R′ space) of the EXAFS data of 2 exhibits a prominent, sharp peak at R′ ≈ 1.5 Å with less prominent peaks at R′ ≈ 1.9, 2.2, and 2.8 Å (Fig. 1C). The first coordination sphere of 2 is fit well with two or three shells of N/O atoms at distances (r) of 1.69, 2.00, and 2.24 Å (Table S4; ESI†). The shell at 1.69 Å, which corresponds to the oxo ligand, is much shorter than the MnII–O (solvent H2O) distance of 2.09 Å observed for 1. The remaining first coordination sphere can be fit with either a single shell of 5 nitrogen scatterers at 1.99 Å or two shells of nitrogen scatterers at 2.00 Å (4 N atoms) and 2.24 Å (1 N atom), representing the nitrogen atoms of the pentadentate N4py ligand. The fit with two shells of N scatterers affords lower GOF and Debye–Waller values than the fit with just one shell of five N scatterers. Fits including outer-sphere features reveal two Mn⋯C shells at 2.82 and 2.97 Å (3 and 5 C atoms, respectively).
Metric parameters from the EXAFS data of 2 are in good agreement with a DFT-computed structure (Fig. 1A, right). This structure has a MnO bond of 1.673 Å (cf. the EXAFS distance of 1.69 Å). The equatorial nitrogens in the DFT-optimized structure have an average Mn–N distance of 2.024 Å, while the trans amine has a longer distance of 2.138 Å, consistent with the two shells of Mn–N scatterers at 2.00 and 2.24 Å.
The ability of 2 to activate C–H bonds was investigated using dihydroanthracene (DHA), diphenylmethane (DPM), ethylbenzene (EtBz), and toluene (Tol), which span a ∼10 kcal mol−1 range of C–H bond strengths. For each substrate, the addition of an excess to 2 at 298 K under an Ar atmosphere led to (i) a disappearance in the optical bands of 2, (ii) formation of a new species, 3, with bands at 460 and 630 nm, and (iii) the appearance of an isosbestic point at 714 nm (Fig. 3A). The decay of 2 and the formation of 3 occurred with the same rate, both following pseudo-first order behaviour to at least four half-lives (see ESI†). Second-order rate constants (k2′, corrected for the number of reactive C–H bonds) determined for all substrates revealed a linear relationship between log(k2′) and substrate bond dissociation enthalpies (BDEs), with a slope of 0.35 (Fig. 3B). Such behaviour is highly suggestive of a rate-limiting H-atom transfer.3a In support, reactions of 2 with deuterated-DHA (d4-DHA) reveal a kinetic isotope effect (KIE) of 11.2, which is larger than that observed for DHA oxidation by other MnIVO adducts (3.1–8).2c,3a,4a,9 Activation energies (E) and Arrhenius prefactors (A) determined from the reaction of 2 with DHA and d4-DHA from 35 to −5 °C in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CF3CH2OH:CH2Cl2 provide evidence for H-atom tunnelling (Table S10; ESI†). Specifically, the difference in activation energies for DHA and d4-DHA (ED − EH) is greater than the difference in zero-point energies of the C–H and C–D bonds (3.6 and 1.26 kcal mol−1, respectively); and the ratio of Arrhenius prefactors (AH/AD = 0.02) is much less than 0.7, and comparable to that of [FeIV(O)(N4py)]2+, where a H-atom tunnelling mechanism was also implicated.10 The reaction of 2 with DHA proceeded with a second-order rate constant (k2′ = 3.6 M−1 s−1) 2–3 orders of magnitude larger than those observed at similar temperatures for nearly all other non-porphyrinic MnIVO complexes (Table S8; ESI†).3a,4a,9,11 An Eyring analysis of DHA activation from 35 to −5 °C, reveal ΔH‡ and ΔS‡ of 9 ± 0.8 kcal mol−1 and −35 ± 3 cal mol−1 K, respectively (Table S9; ESI†). These parameters yield a ΔG‡ (at 25 °C) comparable to that of [MnIV(O)(H3buea)]− but 2 kcal mol−1 smaller than those observed for other MnIVO complexes,3a,4a,b consistent with the greater reactivity of 2.
:1 CF3CH2OH:CH2Cl2 provide evidence for H-atom tunnelling (Table S10; ESI†). Specifically, the difference in activation energies for DHA and d4-DHA (ED − EH) is greater than the difference in zero-point energies of the C–H and C–D bonds (3.6 and 1.26 kcal mol−1, respectively); and the ratio of Arrhenius prefactors (AH/AD = 0.02) is much less than 0.7, and comparable to that of [FeIV(O)(N4py)]2+, where a H-atom tunnelling mechanism was also implicated.10 The reaction of 2 with DHA proceeded with a second-order rate constant (k2′ = 3.6 M−1 s−1) 2–3 orders of magnitude larger than those observed at similar temperatures for nearly all other non-porphyrinic MnIVO complexes (Table S8; ESI†).3a,4a,9,11 An Eyring analysis of DHA activation from 35 to −5 °C, reveal ΔH‡ and ΔS‡ of 9 ± 0.8 kcal mol−1 and −35 ± 3 cal mol−1 K, respectively (Table S9; ESI†). These parameters yield a ΔG‡ (at 25 °C) comparable to that of [MnIV(O)(H3buea)]− but 2 kcal mol−1 smaller than those observed for other MnIVO complexes,3a,4a,b consistent with the greater reactivity of 2.
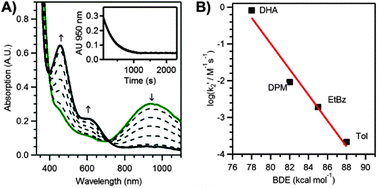 | ||
| Fig. 3 (A) Electronic absorption spectra of 2 upon addition of 200 equiv. EtBz in CF3CH2OH at 298 K. Inset: decay of the 950 nm absorption signal. (B) Corrected second-order rate constant (k2′) versus bond dissociation enthalpies of organic substrates. | ||
The reaction of 2 with DHA yielded 0.56(8) equiv. of anthracene per equiv. of 2. A final Mn oxidation state of 2.7(15) was determined by iodometric titration. This product distribution is consistent with the generation of anthracene by reaction of 1 equiv. DHA with 2 equiv. MnIVO rather than two successive H-atom transfers with a single MnIVO centre. Thus, 2 acts as a one-electron oxidant, which has been observed for other MnIVO compounds.2c,3a,b,4c Such reactivity is consistent with DFT studies by Shaik and Nam that have shown a second H-atom transfer between the nascent organic radical and MnIII–OH centre to be less favorable than diffusion of the organic radical from the MnIII–OH adduct.12 To the best of our knowledge, two-electron oxidation of DHA by a MnIVO has only been observed for [MnIV(O)2(Me2EBC)]+ and [MnIV(O)(OH2)(BQCN)]2+.3c,4c
While the iodometric product analysis gives an average Mn oxidation state following the reaction of 2 with DHA, the nature of the Mn-based products can be better defined on the basis of EPR, electronic absorption, and ESI-MS data. Perpendicular-mode EPR spectra of the product solution showed the strong MnIVO signals replaced by very weak signals. Broad features over a large field range and a sharp multiline signal at g ≈ 2 are respectively attributed to mononuclear MnII and binuclear species (Fig. S5; ESI†). Corresponding parallel-mode EPR spectra are silent. This does not preclude the presence of mononuclear MnIII species, as favourable MnIII zero-field splitting parameters and high-quality glasses are often required to observe the weak six-line signals of mononuclear MnIII centres in X-band experiments. The optical absorption features of product 3 are quite similar to those of [MnIII(OCH2CF3)(Bn-TPEN)]2+ (Bn-TPEN = N-benzyl-N,N′,N′-tris(2-pyridylmethyl)-1,2-diaminoethane), which was the dominant Mn product when [MnIV(O)(Bn-TPEN)]2+ was reacted with hydrocarbons.3b In addition, the dominant molecular ion peak in ESI-MS data of 3 is at m/z 620.1289, consistent with [MnIII(OCH2CF3)(N4py)](OCH2CF3)+ (m/z calc. 620.1293). Thus, we propose a mononuclear MnIII species as the dominant, but not sole, Mn-based product when 2 reacts with DHA.13
The chemical reactivity of 2 is similar to that of [MnIV(O)(Bn-TPEN)]2+.3b Both N4py and Bn-TPEN are N5 aminopyridyl ligands that also support highly reactive FeIVO complexes.10 For the MnIVO adducts, previous DFT computations predicted that 2 has a larger barrier for H-atom abstraction from cyclohexane than [MnIV(O)(Bn-TPEN)]2+.12 Although the addition of a large excess (400–600 equiv.) of cyclohexane increases the decay rate of 2, the reaction does not show pseudo-first order behaviour. In contrast, [MnIV(O)(Bn-TPEN)]2+ reacts with cyclohexane at 25 °C. Thus, we are unable to determine a k2 value to provide a quantitative comparison of reactivity using cyclohexane. However, a comparison can be made using EtBz, with which both compounds react at 25 °C in CF3CH2OH. In the reaction with EtBz, [MnIV(O)(Bn-TPEN)]2+ (1 mM) shows a rate constant five-fold larger than that of 2 (2 mM): k2′ = 2.7 × 10−2 and 5.7 × 10−3 M−1 s−1, respectively. Thus, while 2 is dramatically more reactive towards C–H bonds than most MnIVO adducts, it is less reactive than [MnIV(O)(Bn-TPEN)]2+. This trend holds for the corresponding FeIVO adducts; i.e., [FeIV(O)(Bn-TPEN)]2+ is more reactive towards C–H bonds.10
The origin of the high reactivity of 2 towards C–H bonds is currently unclear. Cyclic voltammetry studies of 2 show a MnIII/IV reduction potential (E1/2) ∼700 mV higher than those of other MnIVO complexes (ESI†).3a,4a,9 Thus, 2 is a significantly more effective one-electron oxidant. Notably the E1/2 of 2 is similar to those of other dicationic MnIV complexes,3a,9 suggesting that the increase in E1/2 is attributed to the +2 total charge of 2versus the +1 and −1 charges of other MnIVO adducts.3a,4a However, rates of H-atom transfer reactions, which are strongly correlated to thermodynamic driving force, depend not only on the reduction potential of the oxidant, but also on the basicity of the metal–hydroxo product.14 Both the MnIII/IV reduction potential and the pKa of the MnIII–OH complex, which for this system is unknown, are necessary for a thermodynamic analysis. While we cannot comment at present on the driving force for C–H bond activation by 2, we note that many other MnIVO adducts have sterically demanding supporting ligands that shield the oxo. In contrast, the oxo ligand in 2 is well-exposed to substrate (Fig. 1A). Reduced steric clash with substrate could contribute to the relatively high reactivity of 2. Future work is needed to determine the role ligand sterics, solvent effects, and thermodynamic driving force play in influencing the H-atom transfer reactivity of 2, and to explore further why MnIVO adducts such as 2 may eschew standard rebound or desaturation mechanisms for C–H activation.
This work was supported by the US NSF (CHE-1056470 to T.A.J.; CHE-1004897 for R.I.; CHE-0946883 and CHE-0079282 supported instrument purchases). XAS experiments were supported by the Center for Synchrotron Biosciences grant, P30-EB-009998, from the National Institute of Biomedical Imaging and Bioengineering (NIBIB). We thank Dr Erik Farquhar at NSLS for outstanding support of our XAS experiments.
Most recently Chen et al. reported the formation and characterization of 2 and described the effects of Sc3+ on oxo and H-atom transfer reactions.12b
Notes and references
- (a) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS; (b) A. J. Wu, J. E. Penner-Hahn and V. L. Pecoraro, Chem. Rev., 2004, 104, 903–938 CrossRef CAS; (c) R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2006, 45, 206–222 CrossRef CAS.
- (a) D. E. Lansky, A. A. Narducci Sarjeant and D. P. Goldberg, Angew. Chem., Int. Ed., 2006, 45, 8214–8217 CrossRef CAS; (b) K. A. Prokop, S. P. de Visser and D. P. Goldberg, Angew. Chem., Int. Ed., 2010, 49, 5091–5095 CrossRef CAS; (c) C. Arunkumar, Y.-M. Lee, J. Y. Lee, S. Fukuzumi and W. Nam, Chem.–Eur. J., 2009, 15, 11482–11489 CrossRef CAS; (d) J. T. Groves, J. Lee and S. S. Marla, J. Am. Chem. Soc., 1997, 119, 6269–6273 CrossRef CAS; (e) T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. Yano, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 1996–1999 CrossRef CAS; (f) T. J. Collins and S. W. Gordon-Wylie, J. Am. Chem. Soc., 1989, 111, 4511–4513 CrossRef CAS.
- (a) I. Garcia-Bosch, A. Company, C. W. Cady, S. Styring, W. R. Browne, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2011, 50, 5648–5653 CrossRef CAS; (b) X. Wu, M. S. Seo, K. M. Davis, Y.-M. Lee, J. Chen, K.-B. Cho, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2011, 133, 20088–20091 CrossRef CAS; (c) S. C. Sawant, X. Wu, J. Cho, K.-B. Cho, S. H. Kim, M. S. Seo, Y.-M. Lee, M. Kubo, T. Ogura, S. Shaik and W. Nam, Angew. Chem., Int. Ed., 2010, 49, 8190–8194 CrossRef CAS; (d) T. H. Parsell, R. K. Behan, M. T. Green, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2006, 128, 8728–8729 CrossRef CAS; (e) G. Yin, A. M. Danby, D. Kitko, J. D. Carter, W. M. Scheper and D. H. Busch, J. Am. Chem. Soc., 2007, 129, 1512–1513 CrossRef CAS.
- (a) T. H. Parsell, M.-Y. Yang and A. S. Borovik, J. Am. Chem. Soc., 2009, 131, 2762–2763 CrossRef CAS; (b) Y. Wang, S. Shi, H. Wang, D. Zhu and G. Yin, Chem. Commun., 2012, 48, 7832–7834 RSC; (c) S. Shi, Y. Wang, A. Xu, H. Wang, Z. Dajian, S. B. Roy, T. A. Jackson, D. H. Busch and G. Yin, Angew. Chem., Int. Ed., 2011, 50, 7321–7324 CrossRef CAS.
- (a) K. Ayougou, E. Bill, J. M. Charnock, C. D. Garner, D. Mandon, A. X. Trautwein, R. Weiss and H. Winkler, Angew. Chem., Int. Ed. Engl., 1995, 34, 343–346 CrossRef CAS; (b) T. Kurahashi, A. Kikuchi, T. Tosha, Y. Shiro, T. Kitagawa and H. Fujii, Inorg. Chem., 2008, 47, 1674–1686 CrossRef CAS; (c) T. Kurahashi, A. Kikuchi, Y. Shiro, M. Hada and H. Fujii, Inorg. Chem., 2010, 49, 6664–6672 CrossRef CAS.
- C. M. Hansel, C. A. Zeiner, C. M. Santelli and S. M. Webb, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12621–12625 CrossRef CAS.
- S. Chattopadhyay, R. A. Geiger, G. Yin, D. H. Busch and T. A. Jackson, Inorg. Chem., 2010, 49, 7530–7535 CrossRef CAS.
- T. M. Rajendiran, J. W. Kampf and V. L. Pecoraro, Inorg. Chim. Acta, 2002, 339, 497–502 CrossRef CAS.
- G. Yin, A. M. Danby, D. Kitko, J. D. Carter, W. M. Scheper and D. H. Busch, J. Am. Chem. Soc., 2008, 130, 16245–16253 CrossRef CAS.
- E. J. Klinker, S. Shaik, H. Hirao and L. Que, Angew. Chem., Int. Ed., 2009, 48, 1291–1295 CrossRef CAS.
- A potential complication for this rate comparison is the difference in solvents (Table S8; ESI†). 2 reacts with DHA at essentially the same rate in 1:1 CF3CH2OH:CH2Cl2 (ESI†), though the KIE drops to 8.7. In other solvents 2 has limited stability or does not form.
- (a) K.-B. Cho, S. Shaik and W. Nam, J. Phys. Chem. Lett., 2012, 3, 2851–2856 CrossRef CAS; (b) J. Chen, Y.-M. Lee, K. M. Davis, X. Wu, M. S. Seo, K.-B. Cho, H. Yoon, Y. J. Park, S. Fukuzumi, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2013 DOI:10.1021/ja312113p.
- In the absence of any substrate, the thermal decay of 2 yields a similar product, as judged by electronic absorption spectroscopy.
- J. M. Mayer, Acc. Chem. Res., 2010, 44, 36–46 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental and kinetic procedures. CCDC 885972. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc00244f |
| This journal is © The Royal Society of Chemistry 2013 |