Development of bioactive hydrogel capsules for the 3D expansion of pluripotent stem cells in bioreactors
Yoji
Tabata
ab,
Ikki
Horiguchi
a,
Matthias P.
Lutolf
b and
Yasuyuki
Sakai
*a
aLaboratory of Organs and Biosystems Engineering, Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba Meguro-ku, Tokyo, Japan. E-mail: sakaiyas@iis.u-tokyo.ac.jp; Fax: (+81)0354526353; Tel: (+81)0354526352
bLaboratory of Stem Cell Bioengineering, Institute of Bioengineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: matthias.lutolf@epfl.ch; Fax: (+41)0216939665; Tel: (+41)0216931876
First published on 1st October 2013
Abstract
Pluripotent stem cells hold great promise for many pharmaceutical and therapeutic applications. However, the lack of scalable methodologies to expand these cells to clinically relevant numbers is a major roadblock in realizing their full potential. To address this problem, we report here a scalable approach for the expansion of pluripotent stem cells within bioactive hydrogel capsules in stirred bioreactors. To achieve rapid crosslinking of cellular microenvironments with tuneable, cell-instructive functionality, we combined calcium-mediated alginate (CaAlg) complexation with crosslinking of poly(ethylene glycol) (PEG) macromers via a Michael-type addition. The resulting hybrid networks have been shown to have very good handling properties and can be readily decorated with biologically active signals such as integrin ligands or Cadherin-based motifs to influence the fate of mouse induced pluripotent stem (iPS) cells. Air-driven co-axial extrusion was used to reproducibly generate gel microcapsules in high-throughput. Furthermore, the gel capsules can be enveloped in a poly(L-lysine) shell to control swelling or molecular permeability independently of the gel composition. iPS cells entrapped within such capsules expanded with limited commitment to the endodermal lineage. Functionalization of gels with an appropriate density of Arg-Gly-Asp (RGD) ligands further increased the iPS cell expansion rate and reduced the spontaneous differentiation. Therefore, the combination of micro-scale instruction of cell fate by an engineered microenvironment and macro-scale cell manipulation in bioreactors opens up exciting opportunities for stem cell-based applications.
Introduction
iPS cells represent a powerful source of cells for clinical applications, because they are expected to help overcome several issues such as the immunological rejection and ethical issues that embryonic stem cells are confronted with.1 However, therapeutic applications demand very large amounts of such cells with strictly regulated quality.2,3 Since the state-of-the-art reprogramming methods are still inefficient,1,4 post-reprogramming in vitro expansion at a clinically and/or industrially relevant scale is crucial. Bioreactor-based expansion systems could be a powerful means of achieving this goal. Recently, several bioreactor culture systems have been used for stem cell expansion, including those based on free suspension, microcarrier and microcapsule culture.5–13 However, without adequate control of stem cell fate in such up-scalable systems, the resulting quality of stem cells may not be sufficient.In vivo, stem cell fate is strongly influenced by various signals from their microenvironment, also termed niche.14 For example, stem cells are often anchored to their niche via adhesion molecules provided by various niche cells and the ECM.15 However, current stirred bioreactor culture systems lack the possibility of exposing stem cells to key niche signals which often results in limited control of stem cell fate. To tackle this problem, we propose to integrate biomimetic mechanisms of stem cell fate control into macro-scale cell culture settings.
During the last decade, a wide variety of synthetic hydrogels have been engineered as artificial niches. In contrast to naturally derived hydrogels, synthetic systems overcome the risk of pathogenic contamination and show a reduced batch-to-batch variability.16 Importantly, synthetic hydrogels such as those based on branched PEG are highly tuneable in terms of their physicochemical and biochemical properties, and as a consequence are increasingly used for in vitro stem cell culture.17 Such gels can be formed under cytocompatible conditions by choosing self-selective cross-linking chemistries.18–20 Moreover, cell-adhesive or proteolytically sensitive domains as well as desired signalling factors can be readily incorporated to rationally control the behaviour of gel-entrapped cells.19–22 On a practical level, a wide variety of PEG-based gel building blocks, such as maleimide-functionalized 4arm-PEG (4MA-PEG),23 are commercially available at reasonable cost which makes these hydrogels particularly attractive for stem cell culture.
Despite the considerable interest in PEG-based hydrogels for stem cell culture, these materials are usually utilized in formats that are not well suited for industrial-scale processes. One notable example is the recent manufacture, in a co-axial extrusion system, of hybrid hydrogel microspheres composed of PEG and calcium alginate (CaAlg).24 These materials are highly promising for stem cell manipulation in bioreactor culture, but, to the best of our knowledge, they have not yet been used for the encapsulation of mammalian cells.
Here we adapted and further developed this method for the microencapsulation and expansion culture of iPS cells in large-scale bioreactors. Using mouse iPS cells as the model system, we specifically demonstrate how the microenvironment can be tailored to achieve optimal expansion.
Results and discussion
Hydrogel capsule preparation and characterization
We used a three-step co-axial extrusion encapsulation system: (i) preparation of gel precursors containing desired amounts of macromers, cells and biologically active signals, (ii) extrusion and subsequent gel formation in baths, and (iii) PLL shell formation to control swelling and molecular permeability independently of the gel composition.A co-axial extrusion mount (Fig. 1a,b) allowed stable generation of 2% (w/v) CaAlg capsules with diameters ranging from approximately 0.7 to 1.5 mm, depending on the N2 flow and on extrusion speed. 1.1 mm diameter droplets were utilized in the present study. Good uniformity in the shape and size was obtained with relative standard deviations below 3%. About 1000 capsules were generated in ca. 5 minutes during this first step. The instantaneous gelation of the CaAlg trapped 20 kDa 4MA-PEG macromers (Fig. 1c) in the alginate network. Placing these gels in a solution containing di-thiolated linear PEG (2S-PEG; mol. weight 3.4 kDa) (Fig. 1d) allowed PEG crosslinking via a Michael addition reaction (Fig. 1e), resulting in hybrid capsules composed of 2% (w/v) CaAlg/2% (w/v) PEG.
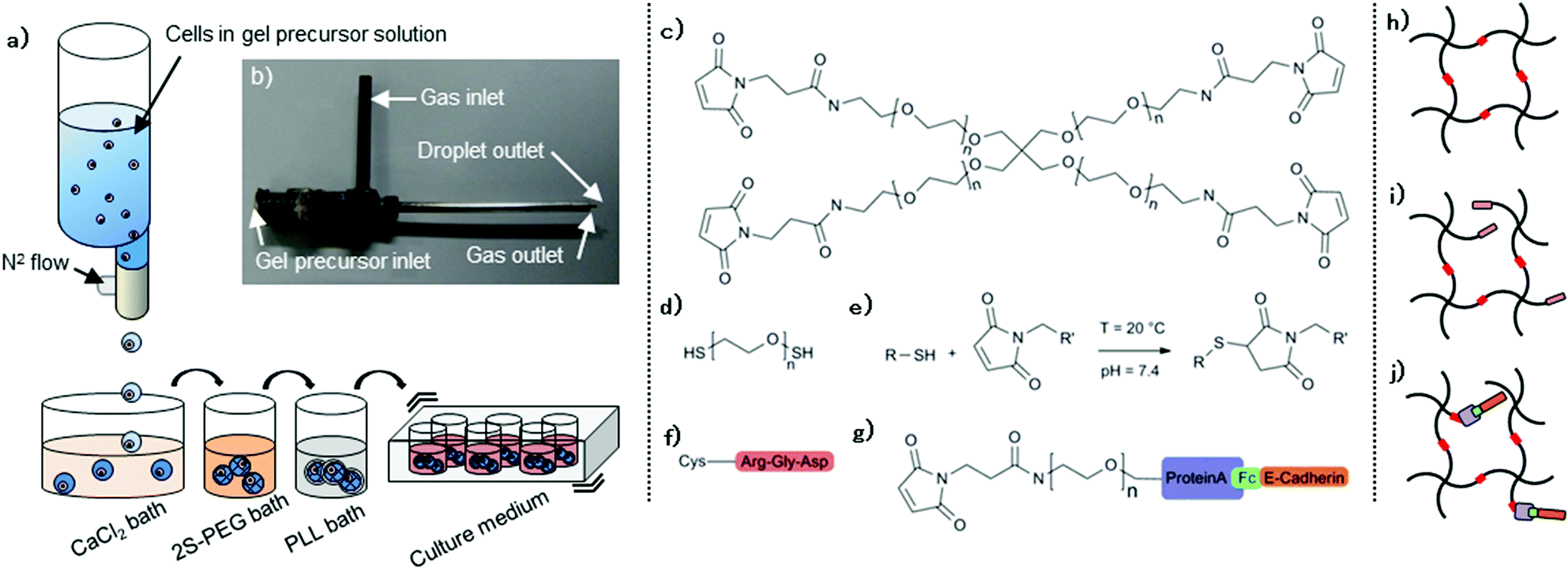 | ||
| Fig. 1 (a) Overall scheme of the cell encapsulation process. Extrusion of the gel precursor solution containing cells, 4MA-PEG and other bioactive ligands into the first CaCl2 bath generates CaAlg capsules. Capsules are then transferred into the second 2S-PEG bath triggering the PEG cross-linking. Capsules are then placed in the third bath to coat capsules with PLL and finally suspended in continuously stirred culture medium for long term culture. (b) Silver soldered custom-made extrusion nozzle. (c) 4MA-PEG. (d) 2S-PEG. (e) Formation of stable and irreversible carbon–sulfur bonds via a Michael addition reaction of maleimide and thiol. (f) Synthetic peptide containing RGD and cysteine. (g) Complex of Fc-tagged E-cadherin and heterobifunctional PEG linker carrying protein A and maleimide. (h) Cross-linked PEG macromers. (i) RGD-functionalized gel is formed by mixing the premixed 4MA-PEG/RGD-peptide with 2SH-PEG. (j) An E-cadherin-functionalized gel is formed by mixing 4MA-PEG, 2S-PEG and the pre-formed complex of E-cadherin/heterobifunctional PEG linker. | ||
Hydrogels are known to have variable volume depending on both their molecular composition and the medium in which they are incubated.25 CaAlg and CaAlg/PEG hybrid capsules suspended in culture medium exhibited limited swelling behaviour, not exceeding volumetric swelling degrees of 50% (Fig. 2a). However, after CaAlg removal by ethylenediaminetetraacetic acid (EDTA) their volume dramatically increased, surpassing 200% swelling. Pure 2% (w/v) PEG, obtained by removing the CaAlg gel phase, was robust enough to keep a round shape even after the CaAlg removal. Conversely, covering capsules with a PLL shell (step 3) significantly restrained swelling, even for the loosest PEG gels. Excess swelling is not problematic because of gel instability but may also be associated with harmful effects on encapsulated cells, exposing them to excessive stress which could reduce viability. Therefore, our coating strategy opens up new possibilities for highly swelling, soft materials in bioreactor cultures that cannot be used otherwise. Diffusion tests on FITC-labelled dextran of controlled molecular weight revealed the expected inverse relationship of molecular weight and diffusivity (Fig. 2b). Stable diffusion was observed after six hours of stirred incubation, in which molecules smaller than 4 kDa could almost freely penetrate into raw CaAlg/PEG capsules, whereas the relative concentration of molecules larger than 20 kDa remained below 70%. PLL coating substantially affected the diffusion of larger macromolecules (>70 kDa). The selective diffusion characteristics allow, for example, capsule uptake or release of nutrients, gases or cellular waste that have small molecular weight. Conversely, it may enable the confinement of larger proteins released by cells that may enhance auto- or paracrine processes. Changing the thickness of the PLL layer had however a minimal influence on the diffusivity of dextran. As alternatives, the PLL molecular weight and concentration could be considered for further controlling the molecular cut-off of the shell.26
 | ||
| Fig. 2 (a) Volumetric swelling degree of the different types of capsules incubated for two days in culture medium. (b) Diffusion of FITC-dextran standards in raw and PLL-coated (20 and 40 μm) CaAlg/PEG hybrid capsules. Fluorescence intensity of the capsules is relative to the bulk signal. (c) Fluorescence intensity of the raw and PLL-coated CaAlg/PEG hybrid capsules, bioconjugated with FITC-hIgG. All the results are presented as mean ± standard deviation (SD) of 2 independent experiments where 10–20 capsules of each group were analyzed (N = 20–40). | ||
Next, we sought to modify the biochemical capsule properties by functionalizing PEG networks with bioactive moieties (Fig. 1f–j). Thiolated biomolecules can be directly tethered to PEG macromers before crosslinking in order to achieve a nearly quantitative incorporation efficiency.18,23 As a first bioconjugation strategy, we therefore used as a model system cysteine-containing Arg-Gly-Asp (RGD) peptide to generate integrin-binding networks (Fig. 1f,i). As a second strategy based on affinity binding, Fc-tagged molecules could be immobilized as well with an intermediate PEG linker bearing both maleimide and ProteinA (Fig. 1g).27 Fc-E-cadherin/PEG linker construct (Fig. 1g) was formed and subsequently added to the gel precursor solution to form E-cadherin-binding networks (Fig. 1j). To validate this affinity-based bioconjugation strategy, capsules were generated from a series of precursor solutions containing different amounts of ProteinA linker and an excess amount of FITC-hIgG. The fluorescence intensity gradually increased as a function of ProteinA content (Fig. 2c), demonstrating that the bioconjugation is well controllable. The signal remains stable for at least one week (data not shown). Of note, we observed that PLL-coated capsules usually showed higher fluorescence intensity, most likely because of their lower swelling-related dilution of tethered proteins. Furthermore, introducing an alternative high-affinity coupling such as Biotin/NeutAvidin offers the possibility of tethering multiple biomolecules in an orthogonal manner.28
Stirred suspension culture of encapsulated iPS cells
Mouse iPS cells were successfully entrapped within capsules without any visible loss. Each capsule contained approximately 200–350 cells that were homogeneously dispersed within the capsule. Trypan blue staining confirmed a post-encapsulation viability of about 78.2% ± 8.5. Capsules were suspended in LIF-containing iPS cell maintenance medium, and stirred at 120 rpm. No cell leakage was observed with PLL-coated capsules, whereas cells tended to escape from uncoated capsules within a few days (data not shown). Consequently, all subsequent experiments were performed with PLL-coated capsules. Cells were able to proliferate in both inert and bioconjugated CaAlg/PEG capsules. Visually identifiable colonies appeared at day 4 and relatively large ones with diameter over 100 μm were observed around day 8 (Fig. 3a). Cells encapsulated in CaAlg or CaAlg/PEG capsules showed similar proliferation characteristics and colony morphologies, suggesting that the presence of PEG networks did not substantially affect cell behavior. The moderate initial viability might be due to the potential pressure during the extrusion and subsequent cross-linking steps. | ||
| Fig. 3 (a) Phase contrast microscopy of mouse iPS cells encapsulated in different formulations of capsules, at days 4 and 8. Fluorescence microscopy indicates the Nanog expression in green at day 8. Scale bar = 200 μm. (b) Fold increase in cell number during eight-day culture under each condition. Results are presented as mean ± standard deviation (SD) of three independent experiments performed in duplicate (N = 6). (c) Evolution of glucose consumption. Standard deviation was mostly maintained below 10% (N = 6). Statistical significance of the compared pairs is presented as *p < 0.5, **p < 0.01 and ***p < 0.001. | ||
To test whether capsule bioconjugation could promote iPS cell expansion and maintenance of pluripotency, capsule formulations containing RGD and E-cadherin were used. Capsules containing 500 μM RGD increased iPS cell expansion by approximately 80% compared to inert gels; lower concentrations (e.g. 100 μM) did not show any significant effect. Furthermore, incorporation of 10 μg mL−1 E-cadherin did not induce any significant change on expansion (Fig. 3b). In order to assess proliferation in a more refined manner, glucose consumption was quantified every two days during the entire culture period. For each group, total glucose consumption gradually increased over time until almost complete depletion of the medium glucose at day 8 (Fig. 3k). Lactose production was monitored in the same way in order to evaluate the lactose to glucose ratio. Lactose to glucose ratios remained between 0.8 and 1.2 (data not shown) over the culture period. These results suggest that both aerobic and anaerobic metabolism were equally present, since the ratio is close to zero when aerobic metabolism is favoured and close to two when anaerobic metabolism dominates.29
To probe iPS cell fate changes in situ, a reporter cell line was used in which green fluorescent protein (GFP) is expressed under the control of the Nanog promoter.30 In each capsule type, the majority of colonies exhibited stable and homogeneous Nanog expression during the entire culture period (Fig. 3a). Additionally, quantitative expression of pluripotency (Nanog, Oct4, Sox2 and Rex1)31,32 as well as primitive endoderm markers (GATA4, HNF4 and AFP)33–35 was evaluated using quantitative RT-PCR (Fig. 4). Encapsulation culture generally enhanced the expression of pluripotency markers compared to conventional static culture. In particular, Rex1, known to be the most stringent pluripotency marker, was significantly up-regulated under all capsule conditions, especially in those modified with bioactive cues. Primitive endodermic GATA4 significantly decreased when cells were exposed to both 100 μM and 500 μM, while HNF4 was uniquely down-regulated in 100 μM capsule. 10 μg mL−1 E-cadherin however did not alter the expression of the latter genes, but strongly supported the expression of AFP, with a ca. 100-fold increase. These data show the possibility of exploiting bioconjugation concepts in capsule engineering in order to control pluripotency cell fate.
 | ||
| Fig. 4 (a) Change in gene expression profile of miPS cells cultured for 8 days under each condition, presented as mean normalized relative quantity (NRQ) ± standard error of the mean (SEM) of three independent experiments performed in duplicate (N = 6). Statistical significance of the compared pairs are presented as *p < 0.5, **p < 0.01 and ***p < 0.001. iPS cells were recovered from CaAlg/PEG capsules at day 8 and reseeded under (b) iPS-non-maintaining conditions and (c) feeder/LIF iPS-maintaining conditions, and cultured for three days. Scale bar: 50 μm. | ||
To further assess their potency, single cells recovered from CaAlg/PEG hybrid capsules after eight days of stirred culture were sub-cultured on either feeder layers in the presence of LIF or tissue-culture plates without LIF for three additional days (Fig. 4b,c). These experiments showed that cells had kept the ability of expanding (as evidenced by strong expression of Nanog-GFP) in conventional 2D pluripotent stem cell culture settings. Some populations nevertheless acquired an epiblast-like morphology (Fig. 4b).36 In contrast, cells cultured under non-iPS-maintaining conditions quickly lost Nanog expression and showed neuronal37 or hepatocyte-like38 morphologies (Fig. 4b). Therefore, iPS cells cultured in hydrogel capsules maintain their potency and the recovered cells can be utilised for subsequent applications.
Encapsulation cultures offer interesting advantages over other methods. For example, colonies do not clump together which is often an issue in both suspension and microcarrier culture.13,40 Secondly, cells are protected from excessive hydrodynamic stresses that may affect both cell viability40 and potency.41 Finally and perhaps most importantly, cells are embedded in 3D microenvironments that can be rationally engineered to instruct stem cell fate.17 Indeed, our study achieved similar cell expansion levels compared to other microencapsulation cultures,42 but offered the possibility of enhancing the maintenance of pluripotency by providing a favourable microenvironment. For example, incorporating 500 μM RGD allowed increasing the expansion yield by about 80% compared to inert capsules, while the pluripotency was better maintained with significantly higher expression of pluripotency markers such as Rex1 and lower expression of primitive endoderm markers such as GATA4. The RGD domain is present in many natural ECM components and culture substrates including laminin, vitronectin or matrigel that are commonly employed to maintain pluripotent stem cells.1,43,44 Indeed, the proliferative-enhancing effect of RGD in 3D encapsulation has been reported in other studies.45 Conversely, E-cadherin did not support cell proliferation despite its important expression in early embryonic development.46 Several research groups reported that preventing pluripotent stem cells from E-cadherin-mediated aggregation by exposing them to competitive E-cadherin ligands47 or by genetic knock-down,48 supports self-renewal and allows cells to grow at a single cell level, increasing access to nutrients, gases or other signals as well as to enough space for efficient proliferation. We believe that in a physically confined 3D context, E-cadherin might not exert its positive effect on pluripotency.
Mouse pluripotent stem cells are routinely cultured with LIF supplement. LIF/STAT signalling inhibits differentiation proceeding from the primitive to visceral endoderm, which is known to be the primary commitment of suspended pluripotent spheroids.49,50 In the present encapsulation culture, the primitive endoderm makers GATA4 and vHNF4 were notably suppressed, suggesting that spontaneous endodermic differentiation might be controlled at an earlier stage than LIF signalling intervenes. Indeed, several studies suggested that stem cell fate decision might be influenced by the visco-elasticity and cell-adhesive characteristics of a substrate.51,52 These factors could have played a role in our 3D culture system. Moreover, the enclosed environment obtained by the PLL-coating might play a role in confining and concentrating certain para- and/or autocrine factors. For example, pluripotent stem cells cultured in microchambers favouring the accumulation of factors produced by cells themselves such as BMP4 showed an increased expression of pluripotency markers.53 The dimension and shape of multicellular spheroids are also known to play a role in stem cell fate decision54 by prompting heterogenization of the environment within the spheroid, in terms of cell–cell or cell–matrix interaction molecules55 as well as oxygen, nutrient or protein distribution.56 It is noteworthy that the colonies we obtain in 3D gels have a flattened, oval or disk-like shape. This unusual colony shape may display different profiles of factors mentioned above, compared to spherical spheroids such as embryoid bodies (EB) that are readily committed.57,58 Colony development and commitment are highly dependent on the stiffness of the substrate. Our hybrid gels supporting iPS cell expansion have an estimated initial Young's modulus, largely determined by the CaAlg gel phase, that surpasses 10 kPa. The stiffness of the gels can nevertheless be readily modulated by the alginate content, allowing further optimization of the substrate.
Conclusions
To achieve a scalable culture of pluripotent stem cells, here we investigated 3D culture within engineered CaAlg/PEG hydrogel capsules. Our approach combines the advantages of the CaAlg gel system, including its fast and upscalable generation, with those of a synthetic PEG-based gel system which is readily amenable to functionalization with cell-instructive signals. Furthermore the use of a CaAlg phase offers the possibility of coating capsules with PLL in order to prevent cells from escaping the carrier and to minimize swelling. Mouse iPS cells encapsulated in such bioengineered capsules expanded with an efficiency comparable to conventional microencapsulation methods. Importantly, cells cultured in capsules modified with an appropriate density of integrin-binding motifs expanded more and with a higher expression of pluripotency genes. Our concept opens up exciting possibilities for the large-scale production of stem cells for clinical applications or drug screening.Experimental
Maintenance of mouse iPS cells
The mouse iPS cell line iPS-MEF-Ng-20D-17 was generously gifted by Riken Cell Bank (Tsukuba, Japan). miPS cells were maintained on monolayers of mitotically inactivated SNL 76/7 cells.59 Cells were cultured in iPS-maintaining medium comprising high glucose DMEM supplemented with 20% (v/v) ES-FBS, 1% (v/v) antibiotic–antimycotic, 1% (v/v) non-essential amino acid and 0.1 mM 2-mercaptoethanol, all obtained from Gibco. The medium was supplemented with 100 U ml−1 of recombinant mouse leukemia inhibitory factor (LIF) (Millipore). miPS cells were sub-cultured every three days with 0.05% trypsin–EDTA (Gibco) treatment followed by depletion of feeder population through incubation onto a gelatin-coated dish (Iwaki). iPS cells of passages 11–25 were used for all the experiments.Cell encapsulation in CaAlg/PEG hydrogel capsules
The gel precursor solution was prepared by dissolving 2% (w/v) 20 kDa 4MA-PEG (NOF) and 2% low viscosity sodium alginate (NaAlg) (Wako) into 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Wako)-buffered 0.45% saline solution. pH was adjusted to 7.4. Hydrogel could be biologically functionalized by introducing the desired amount of biological molecules of interest to the gel precursor solution, at this moment. Here, the integrin-binding peptide comprising an RGD motif Ac-GRCGRGDSPG-NH2 was synthesized (GLBiochem) while the recombinant mouse Fc-E-cadherin, involved in cell–cell interaction, was purchased (R&D Systems). RGD peptide held cysteine residue, so that it directly reacts with PEG macromeres through a Michael-type addition reaction. An appropriate amount of RGD-peptide was added to a 4MA-PEG solution to let them react for 10 minutes at 37 °C. Such pre-mixing allows a nearly 100% peptide incorporation.23 On the other hand, E-cadherin was incorporated into the network with an intermediate hetero-functional PEG linker carrying, on one end, recombinant proteinA for capturing Fc-tagged molecules and maleimide group on the other end, allowing its integration into the polymeric network through the same cross-linking chemistry. The intermediate PEG linker was prepared as described elsewhere.27 10% molar excess of such a linker was pre-mixed with Fc-E-cadherin, and subsequently introduced to the 4MA-PEG precursor solution.Undifferentiated miPS cells were isolated from SNL feeder cells and re-suspended in a gel precursor solution with a cell density adjusted to 5 × 105 cells mL−1. The cell suspension was gently homogenized with a pipette and loaded into an air-driven syringe pump droplet generator, in which both the extrusion and air flow rates were adjustable. A 0.2 μm filter was integrated into the N2 flow path. We used here 100 μL min−1 and 2 L min−1 respectively. Diameters of the outer (gas) and inner (gel solution) outlets were 0.260 and 1.194 mm respectively. The mixture was dropwise dispensed into the first gelation bath containing 150 mM CaCl2 (Wako) and 0.01% polyoxyethylene (20) sorbitan monolaurate (Tween20) (Wako) at room temperature. All solutions were sterile filtered. Instruments, including the dispensing nozzle and tubing, were autoclaved before each use. Extrusion was performed under the laminar flow cabinet. Spherical CaAlg capsules were instantaneously formed upon contact of the droplet with the bath solution via ionotropic gelation. Capsules were then immediately transferred to the second bath solution containing 3.4 kDa linear PEG dithiol (2SH-PEG) dissolved in DMEM, where they were gently stirred for 30 minutes at 37 °C. This solution also contained 30 mM CaCl2 for completing the CaAlg gelation. PEG macromers were covalently cross-linked to each other by forming stable and irreversible carbon–sulfur bonds, resulting in CaAlg/PEG hybrid capsules. The optimal cross-linking for such a system is achieved when the stoichiometric ratio of the reacting groups is close to one.18 In the actual case, because the mixing of the two PEG components was gradually performed by diffusion and thanks to the high reaction velocity, excessive amount, but slightly concentrated 2S-PEG solution was employed. Especially, the maleimide to thiol molar ratio was kept at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 while the total amount ratio was at 1:2.5. Capsules were washed twice with PBS and subsequently incubated in a 0.05% (w/v) 15–30 kDa PLL hydrobromide (Sigma) solution for 10 minutes and then in a 0.075% (w/v) NaAlg solution for 5 minutes for covering up the capsules with a PLL shell. Capsules were optionally treated either with 10 mM EDTA (Sigma) for 10 minutes or with 100 U mL−1 alginate lyase (Sigma) for a few hours to dissolve CaAlg gel resulting in pure PEG capsules. Capsules were finally washed twice with DMEM and transferred into iPS-maintaining medium supplied with 1000 U mL−1 LIF. Encapsulated cells were cultured for eight days in an ultra-low attachment 6-well plate (Corning) placed on an orbital shaker stirring at 120 rpm.
:0.5 while the total amount ratio was at 1:2.5. Capsules were washed twice with PBS and subsequently incubated in a 0.05% (w/v) 15–30 kDa PLL hydrobromide (Sigma) solution for 10 minutes and then in a 0.075% (w/v) NaAlg solution for 5 minutes for covering up the capsules with a PLL shell. Capsules were optionally treated either with 10 mM EDTA (Sigma) for 10 minutes or with 100 U mL−1 alginate lyase (Sigma) for a few hours to dissolve CaAlg gel resulting in pure PEG capsules. Capsules were finally washed twice with DMEM and transferred into iPS-maintaining medium supplied with 1000 U mL−1 LIF. Encapsulated cells were cultured for eight days in an ultra-low attachment 6-well plate (Corning) placed on an orbital shaker stirring at 120 rpm.
Capsule characterization
Swelling behaviour was studied by monitoring the dimensional evolution of the capsules over the incubation time. Raw and PLL-coated CaAlg, CaAlg/PEG and PEG capsules were synthesized as previously described, and were suspended in PBS or iPS-maintaining culture medium for 48 hours at 37 °C; meanwhile they were able to swell freely. The diameter of 20 randomly chosen capsules was measured before and after swelling using image processing software ImageJ. Because of the correct sphericity of the capsules, the swelling degree Sd could be defined as Sd = 100 [(Dc/Ds)3 − 1]24,60 with Dc the capsule diameter after cross-linking and Ds after swelling. Higher Sd corresponds to a highly-swelled, looser polymeric network.Capsule permeability to dissolved molecules was analysed by studying the diffusion of fluorescent molecules. Raw and PLL-coated (20 or 40 μm-thick) capsules were generated and stored in PBS for two days at 37 °C until swelling was completed. A thicker PLL layer was obtained by incubating capsules for a longer time in the PLL solution. Capsules of each type were then incubated in 120 rpm stirred culture medium containing 0.5 mL mg−1 of 4, 20, 70 or 150 kDa FITC-labelled dextran standards (Sigma). The fluorescence within the capsules was measured and normalized to the bulk signal, assumed constant, to estimate the capsule permeability. The fluorescence profile was almost stabilized after 6 hours incubation. 10 randomly chosen capsules were analysed using confocal microscopy.
To determine the incorporation efficiency of Fc-tagged protein via the intermediate heterobifunctional PEG linker, hIgG (Invitrogen) was labelled with FITC using a protein labelling kit (Pierce) following the manufacturers’ instructions. FITC-hIgG and 10% molar excess PEG linker were premixed and added to the gel precursor solution at different final concentrations, 0, 25, 50, 100 and 200 μg mL−1. Capsules were then generated and extensively washed with PBS to remove unreacted molecules and incubated in culture medium for 24 hours. The fluorescence profile of 10 randomly chosen capsules of each group was evaluated using confocal microscopy.
Cell proliferation
The proliferation ability of the encapsulated cells was investigated by quantifying the total DNA amount at the end of the culture period. Capsules were treated with 10 mM EDTA for 10 minutes to dissolve CaAlg gel and were then mechanically ruptured by squeezing them with a 25G syringe needle. The mixture was then ultrasonic-homogenized (UH-300) (SMT) three times for 30 seconds to ensure that capsules and the cellular membrane are completely demolished. Cellular DNA was stained with 100 ng mL−1 4′,6-diamino-2-phenylindole (DAPI) (Dojin) and the fluorescence was measured using a spectrophotometer (RF 5300-PC) (Shimadzu). Fold increase in the cell number was derived by comparing the total amount of the initial and final genetic materials. Additionally, the evolution of glucose metabolism of the cells was investigated in a more progressive manner. To this end, the culture medium was withdrawn every 48 hours and the total amount of glucose and lactic acid was measured using an automated analyser (7100MBS) (LifeSciences).RT-PCR
For preparing the RNA sample, capsules were collected and disrupted as for the previous assay. The mixture was then treated with TRIzol reagent (Invitrogen) according to the manufacturers’ instructions and stored at −80 °C for at least three days. Aqueous phase containing total RNA was isolated, and treated with isopropanol and ethanol to collect and purify RNA molecules. RNA concentration and quality (A260/A280) was analysed using a spectrophotometer (GeneQuant Pro) (Amersham Biosciences). cDNA was synthesized from 2 μg of total RNA using a Super Script III First-Strand reverse transcription system with random hexamer primers (Invitrogen) following the manufacturers’ instructions. cDNA samples were stored at −30 °C until usage. Polymerase chain reaction (PCR) was performed on fast real-time system with TaqMan gene expression assays (StepOnePlus) (Applied Biosystems). PCR was executed with the following conditions: 45 cycles of (i) DNA denaturation and polymerase activation at 85 °C for 5 seconds, (ii) primer annealing at 50 °C for 5 seconds and (iii) extension at 72 °C for 20 seconds. For assessing whether cells had properly sustained their pluripotent properties, the expressions of some major pluripotency markers as well as primitive endoderm and primitive ectoderm markers were evaluated as mentioned in the previous section. miPS cells grown on the conventional SNL feeder layer for 3 days were utilized as the comparative reference group. All the gene expressions were normalized to GADPH and given as relative quantity compared to the reference iPS cells cultured on the feeder layer, according to the ΔΔCT method.61 ANOVA based on the Tukey–Kramer method was performed with the log2-transform of normalized relative quantity (NRQ).62Acknowledgements
The authors thank S. Cosson and A. Negro for help in preparing the Protein APEG-Maleimide linker, as well as the RGD peptide solution. The authors are also grateful to Prof. K. Komori for valuable advice on the experimental setup. This research was partly supported by the S-Innovation project of the Japan Science and Technology Agency (JST).Notes and references
- K. Takahashi and S. Yamanaka, Cell, 2006, 126, 663–676 CrossRef CAS PubMed.
- M. A. Laflamme and C. E. Murry, Nat. Biotechnol., 2005, 23, 845–856 CrossRef CAS PubMed.
- L. Ahrlund-Richter, M. De Luca, D. R. Marshak, M. Munsie, A. Veiga and M. Rao, Cell Stem Cell, 2009, 9, 20–26 CrossRef PubMed.
- J. M. Polo, E. Anderssen, R. M. Walsh, B. A. Schwarz, C. M. Nefzger, S. M. Lim, M. Borkent, E. Apostolou, S. Alaei, J. Cloutier, O. Bar-nur, S. Cheloufi, M. Stadtfeld, M. E. Figueroa, D. Robinton, S. Natesan, A. Melnick and J. Zhu, Cell, 2012, 151, 1617–1632 CrossRef CAS PubMed.
- D. C. Kirouac and P. W. Zandstra, Cell Stem Cell, 2008, 3, 369–381 CrossRef CAS PubMed.
- J. T. Cormier, N. I. zur Nieden, D. E. Rancourt and M. S. Kallos, Tissue Eng., 2006, 12, 3233–3245 CrossRef CAS PubMed.
- M. Schroeder, S. Niebruegge, A. Werner, E. Willbold, M. Burg, M. Ruediger, L. J. Field, J. Lehmann and R. Zweigerdt, Biotechnol. Bioeng., 2005, 92, 920–933 CrossRef CAS PubMed.
- D. E. Kehoe, D. Jing, L. T. Lock and E. S. Tzanakakis, Tissue Eng. Part A, 2010, 16, 405–421 CrossRef CAS PubMed.
- R. Olmer, A. Haase, S. Merkert, W. Cui, J. Palecek, C. Ran, A. Kirschning, T. Scheper, S. Glage, K. Miller, E. C. Curnow, E. S. Hayes and U. Martin, Stem Cell Res., 2010, 5, 51–64 CrossRef CAS PubMed.
- S. K. W. Oh, A. K. Chen, Y. Mok, X. Chen, U.-M. Lim, A. Chin, A. B. H. Choo and S. Reuveny, Stem Cell Res., 2009, 2, 219–230 CrossRef CAS PubMed.
- M. Fernandes, T. G. Fernandes, M. M. Diogo, C. L. da Silva, D. Henrique and J. M. S. Cabral, J. Biotechnol., 2007, 132, 227–236 CrossRef PubMed.
- E. Y. L. Fok and P. W. Zandstra, Stem Cells, 2005, 23, 1333–1342 CrossRef CAS PubMed.
- S. M. Dang, S. Gerecht-Nir, J. Chen, J. Itskovitz-Eldor and P. W. Zandstra, Stem Cells, 2004, 275–282 CrossRef PubMed.
- A. J. Wagers, Cell Stem Cell, 2012, 10, 362–369 CrossRef CAS PubMed.
- S. Chen, M. Lewallen and T. Xie, Development, 2013, 14, 255–265 CrossRef PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- M. P. Lutolf and J. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- M. P. Lutolf and J. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS PubMed.
- M. Ehrbar, S. C. Rizzi, R. Hlushchuk, V. Djonov, A. H. Zisch, J. Hubbell, F. E. Weber and M. P. Lutolf, Biomaterials, 2007, 28, 3856–3866 CrossRef CAS PubMed.
- S. P. Zustiak and J. B. Leach, Biomacromolecules, 2010, 11, 1348–1357 CrossRef CAS PubMed.
- T. P. Kraehenbuehl, P. Zammaretti, A. J. Van der Vlies, R. G. Schoenmakers, M. P. Lutolf, M. E. Jaconi and J. Hubbell, Biomaterials, 2008, 29, 2757–2766 CrossRef CAS PubMed.
- J. Patterson and J. a Hubbell, Biomaterials, 2010, 31, 7836–7845 CrossRef CAS PubMed.
- E. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. Sulchek, T. H. Barker and A. J. García, Adv. Mater., 2012, 24, 64–70 CrossRef CAS PubMed.
- R. Mahou and C. Wandrey, Macromolecules, 2010, 43, 1371–1378 CrossRef CAS.
- T. Canal and N. A. Peppas, J. Biomed., 1989, 23, 1183–1193 CAS.
- G. King, J. Daugulis, P. Faulkner and M. F. Goosen, Biotechnol. Prog., 1987, 3, 231–240 CrossRef CAS.
- M. Charnley, M. Textor, A. Khademhosseini and M. P. Lutolf, Integr. Biol., 2009, 1, 625–634 RSC.
- G. M. Vandenbossche, P. Van Oostveldt, J. Demeester and J. P. Remon, Biotechnol. Bioeng., 1993, 42, 381–386 CrossRef CAS PubMed.
- D. A. Hems and G. Gaja, Biochem. J., 1972, 128, 421–426 CrossRef CAS PubMed.
- K. Mitsui, Y. Tokuzawa, H. Itoh, K. Segawa, M. Murakami, K. Takahashi, M. Maruyama, M. Maeda and S. Yamanaka, Cell, 2003, 113, 631–642 CrossRef CAS PubMed.
- S. Masui, S. Ohtsuka, R. Yagi, K. Takahashi, M. S. H. Ko and H. Niwa, BMC Dev. Biol., 2008, 8, 45 CrossRef PubMed.
- H. Niwa, J. Miyazaki and a G. Smith, Nat. Genet., 2000, 24, 372–376 CrossRef CAS PubMed.
- R. J. Arceci, A. A. King, M. C. Simon, S. H. Orkin and D. B. Wilson, Mol. Cell Biol., 1993, 13, 2235–2246 CrossRef CAS PubMed.
- S. A. Duncan, A. Nagy and W. Chan, Development, 1997, 124, 279–287 CAS.
- G. S. Kwon, S. T. Fraser, G. S. Eakin, M. Mangano, J. Isern, E. Sahr, A. Hadjantonakis and M. H. Baron, Dev. Dyn., 2007, 235, 2549–2558 CrossRef PubMed.
- C. Bernemann, B. Greber, K. Ko, J. Sterneckert, D. W. Han, M. J. Araúzo-Bravo and H. R. Schöler, Stem Cells, 2011, 29, 1496–1503 CrossRef CAS PubMed.
- Q.-L. Ying, M. Stavridis, D. Griffiths, M. Li and A. Smith, Nat. Biotechnol., 2003, 21, 183–186 CrossRef CAS PubMed.
- T. Teratani, H. Yamamoto, K. Aoyagi, H. Sasaki, A. Asari, G. Quinn and H. Sasaki, Hepatology, 2005, 41, 836–846 CrossRef CAS PubMed.
- R. S. Cherry and E. T. Papoutsakis, Biotechnol. Bioeng., 1987, 32, 1001–1014 CrossRef PubMed.
- R. S. Cherry and K. Y. Kwon, Biotechnol. Bioeng., 1990, 36, 563–571 CrossRef CAS PubMed.
- S. Stolberg and K. E. McCloskey, Biotechnol. Prog., 2009, 25, 10–19 CrossRef CAS PubMed.
- D. Jing, A. Prikh and E. S. Tzanakakis, Cell Transplant, 2010, 19, 1397–1412 Search PubMed.
- C. Xu, M. S. Inokuma, J. Denham, K. Golds, P. Kundu, J. D. Gold and M. K. Carpenter, Nat. Biotechnol., 2001, 19, 971–974 CrossRef CAS PubMed.
- S.R. Braam, L. Zeinstra, S. Litjens, D. Ward-van Oostwaard, S. van den Brink, L. van Laake, F. Lebrin, P. Kats, R. Hochstenbach, R. Passier, A. Sonnenberg and C.L. Mummery, Stem Cells, 2008, 26, 2257–2265 CrossRef CAS PubMed.
- I. M. Chung, N. O. Enemchukwu, S. D. Khaja, N. Murthy, A. Mantalaris and A. J. García, Biomaterials, 2008, 29, 2637–2645 CrossRef CAS PubMed.
- Y. Shirayoshi, T. S. Okada and M. Takeichi, Cell, 1983, 35, 631–638 CrossRef CAS PubMed.
- M. Nagaoka, U. Koshimizu, S. Yuasa, F. Hattori, H. Chen, T. Tanaka, M. Okabe, K. Fukuda and T. Akaike, PLoS ONE, 2006, 1, e15 Search PubMed.
- L. Mohamet, M. Lea and C. M. Ward, PLoS ONE, 2010, 5, e12921S Search PubMed.
- T. Hamazaki, M. Oka, S. Yamanaka and N. Terada, J. Cell Sci., 2004, 117, 5681–5686 CrossRef CAS PubMed.
- J. Rodda, S. J. Kavanagh, J. Rathjen and P. D. Rathjen, Int. J. Dev. Biol., 2002, 46, 449–458 Search PubMed.
- F. Chowdhury, Y. Li, Y.-C. Poh, T. Yokohama-Tamaki, N. Wang and T. S. Tanaka, PLoS ONE, 2010, 5, e15655 CAS.
- B. Trappmann, J. E. Gautrot, J. T. Connelly, D. G. T. Strange, Y. Li, M. L. Oyen, M. A. Cohen Stuart, H. Boehm, B. Li, V. Vogel, J. P. Spatz, F. M. Watt and W. T. S. Huck, Nat. Mater., 2012, 11, 642–649 CrossRef CAS PubMed.
- M. M. Chowdhury, T. Katsuda, K. Montagne, H. Kimura, N. Kojima, H. Akutsu, T. Ochiya, T. Fujii and Y. Sakai, Biomed. Microdevices, 2010, 12, 1097–1105 CrossRef CAS PubMed.
- C. L. Bauwens, R. Peerani, S. Niebruegge, K. A. Woodhouse, E. Kumacheva, M. Husain and P. W. Zandstra, Stem Cells, 2008, 26, 2300–2310 CrossRef PubMed.
- L. Cui, K. Johkura, F. Yue, N. Ogiwara, Y. Okouchi, K. Asanuma and K. Sasaki, J. Histochem. Cytochem.: Off. J. Histochem. Soc., 2004, 52, 1447–1457 CrossRef CAS PubMed.
- A. P. Van Winkle, I. D. Gates and M. S. Kallos, Cells, Tissues, Organs, 2012, 196, 34–47 CrossRef PubMed.
- K. H. So, Y. J. Han, H. Y. Park, J. G. Kim, D. J. Sung, Y. M. Bae, B. C. Yang, S. B. Park, S. K. Chang, E. Y. Kim and S. P. Park, Int. J. Cardiol., 2011, 153, 277–285 CrossRef PubMed.
- E. Abranches, M. Silva, L. Pradier, H. Schulz, O. Hummel and E. Bekman, PLoS ONE, 2009, 4, e6286 Search PubMed.
- A. Mcmahon and A. Bradleyt, Cell, 1990, 62, 1073–1085 CrossRef CAS PubMed.
- X. Liu, W. Xue, Q. Liu, W. Yu, Y. Fu, X. Xiong, X. Ma and Q. Yuan, Carbohydr. Polym., 2004, 56, 459–464 CrossRef CAS.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- I. Rieu and S. J. Powers, Plant Cell, 2009, 21, 1031–1033 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |