DOI:
10.1039/C3BM60174A
(Paper)
Biomater. Sci., 2013,
1, 1216-1222
Photo synthesis of protein-based drug-delivery nanoparticles for active tumor targeting
Received
15th July 2013
, Accepted 2nd August 2013
First published on 29th August 2013
Abstract
Recently, nanometer-sized drug-delivery systems (DDSs) with an active targeting function to tumor sites have been cinfirmed to have great efficiency for cancer chemotherapy. However, the use of complicated modification techniques and the poor graft efficiency of the targeting ligands on the surface of the DDS have largely limited the application of most of the nano-DDSs. Especially for protein-based nano-DDSs, grafting of targeting ligands often requires genetic modification of the protein sequences, which is laborious and inefficient. Here, we present a novel method to photo synthesize a protein-based drug-delivery system with an active targeting function to tumor sites. The disulfide bonds in protein bovine α-lactalbumin (BLA) can be ruptured by controlled UV illumination, which triggers the formation of nano-sized protein aggregates and releases free thiol groups for the modification of the active targeting ligand of circular RGD peptide. Moreover, we demonstrated that the anti-cancer drug doxorubicin (DOX) can be loaded onto the protein-based DDS during the photo synthesis step. The synthesis approach is very convenient and cost-effective and can be accomplished under physiological conditions. Both in vitro and in vivo experiments validate that this DDS system possesses a much greater drug-delivery efficiency to the tumor sites and a better inhibition capability of tumor growth than the unmodified counterparts. This novel drug-delivery system can find broad applications in cancer chemotherapy. Moreover, our synthetic strategy can be easily extended to other disulfide-containing proteins and greatly expand the tool-box of protein-based DDSs for active targeting.
1. Introduction
Chemotherapy is the major therapeutic modality for cancers. The efficiency of chemotherapy depends on its capability to specifically eliminate tumors while sparing normal tissues. However, due to its nonspecific distribution in the body, a high-dose, toxic anti-tumor drug is required in clinical cancer therapy, which leads to severe side-effects. There is an urgent need to develop specific cancer-targeting drug-delivery systems (DDSs). In recent years, nanoparticle-based drug-delivery systems merged as effective DDSs.1–7 Nanoparticles can preferentially accumulate in cancerous tissues through the enhanced permeability and retention (EPR) effect, leading to a high drug concentration within a tumor but a low concentration in normal tissues.8–12 Despite the effective transport of nanoparticles to a tumor through the EPR effect, their capability in cell uptake and intracellular drug release has been questioned.13 In addition, many advanced primary cancer cells are prone to spread and form disseminated tumor cells (DTCs), a process known as metastasis.14,15 Although nanoparticles are effective for treating large, well-vascularized tumors through the EPR effect, they may become inadequate for these small clusters of DTCs due to a lack of the EPR. These shortages largely limit nanoparticles as targeting–delivery systems for anti-cancer drugs in cancer therapy.
To circumvent these hurdles, strategies to modify nanoparticles have been developed, including specific ligand–receptor interactions.16–18 This approach is achieved by chemical modification of the nanoparticle surface with ligands that bind to unique moieties presented on cancer cells.19,20 The identity and characteristics of the targeting moiety are important for circulation time, cellular uptake, affinity, and extravasation. Commonly used ligands for this include folic acid, antibodies, peptides, etc.21–25 Despite great success in using such an active targeting mechanism for the treatment of cancers, especially metastatic cancer, the attachment of active targeting agents to nanoparticles is not trivial and straightforward. First, the reaction groups for conjugation with active targeting ligands should be incorporated onto the surface of the nanoparticles with site-specific binding behavior; second, the attachment of the active targeting ligands should not interfere with the assembly of the nanoparticles and the drug loading; third, and more importantly, the amount of active targeting ligands should be adjustable and the side-effect of incorporation of such active targeting ligands should be carefully evaluated. Moreover, conjugation of active targeting ligands to nanoparticles often requires multiple synthetic steps which may jeopardize many unique features of the original nanoparticles, such as good surface properties and the reduction of the active effecting.26–29 For example, maleimide–thiol conjugation is the best approach to realize targeting ligands to protein-based nanoparticles, and must be done by introducing engineered thiol-containing cysteine residues in the proteins. To this extent, the number of nanoparticles that are capable of the active targeting of specific cancers is still very limited.
Here, we report a novel strategy to synthesize protein-based nanoparticles with active tumor targeting capabilities that are suitable for anti-cancer drug delivery. This system can be realized easily by UV illumination under physiological conditions without any redundant and complex processes. Briefly, UV illumination can trigger the rupture of disulfide bonds in disulfide-containing proteins (e.g., α-lactalbumin, BLA)30 leading to the co-assembly of a protein–drug (e.g., doxorubicin, DOX) complex.31 The abundant amount of free thiol groups on the surface of these nanoparticles may be a useful feature for the conjugation of active targeting ligands. In addition, the protection of these abundant thiols can effectively decrease the tendency of these nanoparticles to further aggregate and increase their in vivo stability. Here, we used the maleimide-containing circular RGD peptide for active targeting. Circular RGD can be recognized by over-expressed integrin αvβ3 on the surface of many tumor cells.32–34 The maleimide group allowed this peptide to be conjugated to the thiol groups on the newly synthesized BLA–DOX nanoparticles effectively in a single step. Such modification is simple and controllable, which does not affect the narrow dispersion of the RGD-modified nanoparticles. Both in vitro and in vivo experiments indicated that the RGD-modified nanoparticles showed a much improved cancer cell-targeting efficiency and an enhanced tumor suppression capability compared with the unmodified counterparts. We anticipate that this novel drug-delivery system can find broad applications in cancer therapy, and our synthetic strategy can be extended to other protein-based drug-delivery systems.
2. Experimental section
2 mg mL−1 bovine α-lactalbumin (BLA) in quartz cuvettes with a pathlength of 1 mm was illuminated by a UV-B lamp (Philips, TL20W/12RS) at an intensity of ∼3 mW cm−2. For the case of DOX-loaded nanoparticles, the mixture solution of BLA and DOX (0.2 mg mL−1) was illuminated for 6 min. After illumination, PEG5k-maleimide (BLA–PEG5k-Mal = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, w/w), RGD-Mal (BLA–RGD-Mal = 10:1, w/w), and the mixture of PEG- and RGD-containing peptide (LA–RGD-Mal–PEG5k-Mal = 20:1:20, w/w) was added to the illuminated solution to synthesize PEG–BLA–DOX, RGD–BLA–DOX, and RGD–PEG–BLA–DOX nanoparticles, respectively. Subsequently, the solution was filtrated well under gentle agitation through a dialysis bag with a cutoff of 7000 Da molecular weight to remove un-encapsulated DOX. DOX was quantified by its fluorescence intensity at 580 nm at the excitation wavelength of 480 nm.
About 10 μL of sample was placed on a newly peeled mica wafer for about 10 min, and then rinsed with 50 μL of Milli-Q water to remove unattached sample and salts. The sample on the mica surface was dried at room temperature before imaging using a Nanowizzard II AFM (JPK, Berlin, Germany) operating in the intermittent contact mode with AC-160 cantilevers from Olympus. The mean diameter size of the nanoparticles was obtained by a DLS method using a Brookhaven BI-200SM system (Brookhaven). All DLS measurements were carried out using a wavelength of 640.0 nm at room temperature.
:2, w/w), RGD-Mal (BLA–RGD-Mal = 10:1, w/w), and the mixture of PEG- and RGD-containing peptide (LA–RGD-Mal–PEG5k-Mal = 20:1:20, w/w) was added to the illuminated solution to synthesize PEG–BLA–DOX, RGD–BLA–DOX, and RGD–PEG–BLA–DOX nanoparticles, respectively. Subsequently, the solution was filtrated well under gentle agitation through a dialysis bag with a cutoff of 7000 Da molecular weight to remove un-encapsulated DOX. DOX was quantified by its fluorescence intensity at 580 nm at the excitation wavelength of 480 nm.
About 10 μL of sample was placed on a newly peeled mica wafer for about 10 min, and then rinsed with 50 μL of Milli-Q water to remove unattached sample and salts. The sample on the mica surface was dried at room temperature before imaging using a Nanowizzard II AFM (JPK, Berlin, Germany) operating in the intermittent contact mode with AC-160 cantilevers from Olympus. The mean diameter size of the nanoparticles was obtained by a DLS method using a Brookhaven BI-200SM system (Brookhaven). All DLS measurements were carried out using a wavelength of 640.0 nm at room temperature.
2.3. Detection of in vitro cytotoxicity
ECA109 or WI-38 cells were seeded in 96-well plates and incubated with serial concentrations of the samples under assessment (free DOX, DOX-loaded nanoparticles) and control medium. After 48 h, 20 μL MTT solution per well was added and incubated for 4 h. The absorbance of each well at 570 nm was determined using an ELISA reader. Confocal laser scanning microscopy (CLSM) images were taken using an Olympus System Microscope BX51. ECA109 cells were seeded in a chamber slide and incubated for 2 days (37 °C, 5% CO2). The medium was then replaced with 2 mL culture medium containing DOX or nanoparticles, respectively. After a 4-h incubation, the medium was removed and the cells were rinsed once with PBS, and then the slide was sealed with a microscope cover-slide and subjected to confocal microscopy.
2.4. Detection of in vivo tumor-targeting specificity
2 mL BLA (10 mg mL−1) and 200 μL NIR-797 (10 mg mL−1) were mixed and incubated at 37 °C for 10 h with continuous stirring. The solution was dialyzed for 10 h to obtain NIR-797-labeled BLA (NIR-797-BLA) which can be used to prepare protein BLANIR-797 and its corresponding nanoparticles (PEG–BLANIR-797 nanoparticles, RGD–BLANIR-797 nanoparticles, and RGD–PEG–BLANIR-797 nanoparticles). Then the nanoparticles (300 μL) were injected into ECA109 tumor-bearing mice (ICR mice, 28–32 g) through the tail vein followed by NIR fluorescence imaging using a Maestro Dynamic™ Lumina system (CRI, Woburn, MA). The emission wavelength was 700–800 nm and the exposure time was 2 s. Note that we obtained permission for the animal experiments.
2.5. In vivo therapeutic efficacy
The tumor regression effect of the nanoparticles was investigated in male mice (6–8 weeks old, 18–22 g) that had been inoculated with esophagus tumor ECA109 cells subcutaneously in the right flank. When the tumor volume reached about 100 mm3, the mice were randomly sorted into five groups (5 mice per group), and each group was treated by tail vein injection every 2 days for a total 10 injections with PBS buffer, free DOX, PEG–BLA–DOX nanoparticles, RGD–BLA–DOX nanoparticles, and RGD–PEG–BLA–DOX nanoparticles, respectively. The dose of free DOX and DOX in the nanoparticles was 1.5 mg kg−1. Measurement of the individual tumor volumes (V) was calculated using the formula: V = [length × (width)2]/2.
3. Results and discussion
3.1 Preparation and characterization of RGD-modified nanoparticles
Fig. 1 shows the synthesis scheme for the RGD-modified BLA–DOX nanoparticles. For the first stage, the assembly of the nanoparticles and drug loading can be achieved concurrently in one step by UV illumination with the drug-loading content and the encapsulation efficiency being ca. 8.2% and 70%, respectively.28 During this stage, a large amount of free thiols was produced through reduction of the disulfide bonds under UV illumination.35–38 Lots of these free thiols formed further intermolecular disulfide bonds which play an important role in the assembly and stability of the nanoparticles. Note that there are still abundant free thiols on the surface of these nanoparticles and the amount of the thiols is in direct proportion to the illumination time.31 Thus, during the second stage, the circular RGD peptide with a maleimide group was immediately added to the protein–DOX solution after UV illumination. These RGD peptides were grafted onto the nanoparticles through the reaction between the maleimide groups and the thiol groups on the surface of the nanoparticles. To conjugate more RGD molecules, here we prolonged the illumination time to 6 min to produce more free thiols.31 By using such an approach, we can easily obtain monodispersed RGD–BLA–DOX nanoparticles with diameters of 172 ± 19 nm, as measured by AFM and DLS (Fig. 2Ac, 2B). It is worth mentioning that the attachment of the RGD peptide to the drug-loaded nanoparticles significantly enhanced their stability. This is probably due to both the protection of the redundant free thiols on the surface of nanoparticles and the high solubility of the RGD peptide. To address the effect of RGD modification on the targeting efficiency, we also synthesized a few control nanoparticles for comparison, e.g., BLA nanoparticles (Fig. 2Aa), PEG-modified BLA–DOX nanoparticles (PEG–BLA–DOX, Fig. 2Ab), and RGD and PEG co-modified BLA–DOX nanoparticles (RGD–PEG–BLA–DOX, Fig. 2Ad), respectively. The size and morphology of these control nanoparticles are similar to the RGD–BLA–DOX nanoparticles, as confirmed by both AFM and DLS.
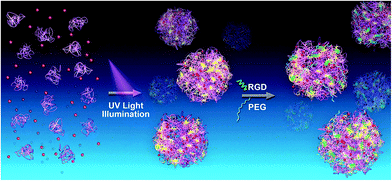 |
| | Fig. 1 Schematic of the photo synthesis of protein-based drug-delivery system with active targeting function. The first stage (left arrow) corresponds to the fast preparation of protein (BLA)–drug (DOX) nanoparticles, and the subsequent stage (right arrow) is the conjugation of the active targeting ligand RGD (green) or hydrophilic molecules PEG (blue) to the free thiols on the surface of the BLA–DOX nanoparticles. | |
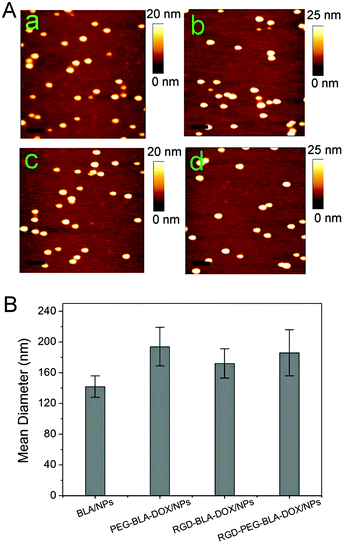 |
| | Fig. 2 (A) AFM images of protein α-lactalbumin (BLA) nanoparticles (a), PEG-modified BLA–DOX nanoparticles (PEG–BLA–DOX/NPs) (b), RGD-modified BLA–DOX nanoparticles (RGD–BLA–DOX/NPs) (c), and RGD and PEG co-modified BLA–DOX nanoparticles (RGD–PEG–BLA–DOX/NPs) (d), respectively. The scale bars are 300 nm. (B) The mean diameter of the four different nanoparticle types measured by DLS. | |
3.2 Passive and active targeting of tumor sites
To determine the tumor-targeting efficiency of the RGD-modified nanoparticles, we performed an experiment in mice bearing human esophageal cancer ECA109 on the armpit. The tumor-targeting efficiency of different samples was monitored through NIR imaging as shown in Fig. 3. For in vivo imaging, the native protein BLA was conjugated with the fluorescent dye NIR-797. The corresponding protein and its nanoparticles are referred to as BLANIR-797, PEG–BLANIR-797, RGD–BLANIR-797, and RGD–PEG–BLANIR-797, respectively. The NIR-797 fluorescence images in mice were obtained 72 h after intravenous injection of different nanoparticles or PBS buffer (control). Isolated BLA proteins were distributed all over the body and not accumulated within the tumor site but mainly in the intestine, probably due to abundant local capillary vessels (Fig. 3B). By contrast, PEG–BLANIR-797 nanoparticles diffused into the body of mice through circulation at the first stage after injection, and gradually accumulated in the tumor due to the enhanced permeability and retention (EPR) effect. After 72 h, the luorescent intensity in the tumor site reached a high level and showed a clear contrast against other tissues as shown in Fig. 3C. Of note, there were still a lot of nanoparticles in the intestines and liver of the mice injected with PEG–BLANIR-797 nanoparticles.
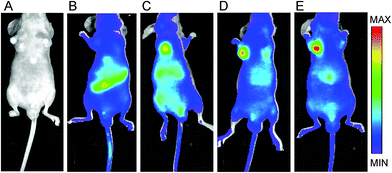 |
| | Fig. 3 The NIR-797 fluorescence images of the ECA109 tumor-bearing mice at 72 h after i.v. injection of PBS (A), BLANIR-797 (B), PEG–BLANIR-797/NPs (C), RGD–BLANIR-797/NPs (D), and RGD–PEG–BLANIR-797/NPs (E), respectively. | |
Due to the introduction of RGD peptide that can specifically bind to integrin αvβ3 over-expressed in tumor cells, the RGD-modified BLA nanoparticles exhibited a distinct bio-distribution pattern as shown in Fig. 3D. The majority of the RGD–BLANIR-797 nanoparticles accumulated in the tumor 72 h post-injection, which is direct evidence showing that the RGD peptide in our protein-based nanoparticle system directed active tumor targeting. This phenomenon becomes even more obvious for the BLA nanoparticles modified combining RGD with PEG molecules (RGD–PEG–BLANIR-797) shown in Fig. 3E. The enhanced tumor-specific targeting may be explained reasonably in that PEG modification on the nanoparticle surface increased the stability of these nanoparticles and prolonged their circulation time in the body of the mice.39,40 Together, these results provided clear evidence that the RGD-modified BLA nanoparticles possess excellent active tumor-targeting efficiency and are suitable for anti-cancer drug-delivery.
3.3. Cytotoxicity to tumor cells and biocompatibility with normal non-cancerous cells
Having tested the active targeting of RGD-modified BLA nanoparticles, we then investigated whether the specific tumor targeting could lead to more profound cytotoxicity. As shown in Fig. 4A, after incubation with RGD–BLA–DOX nanoparticles at a concentration of 400 ng mL−1 DOX for 48 h, only ∼25% of the human esophageal cancer cells ECA109 survived, whereas over 40% viability was observed in cells treated with BLA–DOX nanoparticles without RGD modification. The cytotoxicity was further increased in cells treated with BLA–DOX nanoparticles modified with both RGD and PEG molecules. This is possibly due to the increased nanoparticle stability gained through PEG modification.41,42 The cytotoxicity of different nanoparticles to ECA109 cells can also be determined by the 50% inhibition concentrations (IC50) which were counted by concentration-dependent cell viability curves. As shown in Table 1, the RGD-modified nanoparticles showed a higher efficacy of anti-cancer cytotoxicity compared with other nanoparticles or free DOX. In particular, the IC50 of RGD–PEG–BLA–DOX nanoparticles (0.1 ± 0.02 μg mL−1) was half the IC50 of the PEG-modified BLA–DOX nanoparticles (0.19 ± 0.03 μg mL−1) and only about one-fifth of the IC50 of the free drug DOX. The enhanced anti-tumor effect is mainly provided by the increased accumulation of nanoparticles at the tumor site through the active targeting ligands. It is noteworthy that BLA alone also induced weak cytotoxicity with an IC50 of 380 ± 50 μg mL−1 to tumor cells but not to normal non-cancerous cells. This tumor-specific killing profile may provide a favorable therapeutic window for BLA nanoparticle-based cancer treatment. Furthermore, the cytotoxicity of the RGD-modified BLA nanoparticles without DOX exerted an increased cytotoxicity to tumor cells than did the unmodified particles, which was possibly due to the presence of the active targeting ligand (Fig. 4B).
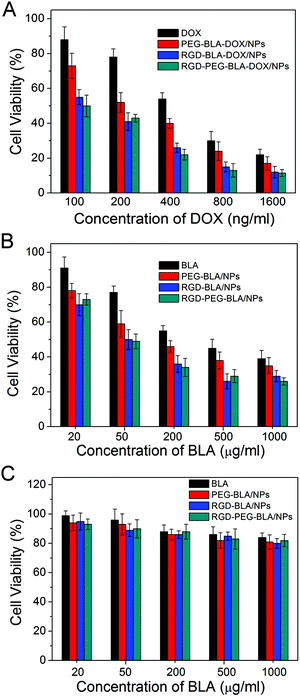 |
| | Fig. 4 (A) Cell viabilities of tumor cells ECA109 in the presence of DOX, PEG–BLA–DOX/NPs, RGD–BLA–DOX/NPs and RGD–PEG–BLA–DOX/NPs containing a series of DOX concentrations. (B) Cell viabilities of tumor cells ECA109 and (C) normal non-cancerous cells WI-38 incubated with BLA, PEG–BLA/NPs, RGD–BLA/NPs, and RGD–PEG–BLA/NPs, respectively. | |
Table 1 IC50 of different DOX formulations with ECA109 cells after 48 h incubation (n = 5)
| Formulations |
IC50 (μg mL−1) |
| Free DOX |
0.48 ± 0.05 |
| PEG–BLA–DOX/NPS |
0.19 ± 0.03 |
| RGD–BLA–DOX/NPS |
0.13 ± 0.03 |
| RGD–PEG–BLA–DOX/NPS |
0.10 ± 0.02 |
We next investigated whether the RGD ligand affected the biocompatibility of the nanoparticles. As shown in Fig. 4C, the RGD-modified BLA nanoparticles remained biocompatible and safe to the health non-cancerous cells (e.g. WI-38), which was similar to cells treated with unmodified protein BLA, even at a concentration of 1 mg mL−1. In addition, our recent study has verified that the photo synthesized BLA–DOX nanoparticles show rapid drug release at low pH and in a reducing environment that are typical in tumor tissues.31 Taken together, the active tumor-targeting RGD–BLA nanoparticles are suitable delivery systems for anti-cancer drugs.
3.4. Improved intracellular uptake by RGD targeting ligand
Subsequently, we verified whether the enhanced cytotoxicity of the RGD-modified nanoparticles was due to their improved intracellular uptake. As shown in Fig. 5, the efficiency of DOX delivery into the tumor cells by different nanoparticle samples was studied using confocal laser scanning microscopy. Under the same conditions, the fluorescence intensity of DOX was much stronger in cells treated with the RGD-modified nanoparticles (Fig. 5D and 5E) than cells treated with free DOX (Fig. 5B) or unmodified BLA–DOX nanoparticles conjugated with PEG only (Fig. 5C). This suggests that the active targeting ligand RGD indeed facilitates the entry of drug-loaded nanoparticles into tumor cells, which presumably is due to increased retention time of the nanoparticles on the tumor cell surface through specific ligand–receptor binding. This increases the chance of RGD-modified nanoparticles being more efficiently taken into the cells through endocytosis. These results are in line with the above in vivo tumor-targeting experiments monitored by NIR imaging (Fig. 3).
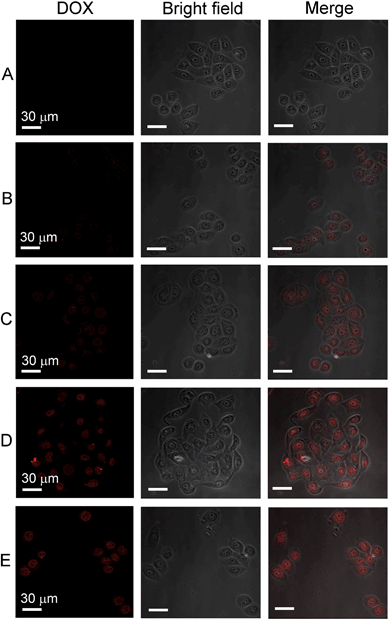 |
| | Fig. 5 Confocal laser scanning microscopy (CLSM) images of ECA109 tumor cells 4 h after treatment with PBS buffer (A), DOX (B), PEG–BLA–DOX/NPs (C), RGD–BLA–DOX/NPs (D), and RGD–PEG–BLA–DOX/NPs (E), respectively. Images in the right-hand column are merged from the DOX fluorescence images (left-hand column) and the light images (middle column). The scale bars are 30 μm. | |
3.5. Tumor suppression study in vivo
To investigate the anti-tumor advantage of the RGD-modified nanoparticles, we performed in vivo experiments with an ECA109 tumor-bearing mouse model as shown in Fig. 6. When tumor volume reached to about 100 mm3, mice were treated with nanoparticles as indicated through i.v. injection every 2 days for 20 days. As shown in Fig. 6A, the tumor growth in mice treated with free DOX (1.5 mg kg−1 per injection) was only a little slower than in PBS-treated or control-treated RGD–PEG–BLA nanoparticles (50 mg kg−1 per injection) mice. After treatment for 20 days, the volume of the tumors following treatment with free DOX (∼900 mm3, Fig. 7C) is comparable to those in PBS-treated (∼1000 mm3, Fig. 7A) or in RGD–PEG–BLA-treated mice (Fig. 7B). Of note, the weight of the mice in the group treated with free DOX decreased slightly as the treatment progressed (Fig. 6B) indicating that the toxicity of free DOX induces a systemic side-effect on the bodies of the mice. When DOX was carried by protein nanoparticles, the tumor growth was significantly inhibited and the tumor volume was about 65% of that in the natural state after treatment for 20 days (Fig. 7D). Moreover, the body weight was not decreased as compared with the PBS-treated controls.
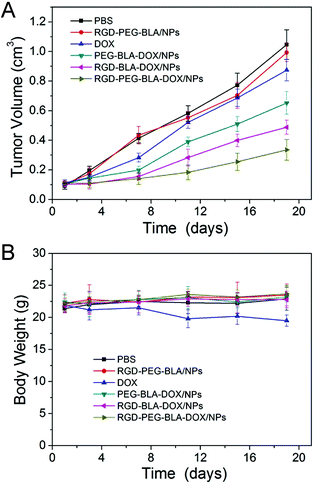 |
| | Fig. 6 (A) In vivo tumor-suppressive capabilities of RGD–BLA–DOX/NPs and controls, such as PBS, RGD–PEG–BLA/NPs, free DOX, PEG–BLA–DOX/NPs, and RGD–PEG–BLA–DOX/NPs, through i.v. injection in EC109 tumor-bearing nude mice (n = 5 of each). (B) The body weights of each group are shown as the mean (n = 5 of each). | |
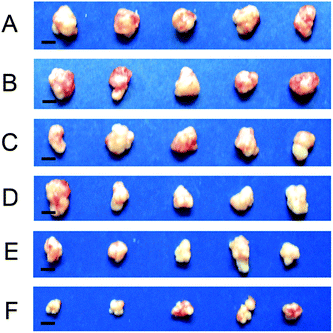 |
| | Fig. 7 Dissected ECA109 tumors from mice treated with PBS (A), RGD–PEG–BLA/NPs (B), free DOX (C), PEG–BLA–DOX/NPs (D), RGD–BLA–DOX/NPs (E), and RGD–PEG–BLA–DOX/NPs (F), respectively, for 20 days. The scale bars are 5 mm. | |
However, when the BLA–DOX nanoparticles were modified with the RGD protein, they exhibited an improved inhibitory capability in tumor growth (Fig. 6A). The tumor volume was less than half that of the natural growth volume 20 days after treatment (Fig. 7E). In particular, nanoparticles co-modified with RGD and PEG exerted a more profound anti-tumor capability. As shown in Fig. 7F, the tumor volume was only 35% of that in the natural growing state after 20 days of treatment. This was possibly due to the increased stability of the nanoparticles in the circulatory system that were protected by the PEG molecules. In line with this, the unchanged mice weight indicated that the systemic toxicity of the anti-tumor drug can be efficiently circumvented by loading with protein nanoparticles. These results further verify that our RGD-modified BLA–DOX nanoparticles possess an excellent tumor-targeting capability and potent tumor-suppressive efficiency.
4. Conclusions
In summary, by a simple and cost-effective approach, we synthesized a novel protein-based drug-delivery system with an active targeting function to tumor sites. The synthesis of the protein-based drug-delivery system can be accomplished in one step through UV illumination and subsequent modification of the BLA–DOX nanoparticles by the active targeting ligand RGD peptide that ingeniously utilized the abundant free thiols on the surface of the nanoparticles produced by the photo-reduced disulfide bonds. The synthesis of such a protein-based DDS can be realized under physiological conditions and does not involve the addition of other chemical agents or introduction of genetic mutations. Importantly, both in vitro and in vivo experiments verified that the RGD-modified BLA–DOX nanoparticles have excellent efficiency in the active targeting of tumor sites and a great capability for inhibiting the growth of tumor tissue. Thus, our RGD-modified BLA–DOX nanoparticles are indeed suitable for the cancer therapy with their active targeting function. In particular, many other disulfide-containing proteins can also be used to synthesize the protein-based DDS with active targeting capabilities. Therefore, our method represents a new direction in the engineering of protein-based DDSs.
Acknowledgements
This work was supported by the NNSF of China (grant no. 91127026, 11374148, 81121062, 11074115, 81071860 and 31170813), the 973 program (2013CB834104), the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Natural Science Foundation of Jiangsu Province of China (BK2010246).
Notes and references
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818 CrossRef CAS PubMed.
- D. A. LaVan, T. McGuire and R. Langer, Nat. Biotechnol., 2003, 21, 1184 CrossRef CAS PubMed.
- Y. Hu, Q. Chen, Y. Ding, R. Li, X. Jiang and B. Liu, Adv. Mater., 2009, 21, 3639 CrossRef CAS.
- W. R. Sanhai, J. H. Sakamoto, R. Canady and M. Ferrari, Nat. Nanotechnol., 2008, 3, 242 CrossRef CAS PubMed.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. Yang, P. L. R. Ee, P. T. Hammond and Y. Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Biomaterials, 2008, 60, 1615 CAS.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638 RSC.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271 CrossRef CAS.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812 CrossRef CAS PubMed.
- B. Haley and E. Frenkel, Urol. Oncol., 2008, 26, 57 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387 CAS.
- Z. Poon, D. Chang, X. Zhao and P. T. Hammond, ACS Nano, 2011, 5, 4284 CrossRef CAS PubMed.
- M. Wang and M. Thanou, Pharmacol. Res., 2010, 62, 90 CrossRef CAS PubMed.
- A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563 CrossRef CAS PubMed.
- L. A. Liotta, P. S. Steeg and W. G. Stetlerstevenson, Cell, 1991, 64, 327 CrossRef CAS.
- G. P. Luo, X. J. Yu, C. Jin, F. Yang, D. L. Fu, J. Long, J. Xu, C. Y. Zhan and W. Y. Lu, Int. J. Pharm., 2010, 385, 150 CrossRef CAS PubMed.
- F. Corsi, L. Fiandra, C. De Palma, M. Colombo, S. Mazzucchelli, P. Verderio, R. Allevi, A. Tosoni, M. Nebuloni, E. Clementi and D. Prosperi, ACS Nano, 2011, 5, 6383 CrossRef CAS PubMed.
- Z. H. Wang, Y. Yu, W. B. Dai, J. K. Lu, J. R. Cui, H. N. Wu, L. Yuan, H. Zhang, X. Q. Wang, J. C. Wang, X. Zhang and Q. Zhan, Biomaterials, 2012, 33, 8451 CrossRef CAS PubMed.
- A. P. Chapman, Adv. Drug Delivery Rev., 2002, 54, 531 CrossRef CAS.
- A. P. Chapman, P. Antoniw, M. Spitali, S. West, S. Stephens and D. J. King, Nat. Biotechnol., 1999, 17, 780 CrossRef CAS PubMed.
- F. Porta, G. E. M. Lamers, J. Morrhayim, A. Chatzopoulou, M. Schaaf, H. den Dulk, C. Backendorf, J. I. Zink and A. Kros, Adv. Healthcare Mater., 2013, 2, 281 CrossRef CAS PubMed.
- G. Destito, R. Yeh, C. S. Rae, M. G. Finn and M. Manchester, Chem. Biol., 2007, 14, 1152 CrossRef CAS PubMed.
- S. P. Wang, N. Mamedova, N. A. Kotov, W. Chen and J. Studer, Nano Lett., 2002, 2, 817 CrossRef CAS.
- Z. Gu, B. E. Rolfe, Z. P. Xu, J. H. Campbell, G. Q. Lu and A. C. Thomas, Adv. Healthcare Mater., 2012, 1, 669 CrossRef CAS PubMed.
- S. Su, H. Wang, X. Liu, Y. Wu and G. Nie, Biomaterials, 2013, 34, 3523 CrossRef CAS PubMed.
- Q. C. Xu, Y. Zhang, M. J. Tan, Y. Liu, S. Yuan, C. Choong, N. S. Tan and T. T. Y. Tan, Adv. Healthcare Mater., 2012, 1, 470 CrossRef CAS PubMed.
- L. S. Lee, C. Conover, C. Shi, M. Whitlow and D. Filpula, Bioconjugate Chem., 1999, 10, 973 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615 CrossRef CAS PubMed.
- H. Kang, M. R. Alam, V. Dixit, M. Fisher and R. L. Juliano, Bioconjugate Chem., 2008, 19, 2182 CrossRef CAS PubMed.
- J. Xie, M. Qin, Y. Cao and W. Wang, Proteins: Struct., Funct., Bioinf., 2011, 79, 2505 CrossRef CAS PubMed.
- J. Xie, Y. Cao, M. Xia, X. Gao, M. Qin, J. Wei and W. Wang, Adv. Healthcare Mater., 2013, 2, 795 CrossRef CAS PubMed.
- R. Pasqualini, E. Koivunen and E. Ruoslahti, Nat. Biotechnol., 1997, 15, 542 CrossRef CAS PubMed.
- M. L. Janssen, W. J. Oyen, I. Dijkgraaf, L. F. Massuger, C. Frielink, D. S. Edwards, M. Rajopadhye, H. Boonstra, F. H. Corstens and O. C. Boerman, Cancer Res., 2002, 62, 6146 CAS.
- R. E. Nisato, J. C. Tille, A. Jonczyk, S. L. Goodman and M. S. Pepper, Angiogenesis, 2003, 6, 105 CrossRef CAS.
- A. Vanhooren, B. Devreese, K. Vanhee, J. Van Beeumen and I. Hanssens, Biochemistry, 2002, 41, 11035 CrossRef CAS PubMed.
- J. J. Prompers, C. W. Hilbers and H. A. Pepermans, FEBS Lett., 1999, 456, 409 CrossRef CAS.
- M. T. Neves-Petersen, Z. Gryczynski, J. Lakowicz, P. Fojan, S. Pedersen, E. Petersen and S. Bjorn Petersen, Protein Sci., 2009, 11, 588 CrossRef PubMed.
- J. B. Xie, Y. Cao, H. Pan, M. Qin, Z. Q. Yan, X. Xiong and W. Wang, Proteins: Struct., Funct., Bioinf., 2012, 80, 2501 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93 CrossRef CAS PubMed.
- X. Zhao, Z. Poon, A. C. Engler, D. K. Bonner and P. T. Hammond, Biomacromolecules, 2012, 13, 1315 CrossRef CAS PubMed.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403 CrossRef CAS.
- D. Bazile, C. Prudhomme, M. T. Bassoullet, M. Marlard, G. Spenlehauer and M. Veillard, J. Pharm. Sci., 1995, 84, 493 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.