Scaffold free fabrication of linear multicellular assemblies by dielectrophoretic hydrogel trapping technique
William R.
Small
a,
Simeon D.
Stoyanov
bcd and
Vesselin N.
Paunov
*a
aSurfactant and Colloid Group, Department of Chemistry, The University of Hull, Hull, HU6 7RX, UK. E-mail: V.N.Paunov@hull.ac.uk; Fax: +44 (0)1482 466410; Tel: +44 (0)1482 465660
bUnilever R&D Vlaardingen, Olivier van Noortlaan 120, 3133 AT Vlaardingen, The Netherlands
cLaboratory of Physical Chemistry and Colloid Science, Wageningen University, 6703 HB Wageningen, The Netherlands
dDepartment of Mechanical Engineering, University College London, Torrington Place, London WC1E 7JE, UK
First published on 1st July 2013
Abstract
We have designed a scaffold-free cell assembly method which can produce linear structures of individual living cells without using templates. The method involves dielectrophoretic assembly of cells suspended in a solution of a gelling agent above the gelling temperature. After the cell assembly in string-like structures was achieved with the AC electric field still applied, we gelled the solution by cooling it below its gelling point. The hydrogel entraps the assembled cell structure, preventing its disassembly and allowing further analysis without the presence of the external electric field. We pre-functionalised the cells with polyelectrolytes before the dielectrophoretic assembly which allowed us to line them up in multicellular strings and bind them together with oppositely charged polyelectrolyte after the gel formation. Finally, by dissolving the hydrogel, we released the linear chains of living cells which were collected and studied by optical and fluorescence microscopy. Cell viability tests with fluorescein diacetate confirmed that the cells in the formed worm-like structures remain viable after the cell assembly procedure. The linear cell aggregates are stable without the electric field and can be further cultured or treated with additional polyelectrolytes which make the method attractive for tissue engineering. We envisage that this technique could find possible applications for assembly of neuron cells in linear structures and more complex cell networks.
Introduction
Engineering of functional structured tissues for replacement of damaged organs and nerves is one of the great challenges of modern medicine. Traditional approaches involve combination of scaffold-like matrix with cells and other biomaterials where the cells are promoted to grow and follow the shape of the matrix.1Various techniques have been developed to assemble cells into controlled biocompatible compartments2 or to deposit and follow the surface of a template.3 The latter often rely on prefabricating surfaces with cell-adhering patterns4 or 3D jet printing of cell suspensions in a layer-by-layer manner,5–8 or by printing prefabricated cell assemblies.6,9 Although these cell assembly technologies are rapidly evolving they are still in their infancy and use expensive and time consuming procedures and templates that can impact the cell viability. There is a scope for using alternative methods based on non-traditional approaches in cell assembly. For example, origami folding techniques10 have recently been employed to produce various 3D scaffolds for artificial tissues11 and cell-loaded containers.12 The folding of these structures is typically driven by surface tension,13 stress,14 and shrinkage of the hinges15 with temperature, electric or chemical triggers.
Recently, a new approach was employed by Krebs et al.16 which allowed the formation of ordered cellular structures in suspension by using negative magnetophoresis and inert cytocompatible magnetic particles, navigating the cell assembly without relying on cell binding or uptake. The common problem with this approach is the cell compatibility with the magnetite particles which requires careful design of the magnetite particle coating and the functionalisation of the cells with magnetic particles17–23 which may also compromise the cell viability.14,24
Using electric fields is a viable alternative for the controlled assembly and alignment of cells.25,26 Rapid, controllable and scalable cell assembly can also be achieved by using alternating current (AC) electric fields.27 Dielectrophoresis (DEP) is due to the interactions of assembled particles induced by AC electric field.28,29 As DEP does not require that the particles possess any specific property or attribute, it has proved to be a very versatile technique suited to different materials including cell manipulation and assembly. One of the most widely used applications that employs DEP exploits the negative dielectrophoretic force, where particles move away from regions of high field strength. Dielectrophoretic-field-flow-fractionation is a technique widely used for the sorting and separating of cells.28 DEP is however in many cases only a method for temporary assembling of the particle structures, as the dielectrophoretic force which causes the particles to aggregate is lost when the AC field is switched off. The assembled building blocks may then undergo Brownian motion, electrostatic repulsion or gravity induced sedimentation, depending on the properties of the particles (or cells) involved, resulting in disassembly of the formed structures. As Gupta et al.27 point out, “the challenge is thus to bind the live cells together in cohesive structures that are robust enough to be manipulated by external fields.” They developed a technique for permanent assembly of cells by DEP using functionalized synthetic microparticles binding to the cells through biospecific interactions.27
Several methods have been proposed to facilitate the preservation of these structures formed by DEP. One such method is reported by Shang and co-workers,30 who use chemical functionalisation to achieve permanent assemblies. They functionalise their electrodes with biotin and then wash with avidin which has multiple binding sites for the biotin molecules. The gold nanorods that they assemble are also functionalised with biotin, and when DEP is performed the rods are pulled into position between the electrodes, with the avidin bound to the substrates also binding to the biotin on the gold nanorods. This locks the assemblies in place, even when the field is removed. Another route to permanent assemblies of dielectrophoretically assembled structures is presented by Snoswell and co-workers,31 who fabricated strings of colloidal latex and microgel particles. These assemblies remain intact due to the use of oppositely charged particles, which are held together through attractive forces. These methods, although undoubtedly resulting in the fixation of dielectrophoretically assembled structures, do not constitute a generic method for DEP induced assembly. For example, not all particles can be functionalised with biotin or other binding agents. In addition, the use of oppositely charged colloidal particles is suitable only for the fabrication of “strings” of cells conjugated with oppositely charged colloidal particles. There remains therefore no general method to “arrest” the structures formed through dielectrophoretic assembly, which does not rely on any specific property of the building block, solvent or substrate.
In this paper we demonstrate a generic approach for assembly of cells in linear structures based on dielectrophoresis and a combination of hydrogel trapping and polyelectrolyte mediated biding. A similar approach was used by Small and Paunov32 for fabrication of electrically anisotropic agarose gels by dielectrophoretic assembly and encapsulation of silver nanowires. Fig. 1 shows a schematic to represent the proposed method for the fabrication of cell strings, utilising the technique we have developed for dielectrophoretic assembly with a hydrogel trapping. We chose yeast as model cells to prove the concept due to their robustness and easiness to culture and maintain. The cells were first pre-coated with polyelectrolytes24 which acted as an anchor to which an oppositely charged polyelectrolyte could be used to lock the cells together, following the cell assembly. We treated the hydrogel-trapped cell assemblies with oppositely charge polyelectrolyte after their alignment and then dissolved the hydrogel matrix, which produced free-standing yeast cell strings. We tested the viability of the cells in these by incubation in fluorescein diacetate solution.33 We demonstrate that cells remain viable after their treatment with the polyelectrolytes and subsequent dielectrophoretic assembly in linear strings. We provide more details on the practical application of this technique in the following sections.
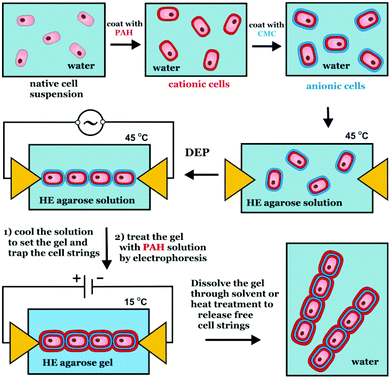 | ||
| Fig. 1 Schematics of the preparation of cell ‘strings’, using dielectrophoretic assembly combined with a hydrogel trapping method and polyelectrolyte mediated binding. | ||
Experimental
Materials
Agarose (type D5) was a gift from Hispanagar, Spain. This agarose forms gels on cooling at 32–40 °C depending on concentration, with the lowest gelling concentration at ∼0.5% w/v. D5 is a ‘high strength’ agarose gel, with a strength of 3200 g cm−2 at a concentration of 1.5% w/v. SeaPrep® agarose (hydroxyethylated)34 was purchased from Lonza, UK. This has a lower strength than the D5 agarose, ∼75 g cm−2 at a concentration of 2.0% w/v, and also has a low gelling and melting temperature. Baker’s yeast cells were obtained from Allinson's Dried Active Yeast, available from a local supermarket. All aqueous solutions were prepared with de-ionized water that had been passed through a Millipore Milli-Q Plus water purification system. The other materials used in this study are summarized in Table 1.| Material | Formula | Purity | Source |
|---|---|---|---|
| Sodium chloride | NaCl | 99.5% | BDH |
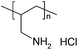
Polyallylamine hydrochloride (PAH) Mw ∼ 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
|
— | Aldrich |
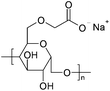
| Carboxymethyl cellulose (CMC) |
|
— | Fluka |
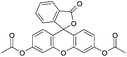
| Fluorescein diacetate (FDA) |
|
≥98% | Fluka |
Methods
Results and discussion
One of the advantages of using a gelation process to arrest the structures formed through DEP is that no functionalisation of the building blocks or substrates is required. This means that this method should find widespread use for any number of building blocks that are to be assembled into organised structures. Here, we demonstrate the viability of this method as we assemble ‘strings’ of living yeast cells. In order to produce free-standing strings of yeast cells, we first had to coat the cells with polyelectrolytes using the layer-by-layer (LbL) approach.22–24 As the yeast cells carry a native (weak) negative charge (zeta potential approx. −7 ± 5 mV), the cells were first coated in poly(allylamine hydrochloride) (PAH) and then with carboxymethyl cellulose (CMC), using an incubation, centrifugation and washing cycle. The coated cells were then assembled into strings in a solution of hydrogel using DEP, rather than using a template. After the hydrogel has set, holding the cells together, the hydrogel was exposed to a solution of the oppositely charged polyelectrolyte to the one used last to coat the cells. The polyelectrolyte was transferred through the hydrogel by applying DC voltage between the electrodes. The polyelectrolyte moving electrophoretically though the weak agarose gel was expected to act as an adhesive for the oppositely charged cells, fixing the cells together so that when the gel is removed, the cells remain bond into string-like assemblies.Strategy for the assembly of yeast cell strings through DEP
The PAH/CMC coated yeast cells were then suspended in molten agarose solution brought at a temperature of 45 °C before mixing with the cells. Note that the yeast cells lose their viability after prolonged exposure to temperatures higher than 45 °C. Therefore we were constrained with the choice of gelling agent as its gelling point had to be significantly below this temperature range. For D5 agarose the gelling point is around 37 °C which means that the DEP cell temperature had to be maintained in the range 40–45 °C. A few drops of the yeast cell suspension in agarose solution were then introduced to the DEP cell, which was also held at 45 °C, and DEP was performed as described earlier, resulting in the yeast cells aligning to form strings. The AC field was then lowered while the temperature was lowered below the gelling temperature of the gel. In one version of this experiment, the sample was then incubated in a solution of PAH for three hours, allowing the electrolyte to migrate through the gel and adsorb onto the assembled yeast cell strings. The weak gel was then washed in water, before it was dissolved with gentle heating to release the cell strings from the gel. The strings were collected and washed in water, and then tested with fluorescein diacetate (FDA),33 again employing a cycle of incubation, centrifugation and washing. The strings were analysed with fluorescence microscopy, which allows the viability of the cells to be determined. FDA is a non-fluorescent derivative of fluorescein, which readily crosses the cell membrane due to its non-polar nature. Once inside the cytoplasm, FDA is hydrolysed by esterase enzymes and produces a fluorescein which is fluorescent. This happens only if the cellular membrane is intact. This results in the accumulation of fluorescein by-product in the living cells, which can then be visualised through fluorescence microscopy if the fluorescein is present inside the cell in sufficient concentration.33DEP of yeast cells with unmodified agarose gel
The concentration of yeast cells was 0.20% w/v, in an agarose gel at a concentration of 0.50% w/v agarose. The frequency of the field was fixed at 20 kHz, while the voltage between the electrodes was increased until the cells started to assemble into strings. Typical optical microscopy images of the yeast cells, aligned with DEP, are shown in Fig. 2 where the yeast cells align to form strings in a dielectrophoretic field. | ||
| Fig. 2 Optical microscopy images of yeast cells (0.20% w/v) in agarose solution (0.50% w/v) undergoing dielectrophoretic assembly (20 kHz, 600 V cm−1). Images captured: (a) 30 seconds and (b) 2 minutes after the field was applied. Scale bars are 100 μm in both images. | ||
The field strength required for assembly to take place, at a frequency of 20 kHz, was approximately 600 V cm−1, although raising the voltage further resulted in more rapid assembly. The string assembly started with the cells aggregating into small strings comprising two or three cells, with these small strings gradually joining together to form increasingly large assemblies.
The length of the string was dependent on the length of time that the field was applied for, as shown by the longer assemblies observed in Fig. 2b after the field had been applied for two minutes. Eventually, two or more strings joined together to form parallel ‘belts’ of yeast cells. Yeast cells that were not coated in any polyelectrolyte also assemble but at much higher field strengths. A possible explanation for this is that the formation of the strings is a result of the dielectrophoretic effect on the electric double layers (EDL) of the cell surface. The presence of two polyelectrolyte layers deposited on the cell surface serve to enhance the polarisation of the EDL and the magnitude of the effective dipole–dipole interactions, resulting in string formation.
After the cells assembled in chains, the field was lowered to 100 V cm−1 and the system was cooled to 25 °C. This resulted in gelling of the agarose, which prevented the cells from moving away from each other when the AC field was finally switched off. Such disorganisation of the yeast cells was observed in water and in the hydrogels above the gelling temperature, if the AC field was removed. The weak agarose gel maintained its structural integrity and did not break apart when it was introduced to a solution of PAH, and optical microscopy of the washed hydrogel after three hours incubation in PAH solution revealed that the yeast cell strings remained intact. A problem was encountered in the next stage however, as even at a concentration as low as 0.50% w/v agarose gel, the gel would not undergo a transition to its ‘liquid’ state unless heated in excess of 55 °C. This meant that the yeast cells could not be removed from the agarose gel without heating them up to a temperature that results in their death. Another attempt was made to dissolve the agarose in a solution of urea, but the concentrations required (>6 M) also resulted in a total loss of viability of the yeast cells. It seemed therefore that ordinary agarose, although providing a good medium for dielectrophoretic assembly to take place and sufficient structural integrity to stop the assemblies from disbanding, was not suitable for the eventual release of the cell assemblies from the gel.
DEP of yeast cells with a hydroxyethylated agarose gel
The dielectrophoretic assembly of yeast cells was then attempted in another type of agarose gel, which had been modified by substituting some of the agarobiose units with a hydroxyethyl group (HE-agarose).9 Depending on the degree of substitution, the presence of hydroxyethyl groups results in a lower gelling temperature (and subsequent melting temperature, though some hysteresis remains), reduction in gel strength and an increase in the optical transparency of the gel. Dielectrophoretic assembly was carried out at a yeast cell concentration of 0.20% w/v, and at several HE-agarose concentrations (0.5%, 1.0% and 1.5% w/v). Again, the cells were first coated in PAH and then CMC before suspending them in a warm HE agarose solution at 40 °C. The field frequency was fixed at 20 kHz, and the temperature of the cell was held at 45 °C during the DEP. At an HE-agarose concentration of 0.50% w/v the yeast cells assembled into long strings, but because of the reduction in mechanical strength, the gel at this concentration could not prevent the cells from disassembling when cooled to below the gelling temperature. At HE-agarose concentration of 1.50% w/v however, the hydrogel appeared to be too viscous to allow dielectrophoretic assembly to take place even at quite high field strengths and for long time periods. Only very small assemblies, two to three yeast cells long, were assembled, though the gel now had sufficient mechanical strength to prevent the assemblies from disbanding. A compromise between the length of the assemblies and the gel strength was reached at an HE-agarose concentration of 1.00% w/v but at significantly higher field strength (see Fig. 3).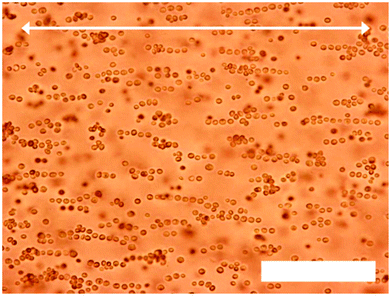 | ||
| Fig. 3 Optical microscopy images of yeast cells (0.20% w/v) in HE-agarose solution (1.00% w/v) undergoing dielectrophoretic assembly (20 kHz, 1250 V cm−1). The field was applied for 20 minutes. Arrow denotes direction of electrodes in relation to the yeast cell assemblies. Scale bar is 100 μm. | ||
Here, the cells formed strings that were on average 5–15 yeast cells in length, as shown in Fig. 3. The reduction in length of the strings is attributed to the different type of agarose gel in which the dielectrophoretic assembly is being carried out. The concentration here (1.00% w/v) is higher than that used previously (0.50% w/v), hence the medium viscosity is higher, which inhibits movement of the yeast cells towards each other. In addition, the hydroxyethylation of the HE-agarose may alter its electrical properties, which would change the dielectrophoretic force required for the yeast cells to assemble. At a concentration of 1.00% w/v the gelling temperature of the HE-agarose was approximately 17 °C. The sample was cooled to below this temperature to set the gel while the voltage was lowered to 100 V cm−1. When the field was then removed, the cells remained chained together and did not undergo disassembly. The binding of the assembled cells in string was done in two alternative ways: (i) we incubated the sample in a PAH solution for three hours. (ii) Alternatively, we deposited a drop of PAH solution close to one of the electrodes and conducted electrophoreses to spread the polyelectrolyte throughout the gel with the yeast structure. Both approaches yielded similar results. The hydrogel was washed and then dissolved by gently heating to 45 °C, resulting in the cell strings freeing themselves from the hydrogel. The yeast cell strings were then separated from the agarose solution by centrifugation, and washed several times with water before incubating in an FDA solution. The cell strings were then washed with water to remove excess FDA, and re-suspended in water. Fig. 4 shows optical microscopy of some of the yeast cell strings, re-suspended in Milli-Q water. The images in Fig. 4 are typical of the cell strings that were recovered from the HE agarose gels.
 | ||
| Fig. 4 Optical microscopy images of the yeast cell strings re-suspended in Milli-Q water. Scale bars are 25 μm in both images. | ||
These particular strings contained 6–7 yeast cells (Fig. 4a and 4b, respectively), and the average length of the strings recovered was in good agreement with the assembly lengths that were being formed in Fig. 3. This suggests that the strings are not undergoing any significant shortening, or cutting, during any part of the process (incubating in PAH solution, dissolving the hydrogel, washing, incubating with FDA) after the AC electric field is switched off. Applying light pressure to the microscope cover slip caused the cell strings to move through the sample, revealing two interesting observations. One observation was that the strings were quite flexible, as they bent and wrapped around static objects that they encountered as they moved. This is demonstrated in Fig. 4a, where the string has undergone a change in its orientation from linear to ‘L-shaped’ which indicates that the strings of cells are flexible. The second observation is that the strings also did not break apart under low shear, suggesting that they are well adhered together by the polyelectrolytes. Fluorescence microscopy images of the yeast cell strings, which are 6–8 yeast cells in length, are shown in Fig. 5. There one can see that the yeast cells in the strings are all emitting a green fluorescence, indicating the accumulation of fluorescein within the cells as a result of internal FDA hydrolysis by the cell esterases. This in turn suggests that the cells are still viable, and have not been harmed by the AC electric field or at any other stage in the process of assembling the cells together into strings. We have thus demonstrated that the technique developed for assembling and encapsulating building blocks in agarose gel can be applied to the assembly of strings of yeast cells. We achieved DEP assembly of very long cell strings (30–40 cells) in the D5 agarose solution; however, we were not able to release them from the agarose gel without compromising cell viability. We succeeded in releasing viable cell assemblies released from the HE agarose gel but they were significantly shorter and contained an average of 7–10 yeast cells. Further optimisation with the HE agarose would be needed to preserve longer cell strings and release them from the hydrogel successfully. In future work we are planning to explore the assembly of other cells types and polyelectrolyte coating which will be reported in follow up publications.
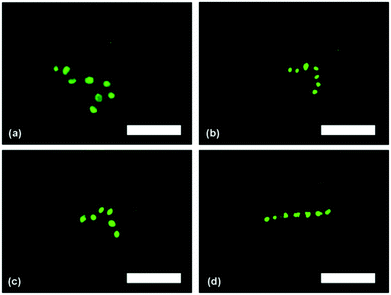 | ||
| Fig. 5 Fluorescence microscopy images of yeast cell ‘strings’ after treatment with FDA and re-suspending in water. Scale bars are 25 μm in all images. | ||
Conclusions
In summary, we have developed a novel method of assembling linear cell structures within a low melting point agarose solution using dielectrophoresis followed by setting of the hydrogel. This technique was applied to polyelectrolyte pre-treated yeast cells, which have been assembled into ‘strings’ of linearly connected living cells. The cells were locked into linear structures by setting the hydrogel which allowed additional treatment with oppositely charged polyelectrolyte for their permanent binding. We showed that the method could be used to align and encapsulate yeast cells, so that they form strings of cells entrapped in the hydrogel. After dissolving the hydrogel matrix, we released free-standing yeast cell strings, in which the cells remained viable following this treatment. This demonstrates the applicability of this method and possible applications for other cell systems, and is also an interesting development towards producing living multi-cellular assemblies. This cell assembly approach can find applications for tissue engineering research and has the ability to organise cells in a cheap, easily applicable technology that can be widely implemented. This method can potentially be used in assembly of tissue structures of linear architecture, e.g. assembly of artificial neuron strings and more complex cell networks for tissue regeneration applications.Acknowledgements
The authors acknowledge the PhD studentship of William Small from the University of Hull and partial financial support from Unilever and EU COST actions MP1106 and CM1101.Notes and references
- E. Alsberg, E. E. Hill and D. J. Mooney, Crit. Rev. Oral Biol. Med., 2001, 12, 64–75 CAS.
- A. Folch and M. Toner, Annu. Rev. Biomed. Eng., 2000, 2, 227–256 CrossRef CAS.
- D. Falconnet, G. Csucs, H. M. Grandin and M. Textor, Biomaterials, 2006, 27, 3044–3063 CrossRef CAS.
- P. G. Campbell and L. E. Weiss, Expert Opin. Biol. Ther., 2007, 7, 1123–1127 CrossRef CAS.
- V. Mironov, T. Boland, T. Trusk, G. Forgacs and R. R. Markwald, Trends Biotechnol., 2003, 21, 157–161 CrossRef CAS.
- V. Mironov, V. Kasyanov and R. R. Markwald, Trends Biotechnol., 2008, 26, 338–344 CrossRef CAS.
- V. Mironov, V. Kasyanov and R. R. Markwald, Curr. Opp. In Biotechnol., 2011, 22, 667–673 CrossRef CAS.
- G. Villar, A. D. Graham and H. Bayley, Science, 2013, 340, 48–52 CrossRef CAS.
- B. Derby, Science, 2013, 338, 921–926 CrossRef.
- K. Kuribayashi-Shigetomi, H. Onoe and S. Takeuchi, PLoS One, 2012, 7, e51085.1–e51085.8 Search PubMed.
- N. Bassik, G. M. Stern, M. Jamal and D. H. Gracias, Adv. Mater., 2008, 20, 4760–4764 CrossRef CAS.
- T. G. Leong, C. L. Randall, B. R. Benson, A. M. Zarafshar and D. H. Gracias, Lab Chip, 2008, 8, 1621–1624 RSC.
- B. Gimi, T. Leong, Z. Y. Gu, M. Yang, D. Artemov, Z. M. Bhujwalla and D. H. Gracias, Biomed. Microdevices, 2005, 7, 341–345 CrossRef.
- M. Jamal, N. Bassik, J. H. Cho, C. L. Randall and D. H. Gracias, Biomaterials, 2010, 31, 1683–1690 CrossRef CAS.
- S. Zakharchenko, N. Puretskiy, G. Stoychev, M. Stamm and L. Ionov, Soft Matter, 2010, 6, 2633–2636 RSC.
- M. D. Krebs, R. M. Erb, B. B. Yellen, B. Samanta, A. Bajaj, V. M. Rotello and E. Alsberg, Nano Lett., 2009, 9, 1812–1817 CrossRef CAS.
- R. F. Fakhrullin, J. Garcia-Alonso and V. N. Paunov, Soft Matter, 2010, 6, 391–397 RSC.
- D. Zhang, R. F. Fakhrullin, M. Ozmen, H. Wang, J. Wang, V. N. Paunov, G. Li and W. E. Huang, Microb. Biotechnol., 2011, 4, 89–97 CrossRef CAS.
- R. F. Fakhrullin, L. V. Shlykova, A. I. Zamaleeva, D. K. Nurgaliev, Y. N. Osin, J. Garcia-Alonso and V. N. Paunov, Macromol. Biosci., 2010, 10, 1257–1264 CrossRef CAS.
- S. A. Konnova, M. Kahraman, A. I. Zamaleeva, M. Culha, V. N. Paunov and R. F. Fakhrullin, Colloids Surf., B, 2011, 88, 656–663 CrossRef CAS.
- M.-L. Brandy, O. J. Cayre, R. F. Fakhrullin, O. D. Velev and V. N. Paunov, Soft Matter, 2010, 6, 3494–3498 RSC.
- R. F. Fakhrullin and V. N. Paunov, Chem. Commun., 2009, 2511–2513 RSC.
- R. F. Fakhrullin, M.-L. Brandy, O. J. Cayre, O. D. Velev and V. N. Paunov, Phys. Chem. Chem. Phys., 2010, 12, 11912–11922 RSC.
- R. F. Fakhrullin, A. I. Zamaleeva, R. T. Minullina, S. A. Konnova and V. N. Paunov, Chem. Soc. Rev., 2012, 41, 4189–4206 RSC.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS.
- M. Yang and X. Zhang, Sens. Actuators, A, 2007, 135, 73–79 CrossRef CAS.
- S. Gupta, R. G. Alargova, P. K. Kilpatrick and O. D. Velev, Soft Matter, 2008, 4, 726–730 RSC.
- H. A. Pohl, J. Appl. Phys., 1951, 22, 869 CrossRef CAS.
- H. A. Pohl, J. Appl. Phys., 1958, 29, 1182 CrossRef.
- L. Shang, T. L. Clare, M. A. Eriksson, M. S. Marcus, K. M. Metz and R. J. Hamers, Nanotechnology, 2005, 16, 2846 CrossRef CAS.
- D. R. E. Snoswell, R. K. Brill and B. Vincent, Adv. Mater., 2007, 19, 1523 CrossRef CAS.
- W. R. Small and V. N. Paunov, J. Mater. Chem., 2008, 18, 2082–2084 RSC.
- P. Breeuwer, J.-L. Drocourt, N. Bunschoten, M. C. Zwietering, F. M. Rombouts and T. Abee, Appl. Environ. Microbiol., 1995, 61, 1614–1619 CAS.
- SeaPrep® Agarose product information sheet, Lonza, http://www.lonza.com.
| This journal is © The Royal Society of Chemistry 2013 |