DOI:
10.1039/C2BM00184E
(Paper)
Biomater. Sci., 2013,
1, 434-442
Enhanced uptake of nanoparticle drug carriers via a thermoresponsive shell enhances cytotoxicity in a cancer cell line†
Received
6th December 2012
, Accepted 5th January 2013
First published on 14th January 2013
Abstract
Polymer particles consisting of a biodegradable poly[lactide-co-glycolide] (PLGA) core and a thermoresponsive shell have been formulated to encapsulate the dye rhodamine 6G and the potent cytotoxic drug paclitaxel. Cellular uptake of these particles is significantly enhanced above the thermal transition temperature (TTT) of the polymer shells in the human breast carcinoma cell line MCF-7 as determined by flow cytometry and fluorescence microscopy. Paclitaxel-loaded particles display reduced and enhanced cytotoxicity below and above the TTT respectively compared to unencapsulated drug. The data suggests a potential route to enhanced anti-cancer efficacy through temperature-mediated cell targeting.
Introduction
A major hurdle in the development of an efficacious drug is delivery of an adequate dose to the target site while minimising off-target effects. This is particularly problematic for anti-neoplastic drugs, which are typically hydrophobic compounds with poor pharmacokinetic profiles and thus requiring repeat administration to achieve a suitable dose. In combination with the inherent cytotoxicity of such compounds this can lead to significant side effects and systemic toxicity.
To overcome these limitations various methodologies have been developed for enhanced delivery to disease sites. These include conjugation to synthetic polymers or encapsulation within colloidal carriers such as micelles, liposomes and nanoparticles. One area that has been recently explored is the use of thermoresponsive colloidal systems.1–6 These aim to exploit the slight hyperthermia (1–2 °C) displayed by certain tumour pathologies compared to surrounding healthy tissue.7 Additionally, induced local hyperthermia is now becoming an accepted adjunct treatment with chemo- and radiotherapy.8 By delivering a drug in a thermoresponsive vehicle it is possible to target the therapy to tissue at elevated temperature while leaving other tissues unaffected.9 This is because the hyperthermic region can be defined by the application of external ultrasound, or by near-infra-red irradiation.10 Accordingly, as long as a formulation can respond to the increased temperature at the target site, there is the possibility to localise therapy to this specific region through the externally directed hyperthermia as well as through intrinsic temperature variations. An example of a thermoresponsive liposomal formulation of doxorubicin is ThermoDox (Celsion Corporation), which is currently entering Phase III trial for the treatment of hepatocellular carcinoma. At 41–42 °C its lipid membrane undergoes a phase transition resulting in rapid release of the encapsulated doxorubicin. In combination with local thermal ablation treatment the formulation response limits drug release to the target site. Over the last few years a number of studies have indicated possible efficacy of thermoresponsive polymers in local drug delivery, either via controlled cell uptake in vitro11,12 or by localisation at hyperthermic regions in well-characterised in vivo models.13–16 We have recently described the synthesis and characterisation of PLGA particles with a thermoresponsive coat, via a synthesis procedure that enables easy modification of core components as well as response-tuning of the outer shell.17 Here we describe temperature dependent cellular uptake of such particles and the applicability to the delivery of paclitaxel in vitro (Fig. 1).
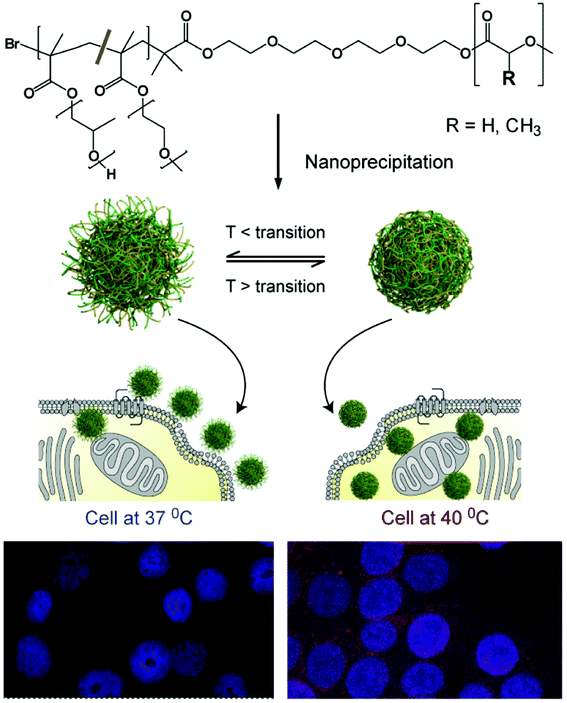 |
| | Fig. 1 Schematic of the formulation of thermoresponsive nanoparticles and the proposed enhancement in cellular uptake due to change in surface corona with thermal response. Particles at temperatures below the thermal transition temperature of their outer shell; display hydrophilic chain-extended polymers (left-hand side) and enter cells less readily than the same particles above their polymer thermal transition temperature (TTT) (right-hand side). An increased signal from nanoparticle-encapsulated fluorescent dye is indicative of the increased uptake of particles above their TTT in cells at 40 °C (bottom right) compared to 37 °C (bottom left). | |
Materials and methods
Materials
Tetraethylene glycol mono bromoisobutyrate was synthesised as previously described.17 Lactide (Aldrich, >98%), glycolide (Sigma, >99%), poly(ethylene glycol) methyl ether methacrylate (PEGMEMA, Mnca. 475, Aldrich), poly(propylene glycol)methacrylate (PPGMA, Mnca. 375, Aldrich), copper(II) bromide (Alfa Aesar, 99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA, Aldrich, 99%), tin(II) 2-ethylhexanoate (Sn(EH)2, Aldrich, 95%), rhodamine 6G (Fluka), 2-butanone (Alfa Aesar, 99%) and paclitaxel (PTX, Sigma) were used as received. All solvents were Fisher Analytical or HPLC grade and used as received.
Instrumentation
Number average molecular weight (Mn), weight average molecular weight (Mw) and polydispersity index (Mw/Mn) of the prepared copolymers were determined by gel permeation chromatography (GPC) on either a Polymer Labs PL-120 or GPC50 instrument fitted with a refractive index detector. The columns (7.5 × 300 mm Resipore Mixed-D, 2 in series) were eluted by chloroform and calibrated with polystyrene standards. All calibration and analysis were performed at 40 °C and a flow rate of 1 mL min−1. Molecular weight averages and polydispersity indices were calculated with Polymer Labs Cirrus 3.0 software. Samples were prepared by dissolution in chloroform and filtered (0.2 μm) prior to injection.
1H NMR spectra were recorded at 20 °C on a Bruker-DRX instrument operating at 400 MHz. Chemical shifts (δ) are reported in parts per million (ppm), referenced to CDCl3 (δ 7.26). The data was processed using the TOPSPIN 2 software.
Synthesis of thermoresponsive polymers
Synthesis of PLGA macroinitiator (PLGA-Br).
To a dry 100 mL round-bottom flask equipped with a magnetic stir bar (0.18 g, 0.52 mmol), lactide (3.0 g, 21.0 mmol) and glycolide (2.42 g, 21.0 mmol) were added and purged with argon for 20 min. The mixture was melted by heating up to 140 °C. The melt was degassed and purged with Ar for a further 20 min. This was followed by the addition of tin(II) 2-ethyl hexanoate (42 mg, 0.1 mmol). The reaction was left stirring overnight at 140 °C. The resulting polymer was purified by dialysing against acetone (3 × 500 mL) to afford PLGA-Br copolymer. Mn = 12 kDa, Mw/Mn = 1.28 as determined by GPC against polystyrene standards. Molar composition was calculated by 1H NMR, which indicated a lactic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolic acid ratio of 1:1. Analytical data were in accord with previous studies.17
:glycolic acid ratio of 1:1. Analytical data were in accord with previous studies.17
Synthesis of PLGA-block-(PEGMEMA-co-PPGMA).
A round-bottom flask fitted with a three-way stopcock was charged with PLGA-Br (1.2 g, 0.1 mmol), PMDTA (17.3 mg, 0.1 mmol) and CuBr2 (22.3 mg, 0.1 mmol). To the mixture, varying amounts of the two co-monomers, PPGMA and PEGMEMA were used as described previously17 to give polymers with different properties (Table S1, ESI†). All the materials were dissolved in 15 mL of 2-butanone. The mixture was degassed by flushing with Ar and stirred for 1 h. Tin(II) 2-ethyl hexanoate (40.5 mg, 0.1 mmol) was added under Ar. The flask was transferred to an oil bath at 60 °C. The polymerization was terminated by opening the flask and exposing the catalyst to air. The reaction mixture was triturated three times using an excess of hexane:diethyl ether (1:1) mixture. The precipitate was then dissolved with chloroform and passed through an aluminum oxide column. The resulting co-polymer was obtained by evaporating to dryness in vacuo. The resulting copolymers were all found to have similar overall molar masses as determined by GPC. Polymerisations were terminated at 60% conversion to reduce polydispersity as commercially supplied PPGMA monomer can contain small amounts of dimethacrylate impurity that leads to markedly increased polydispersity and cross-linking at higher conversion. Spectroscopic and other analytical data were consistent with samples of these polymers reported in our previous study.17
Formulation of R6G-loaded NPs
R6G-labelled NPs were prepared using an interfacial polymer deposition (IPD) or nanoprecipitation, method based on that of Fessi et al.18 Briefly, PLGA-b-(PEGMEMA-co-PPGMA) copolymer (100 mg) was dissolved in acetone (5 mL) containing rhodamine 6G (0.1, 0.25 or 0.4 mg). The resulting solution was added dropwise into phosphate-buffered saline (10 mM, pH 7.4, 10 mL) with vigorous magnetic stirring. The solution was covered with aluminium foil and left stirring in a hood overnight at room temperature to evaporate acetone. The NP suspension was purified by filtration (1.2 μm filters) followed by size exclusion chromatography using a PD-10 desalting column (GE Healthcare) to remove unencapsulated dye. Blank NPs were prepared similarly for comparison purposes.
Formulation of paclitaxel (PTX)-loaded NPs
Conventional IPD method.
Paclitaxel-loaded nanoparticles were prepared using the IPD method.18 Briefly, a specific amount of PLGA-b-(PEGMEMA-co-PPGMA) copolymer (P2) (50 or 100 mg) was dissolved in acetone (5 mL) containing a specific amount of PTX (0.5, 1 or 2 mg). The organic solution was added dropwise into deionised water under magnetic stirring. The solution was left in an Isomat® negative pressure isolator at room temperature to evaporate acetone. The particle suspension was purified as described above.
Modified IPD method.
PLGA-b-(PEGMEMA-co-PPGMA) copolymer (P2) (100 mg) was dissolved in PTX solution (1 mg mL−1) in acetone (1 mL). The organic solvent was allowed to evaporate and the dried mixture was reconstituted in 5 mL of acetone, which was then added dropwise to 10 mL water followed by gentle magnetic stirring overnight. The solution was left in the Isomat® negative pressure isolator at room temperature to evaporate acetone. The particle suspension was purified as described above.
Physicochemical characterisation of nanoparticles
Dynamic light-scattering.
The hydrodynamic radii of the nanoparticles were determined via scattered light recorded at 90° angle to incident radiation using a Viscotek Model 802 instrument equipped with an internal laser (825–832 nm) with a maximum radiation power of 60 mW. The samples were diluted with filtered, deionised water and at least five measurements of each sample were taken. The mean and standard deviation were calculated. Data processing was performed with the software program OmniSize2.
Laser Doppler anemometry.
Zeta potential (ζ) measurements of the nanoparticles were performed by laser Doppler anemometry using a Malvern Zetasizer 2000 equipped with a 10 mW He–Ne laser operating at a wavelength of 633 nm. Measurements were performed in a low ionic strength buffer, 1.0 mM HEPES buffer adjusted to pH 7.4. The mean value and standard deviation for each sample was calculated from at least five measurements.
Transmission electron microscopy.
Morphology of the particles was examined using a transmission electron microscope (TEM) (Jeol Jem 1010 electron microscope, Japan). A sample of particle suspension was diluted with a solution of phosphotungstic acid (3% w/v, pH 7.4) and observed under TEM. One drop of sample was placed for 1 minute on a copper grid coated with a formvar carbon film. The excess of sample was wicked away with the aid of filter paper prior to imaging by TEM.
Thermal transition temperatures.
Thermal transition temperatures (TTTs) of NPs were determined via turbidity. The change of turbidity of nanoparticle suspensions (in cell culture media) in response to change of temperature was investigated using UV/VIS spectrophotometer (Beckman, DU 800). Absorbance measurements were taken at 550 nm. Cycles were programmed to run from 20–90 °C (ramp rate of 1 °C min−1).
Determination of R6G encapsulation efficiency and loading in nanoparticles
The amount of R6G encapsulated was determined by a direct method. Freeze-dried R6G-labelled NPs were dissolved in DMSO, and the absorbance at 540 nm were determined using a UV/VIS spectrophotometer (Beckman, DU 800). With the aid of a standard curve of known concentrations of R6G in the same organic solvent, the amount of R6G was determined.
The influence of different theoretical drug loadings on the drug incorporation efficiency was investigated. Encapsulation and recovery were calculated using the equations below.19
| used in formulation/Mass of copolymer used in formulation) × 100 |
| loading (wt%) = (Mass of drug in NPs/Mass of NPs recovered) × 100 |
| Encapsulation efficiency (%) = (Mass of |
| in NPs/Mass of drug used in formulation) × 100 |
| NP recovery (yield) (%) = (Mass of NPs recovered/Theoretical mass of |
| , drug and any other stabilisers used in formulation) × 100 |
In vitro release of R6G
The R6G-labelled NP suspension (1 mL, about 4 mg mL−1) and 1 mL of PBS (pH 7.4) were added to a dialysis bag (molecular weight cut-off 6–8 kDa), and then placed in a beaker with phosphate buffered saline (PBS) (pH 7.4, 15 mL). Dialysate (15 mL) was removed from the beaker and an equal volume of fresh PBS was added at regular time intervals. The fluorescent solution was measured at λex = 525 nm, λem = 550 nm, slit widths (ex./em.) = 2.5/2.5 nm (Hitachi F-4500 fluorescence spectrophotometer, Hitachi Scientific Instruments, Finchampstead, UK). The amount of fluorescent dye was calculated by comparison with a standard curve of R6G fluorescence in PBS (pH 7.4). Effect of temperature was investigated by studying the release at 37 and 42 °C.
Cell culture
Human breast adenocarcinoma MCF7 cells were maintained on RPMI-1640 media supplemented with L-glutamine, sodium bicarbonate and 10% fetal bovine serum (FBS) at 37 °C and 5% CO2.
Cellular uptake of NPs by MCF7 cells
To investigate the effect of temperature on cell uptake, three R6G-loaded NPs with different thermal transition temperatures (TTTs) were incubated with MCF7 cells at 37 and 40 °C.
Fluorescence microscopy
Cells were seeded in four 12-well plates that contained sterilised cover-slips at a concentration of 100000 cells per well. A day after seeding, the media was replaced with media containing 200 μg mL−1 of R6G-loaded nanoparticles (NP1, NP2, NP3). The cells were incubated at 37 °C or 40 °C for 2 h. Subsequently, media was removed and cells were washed three times with PBS, fixed for 10 min in 1% paraformaldehyde and again washed three times. For nuclei staining, DAPI standard solution (Invitrogen, 1 drop) was added to each well and the stain allowed to enter the cells over a period of 10 min followed with washing. Cover-slips were then mounted onto microscopy slides to protect the samples. Microscopy images were obtained with a Leica DM IRB instrument. Images were recorded using a Leica DC200 camera and processed using DC viewer, version 3.2.0.0 software (Leica Microsystems, Germany). All microscopy gain and offset settings were maintained constant throughout the study.
Cells were seeded at a concentration of 500000 cells per well in 6-well plates for 24 h. Growth media was replaced with formulations containing 200 μg mL−1 of R6G-loaded nanoparticles (NP1, NP2, or NP3). The cells were incubated at 37 or 40 °C for 2 h. Cells were then washed three times with PBS and a cell suspension was created using trypsin/EDTA. Cell suspensions were diluted in growth media to stop the trypsinisation process, centrifuged and washed three times with PBS. Cells were finally re-suspended in 500 μL of PBS and immediately analyzed with a flow cytometer (Beckman Coulter FC500; High Wycombe, Buckinghamshire, UK) with XL SYSTEM II™ software using a 488 nm laser for excitation of R6G and a band centred at 585 nm for detection of fluorescence. Flow cytometer channel voltage and gain were maintained constant throughout the analysis. Approximately 10000 cell detection events were evaluated for forward and side scatter characteristics to determine the trend of R6G-labelled NP taken up by cells.
The amount of drug encapsulated was determined by a direct method. Freeze dried nanoparticles were weighed and dissolved in acetonitrile and vigorously vortexed to get a clear solution. Samples were analysed by HPLC at room temperature using a Hewlett-Packard HP1050 (Hewlett-Packard, Milano, Italy) on a Lichrosphere 100 RP-18 (5 μm) end-capped column equipped with a Hypersil 100 RP-18 (5 μm) guard cartridge (4 mm × 4 mm i.d.) and eluted isocratically with acetonitrile/water (70/30 v/v). The flow rate was fixed at 1 mL min−1 and operating pressure 1280 psi. The effluent was monitored at 227 nm. PTX solutions of known concentrations in the range 2.5–100 μg mL−1 were used to produce a standard curve from which concentrations of unknowns were determined.
In vitro drug release study
PTX-loaded NPs prepared by the modified IPD method (with 1% theoretical drug loading) were used for in vitro release study. The PTX-loaded NP suspension (1 mL, about 4 mg mL−1) and 1 mL of the media were added to a dialysis bag (molecular weight cut-off 6–8 kDa), and then placed in a beaker with PBS (pH 7.4 or 4.5, 10 mL). Dialysate (10 mL) was removed from the beaker and an equal volume of fresh media was added at regular time intervals. The collected dialysates were analysed with HPLC as described previously. The effect of temperature was investigated by studying the release at 37 and 42 °C.
In vitro anti-tumour activity (MTT assay)
Human breast adenocarcinoma MCF7 cells were seeded in 96-well plates at the density of 10000 viable cells per well and incubated for 24 h in growth media to allow cell attachment. The cells were then incubated with aqueous dispersions of free PTX (control), PTX-loaded nanoparticles (NP4) (experimental) or drug-free nanoparticles (4 mg mL−1) for 24 h at 37 or 40 °C in PBS (pH 7.4). At pre-determined times, the formulations were replaced with media containing 3-[4,5-dimethylthiazol-2-yl]-3,5-diphenyl tetrazolium bromide (MTT) and cells were then incubated for additional 4 h. The MTT-containing solution was aspirated off and DMSO was added to dissolve the formazan crystals. Absorbance was measured at 570 nm using an absorbance microplate reader (Dynex Technologies, USA). Untreated cells were taken as control with 100% viability. Cell viability was expressed using the following equation:
| Cell viability (%) = (Absexp./Abscontrol) × 100 |
where Absexp. and Abscontrol represent the amount of formazan determined for cells treated with the different formulations and for control cells (non-treated), respectively. The experiment was performed in triplicates and the results are expressed as mean values ± standard deviation.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 5.04 (GraphPad Software Inc.).
Results and discussion
Synthesis of PLGA-b-(PEGMEMA-co-PPGMA)
Poly[lactide-co-glycolide]-block-poly[poly(ethylene glycol)monomethyl ether methacrylate-co-poly(propylene glycol) methacrylate] (PLGA-b-(PEGMEMA-co-PPGMA)) was synthesised as previously described.17 Briefly, a PLGA block was synthesised by ring-opening polymerisation of lactide and glycolide in melt using tin(II) 2-ethyl hexanoate as catalysis and tetraethylene glycol mono 2-bromoisobutyrate as an initiator. The resulting bromide terminated PLGA was then used as the initiator in AGET ATRP of PEGMEMA/PPGMA.20 The PEGMEMA/PPGMA feed ratio (Table S1, ESI†) was varied to produce polymers with varying thermal transition temperatures (TTT). Molecular weights (Mn) and polydispersity indices (Mw/Mn) for the polymers used are given in Table 1.
Table 1 Characterisation of R6G-and PTX-loaded nanoparticles studied in vitro
| Particle |
Polymer used |
M
na (kDa) |
M
w/Mna |
R
h ± SDb (nm) |
ζ potential ± SD (mV) |
Theoretical drug loading (wt%) |
Practical drug loading (wt%) |
Encapsulation efficiency (%) |
NP recovery (yield) (%) |
Transition temperaturec (°C) |
|
Determined by gel permeation chromatography against polystyrene standards.
Determined by dynamic light-scattering after dye/drug incorporation.
Determined by cloud point measurements.
|
| NP1 |
P1 |
20.4 |
1.3 |
26.1 ± 9.1 |
−0.6 ± 2.2 |
0.25 |
0.113 ± 0.007 |
45.0 ± 2.9 |
96.1 ± 1.7 |
33 |
| NP2 |
P2 |
19.8 |
1.3 |
25.4 ± 12.0 |
−1.2 ± 2.2 |
0.25 |
0.112 ± 0.006 |
44.8 ± 2.3 |
96.0 ± 1.1 |
39 |
| NP3 |
P3 |
17.2 |
1.3 |
26.3 ± 12.8 |
−1.9 ± 3.2 |
0.25 |
0.117 ± 0.005 |
46.8 ± 1.8 |
96.8 ± 3.8 |
56 |
| NP4 |
P2 |
19.8 |
1.3 |
31.5 ± 11.7 |
N.D. |
1.0 |
0.76 ± 0.01 |
76.2 ± 1.2 |
80.4 ± 0.03 |
39 |
Formulation and physicochemical characterisation of PLGA-b-(PEGMEMA-co-PPGMA) particles encapsulating rhodamine 6G
Particles encapsulating the fluorophore rhodamine 6G (R6G) were formulated using the interfacial polymer deposition (IPD, or “nanoprecipitation”) method described by Fessi et al.18 R6G loading was targeted at 0.1, 0.25 and 0.4 wt%. As expected increasing theoretical loading resulted in an increase in practical loading (encapsulation) from 0.05 to 0.13 wt%, albeit with a reduction in encapsulation efficiency from 48% to 32% presumably due to a limiting encapsulation capacity of the particles for R6G (Table S2, ESI†). Consequently, subsequent formulations of R6G-loaded particles were produced using a theoretical loading of 0.25 wt%. Particles with varying thermal transition temperatures were formulated by using polymers with different PEGMEMA/PPGMA ratios. Particle size distributions, surface charge and dye loading were found to be comparable between formulations (NP1–3; Table 1, entries 1–3).
In vitro release of rhodamine 6G from PLGA-b-(PEGMEMA-co-PPGMA) particles
In order to test any intrinsic changes in drug release due to nanoparticle thermal responses over an approximate physiological/hyperthermia range, the release of rhodamine 6G from NP2 was studied at 37 and 42 °C, i.e. above and below these nanoparticles’ TTT, in PBS at pH 7.4. Within the first 8 hours, approximately 30% of encapsulated dye was released both above and below the thermal transition temperature. After 24 h the cumulative dye release only increased slightly to approximately 34% (Fig. 2A). This rapid early release was likely to derive from release of dye entrapped within the polymer corona, or loosely physisorbed to the surface of the PLGA core, rather than truly encapsulated within the core. This assertion was supported by the slightly faster release in the first 3 h at 42 °C compared to 37 °C, where dye entrapped in the corona would be rapidly excluded upon chain collapse.
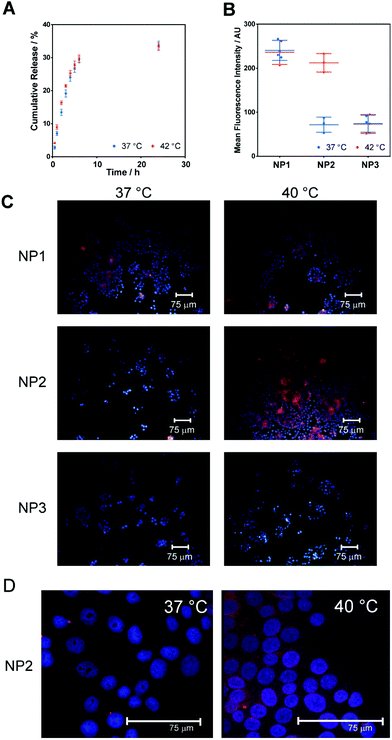 |
| | Fig. 2 (A) Cumulative release of rhodamine 6G from NP2 nanoparticles (formulated from polymer 2) at below (37 °C) and above TTT (42 °C). Points are mean ± one standard deviation for 2 replicates. (B) Mean of the fluorescence intensity of MCF7 cells exposed to R6G-loaded nanoparticles for 2 h as a function of temperature (below and above TTT) obtained by flow cytometry. Circles represent individual measurements, lines represent mean values and error bars represent one standard deviation. ***P < 0.001 for NP2 at the two temperatures. (C) Fluorescence micrographs of MCF-7 cells treated with thermoresponsive nanoparticles over clinically relevant temperature range (37 vs. 40 °C). (D) Higher resolution confocal micrographs of MCF-7 cells treated with R6G-loaded NP2 at 37 °C and 40 °C, showing enhanced signal from NP2-encapsulated R6G in cells. Nuclei are stained with DAPI (blue) while particles carry rhodamine 6G (red). | |
Effect of temperature on cellular uptake of thermoresponsive nanoparticles with different thermal transition temperatures
The effect of temperature on cellular uptake of nanoparticles NP1–3 was determined in the MCF-7 breast carcinoma cell line. All particle batches had similar sizes, zeta potentials and dye loading, such that the most important differences in their properties were likely to be their various thermal transition temperatures (33, 39 and 56 °C respectively). These transition temperatures were chosen to provide the three possible hydration states under physiological conditions, i.e. (i) permanently collapsed, (ii) hydrated under normal conditions but collapsed under mildly hyperthermic conditions, and (iii) permanently hydrated. Cells were incubated with the particles at 37 and 40 °C for 2 h before washing and analysis by flow cytometry and fluorescence microscopy.
Mean fluorescence intensity (MFI), measured by flow cytometry of cells treated with NP1–3 is given in Fig. 2B. It is evident that temperature had no effect on MFI after treatment with NP1 or NP3. However, the MFI of cells treated with NP1 was significantly higher than for those treated with NP3 at both temperatures (P < 0.001, 2-sided unpaired Student's t-test). In contrast, MFI of cells treated with NP2 was significantly affected by temperature (P < 0.001, 2-sided paired Student's t-test) with greatly enhanced fluorescence for cells incubated above the TTT of the particles. The temperature-dependent nature of cellular fluorescence was confirmed by microscopy (Fig. 2C). Again it is evident that fluorescence of cells treated with NP1 or NP3 was independent of incubation temperature while fluorescence of cells treated with NP2 was highly dependent on the changes temperature, specifically 37 and 40 °C, which are in fact below and above the TTT for NP2.
As the release profile of R6G from NP2 was only slightly affected by temperature the greatly enhanced fluorescence in cells treated above the thermal transition temperature was unlikely to be due to increased uptake of free R6G but instead due to increased particle uptake by the cells. This hypothesis was supported by the observation that cells treated with NP1 and NP3 showed fluorescence intensities correlating to treatment with particles above their TTT; i.e. cells treated with NP1 at 37 °C and 40 °C exhibited high rhodamine fluorescence as both temperatures were above the TTT for NP1, whereas low fluorescence was observed for cells treated with NP3 at both temperatures (TTT for NP3 is 56 °C). The enhanced uptake of particles above their thermal transition temperature was thus in agreement with the report of Salmaso et al. for non-ligand targeted pNIPAM-coated gold nanoparticles with MCF-7 cancer cell lines.12 We then proceeded to evaluate the potential of our nanoparticle assembly for the targeted delivery of a clinically pertinent cytotoxic agent, paclitaxel.
Paclitaxel (PTX) containing particles were formulated in a similar manner to the R6G particles (vide supra). Polymer solutions (50 or 100 mg in acetone) containing PTX (0.25, 0.5 or 1 mg mL−1) were precipitated into buffer as before (Table S3, ESI†). A PTX concentration of 0.5 mg mL−1 resulted in relatively high drug loading (0.4 and 0.58 wt%) with good particle recovery. In contrast, increasing PTX concentration to 1 mg mL−1 resulted in poor particle recovery (42 and 64%).
In order to increase encapsulation efficiency, formulation with a modified IPD method was attempted. Here, polymer and PTX were dissolved in acetone and the solvent was allowed to evaporate. The residual material was redissolved in acetone and precipitated into buffer as before. This method increased encapsulation efficiency from 50 to 76% while increasing particle recovery from 64 to 80% (Table S3, ESI†) and was subsequently used for formulation of particles, i.e.NP4, tested here.
The release of encapsulated PTX was studied at pH 7.4 and 4.5 as these are representative of extracellular and lysosomal pH, respectively. In both cases, release above and below the TTT was quantified (Fig. 3A). At pH 7.4, less than 10% of encapsulated PTX was released after 24 h and temperature had no effect on release rate. At pH 4.5, PTX release was more rapid with approximately 25% release after 24 h; again little effect of temperature was observed. The increased release rate at lower pH is expected as under these conditions the degradation of the PLGA core is considerably faster than at neutral pH allowing more PTX to diffuse into solution. The differences in release rates also imply that these particles would be suited to selective delivery as significant drug release would only occur at low, i.e. lysosomal, pH rather than the near neutral pH of the circulatory system thus reducing off-target effects.
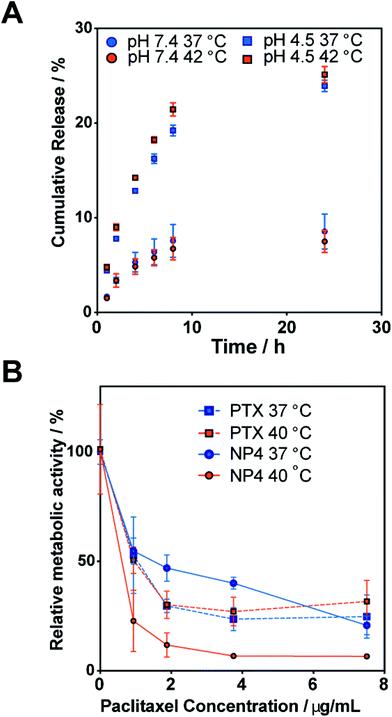 |
| | Fig. 3 (A) Release of paclitaxel from nanoparticles NP4 at pH 4.5 and 7.4, and above and below the thermal transition temperature. (B) Relative metabolic activity as measured by an MTT assay of MCF-7 cells after treatment with free paclitaxel or NP4 for 24 h above and below the thermal transition temperature. | |
When considered together, the data is strongly suggestive of a thermoresponsive polymer-mediated mechanism for cell entry, and an enhanced cytotoxicity through increased uptake above the thermal transition temperature of the nanoparticles. Similar thermoresponsive cytotoxicity was reported for thermo-sensitive micelles21 formed by coupling dicarboxylated poly(ethylene glycol) (PEG; Mw 2000) with poly(L-lactic acid)/poly(ethylene glycol)/poly(L-lactic acid) (PLLA/PEG/PLLA) triblock copolymers. The PLLA–PEG–PLLA micelles, in this case with encapsulated doxorubicin (DOX), showed 20-fold enhanced cytotoxicity at 42 °C against Lewis lung carcinoma cells when compared to that at 37 °C although the total amount of released DOX at 42 °C was only two times higher than that at 37 °C. This result was attributed to thermal switching of the PLLA–PEG–PLLA micelle to a more hydrophobic surface at the higher temperature, facilitating internalization of DOX-loaded micelles into the cells. Accordingly, there was clear precedent for a polymer-response-mediated uptake effect, though in our case we used responsive core–shell nanoparticles rather than thermoresponsive liposomes22 and thermo-responsive micelles21 used previously.
It is important to note that cytotoxicity effects observed in the variable temperature studies were not due to intrinsic temperature effects on the cells. Prior studies have shown that MCF-7 cells can tolerate temperatures up to 43 °C, i.e. above those used in our assays, without marked changes in viability or activity.23–25 The MTT assays in our study (Fig. S3, ESI†) showed that MCF7 cells showed similar viability at both incubation temperatures (i.e. 37 and 40 °C), a result consistent with prior literature showing that moderate hyperthermia (i.e. 38–42 °C) has no acute cytotoxic effect on cells.26,27 In addition, no cytotoxic activity was observed for the drug-free nanoparticles as cell viability was found to be in the range of 90–100% for applied polymer concentrations up to 4 mg mL−1 across both investigated temperatures (Fig. S3, ESI†). The biocompatibility of these NPs was in good accord with studies of related polymers.28–31 The results also demonstrated, as with previously published reports,21 that at 37 °C comparable cytotoxicity was observed for both free PTX and PTX-loaded NPs. These findings suggest that reduced side effects would be expected for these thermoresponsive NPs under normal physiological conditions. However, the incorporation of PTX into nanoparticles may enhance its anti-tumoural activity compared to the free PTX, which is attributed to the higher cellular uptake of NPs compared to the free drug.32 For example, Westedt et al. evaluated the anti-tumoural activity of free PTX and PTX-loaded PVA-g-PLGA NPs using primary RbVSMC cells in culture.33 Their results demonstrated that at high drug loading levels, free PTX was less toxic than when incorporated into nanoparticles. Similarly, the Preat group evaluated the in vitro cytotoxic activity of Taxol® and PTX-loaded PEG–PLGA NPs by the MTT assay using HeLa cell lines.34 In this example the IC50 value for HeLa cells decreased from 15.5 μg mL−1 for Taxol® to 5.5 μg mL−1 for the PTX-loaded NPs. Thus, in certain cases and for specific cell lines, one could anticipate an extra advantage for incorporation of PTX into responsive nanoparticles, as further enhanced uptake into tumour cells could occur relative to normal cells.
Although not investigated directly in this study, the results are encouraging for future in vivo applications since for delivery to tumours, PTX-loaded nanoparticles are likely to be administered intravascularly to circulate in a pH 7.4 environment. At this pH, minimal release of PTX is desired to minimise off-target effects. However, in the acidic (pH 5–6) environment typical of lysosomes (to which the NPs would probably be routed during the cellular uptake) the data showed enhanced release of PTX from the NP4 materials. This effect would have contributed to the enhanced cytotoxicity of nanoparticle delivered PTX compared to free drugs as apparent in the MTT assay, since the endolysosomal pathway is the ‘default’ route for nanoparticles of this type.35 Thus, in addition to the temperature-mediated localisation due to the thermoresponsive shell, the PTX-loaded pPLGA-b-(PEGMEMA-co-PPGMA) NPs have the properties of enhanced release in response to the physiological changes in the local environment. Similar pH dependent release profiles have been observed previously with PLGA nanoparticles encapsulating doxorubicin36 and rhodamine 6G37 due to accelerated degradation of the polymer at acidic pH. The fact that these profiles have been obtained with both hydrophilic compounds (DOX, R6G) and hydrophobic drugs (PTX) points to the key advantage of nanoprecipitated drug delivery systems, i.e. their ability to incorporate both hydrophilic and hydrophobic drugs.38,39
For practical application and regulatory requirements, drug delivery systems need to be well-defined. The thermoresponsive systems in this work were prepared via controlled polymerisation techniques (ATRP and ROP), enabling good control over molar masses and polydispersities, as well as well-defined thermal phase transitions.40 Clearly, these thermal transitions are highly sensitive to the molar ratios of the co-monomers which can be defined through monomer feed and order of addition. Variations in molecular weight could, in principle, be used to manipulate the encapsulation efficiency of drug compounds as well as their release profiles. Depending on the desired target and clinical applications of these systems, “On–Off” release or thermoresponsive release profiles could thus be tailored by varying the ratios of the monomers or by incorporation of different functionalities. Recent studies by the Chilkoti group have shown that precisely engineered biopolymers based on elastin-like-polypeptides (ELPs) can be tuned to self-assemble into nanoparticles in vivo over narrow temperature ranges, and that conjugates of these polypeptides with doxorubicin can target tumours effectively.13,41 These impressive data show that extension of the thermally-targeted nanoparticle delivery mechanism can operate in an animal model. We envisage that our modular core–shell synthetic polymer nanoparticles delivery can be used in an analogous means to ELP-derived responsive nanoparticles, but across a wider range of therapeutic compounds and broader formulation conditions. Experiments to evaluate these hypotheses are in progress.
Conclusions
In conclusion, we have demonstrated that particles formulated from PLGA-block-(PEGMEMA-co-PPGMA) polymers are suitable for encapsulation and temperature controlled delivery of drugs. The reduced cytotoxicity of encapsulated paclitaxel below the thermal transition temperature indicates the suitability of these particles for systemic delivery where minimal drug release to non-target sites is essential. As cellular uptake may be triggered by only a small increase in temperature, these particles hold promise for delivery to hyperthermic tissues such as those found at sites of inflammation or in tumours. Additionally, induced hyperthermia using thermal ablation techniques such as high-intensity focused ultrasound, should enable these materials to be activated for uptake by an external trigger, thus enabling targeting even where natural hyperthermia is absent. Current studies are investigating the potential for these systems to be used in vivo for targeted delivery of anticancer agents.
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (Grant EP/H005625/1) and by a scholarship to SRA from the University of Jordan. KJT thanks the Australian Research Council for funds under programs DP0880032, DP1094205, and FT110100284. We also thank Christine Grainger-Boultby, University of Nottingham for technical help.
Notes and references
- M. Nakayama and T. Okano, J. Drug Deliv. Sci. Technol., 2006, 16, 35–44 CAS.
- R. R. Sawant and V. P. Torchilin, Soft Matter, 2010, 6, 4026–4044 RSC.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- S. R. Abulateefeh, S. G. Spain, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Macromol. Biosci., 2011, 11, 1722–1734 CrossRef CAS.
- J. Xu and S. Liu, Soft Matter, 2008, 4, 1745–1749 RSC.
- K. T. Oh, H. Yin, E. S. Lee and Y. H. Bae, J. Mater. Chem., 2007, 17, 3987–4001 RSC.
- C. Stefanadis, C. Chrysochoou, D. Markou, K. Petraki, D. B. Panagiotakos, C. Fasoulakis, A. Kyriakidis, C. Papadimitriou and P. K. Toutouzas, J. Clin. Oncol., 2001, 19, 676–681 CAS.
- P. Wust, B. Hildebrandt, G. Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix and P. M. Schlag, Lancet Oncol., 2002, 3, 487–497 CrossRef CAS.
- S. J. Grainger, J. V. Serna, S. Sunny, Y. Zhou, C. X. Deng and M. E. H. El-Sayed, Mol. Pharm., 2010, 7, 2006–2019 CrossRef CAS.
- R. L. Manthe, S. P. Foy, N. Krishnamurthy, B. Sharma and V. Labhasetwar, Mol. Pharm., 2010, 7, 1880–1898 CrossRef CAS.
- F. Mastrotto, P. Caliceti, V. Amendola, S. Bersani, J. P. Magnusson, M. Meneghetti, G. Mantovani, C. Alexander and S. Salmaso, Chem. Commun., 2011, 47, 9846–9848 RSC.
- S. Salmaso, P. Caliceti, V. Amendola, M. Meneghetti, J. P. Magnusson, G. Pasparakis and C. Alexander, J. Mater. Chem., 2009, 19, 1608–1615 RSC.
- J. R. McDaniel, S. R. MacEwan, M. Dewhirst and A. Chilkoti, J. Controlled Release, 2012, 159, 362–367 CrossRef CAS.
- A. J. Simnick, M. Amiram, W. Liu, G. Hanna, M. W. Dewhirst, C. D. Kontos and A. Chilkoti, J. Controlled Release, 2011, 155, 144–151 CrossRef CAS.
- W. Liu, J. A. MacKay, M. R. Dreher, M. Chen, J. R. McDaniel, A. J. Simnick, D. J. Callahan, M. R. Zalutsky and A. Chilkoti, J. Controlled Release, 2010, 144, 2–9 CrossRef CAS.
- A. J. Gormley, K. Greish, A. Ray, R. Robinson, J. A. Gustafson and H. Ghandehari, Int. J. Pharm., 2011, 415, 315–318 CrossRef CAS.
- S. R. Abulateefeh, A. O. Saeed, J. W. Aylott, W. C. Chan, M. C. Garnett, B. R. Saunders and C. Alexander, Chem. Commun., 2009, 6068–6070 RSC.
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, R1–R4 CrossRef CAS.
- T. Govender, S. Stolnik, M. C. Garnett, L. Illum and S. S. Davis, J. Controlled Release, 1999, 57, 171–185 CrossRef CAS.
- J. K. Oh, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 3161–3167 CrossRef CAS.
- K. Na, K. H. Lee, D. H. Lee and Y. H. Bae, Eur. J. Pharm. Sci., 2006, 27, 115–122 CrossRef CAS.
- D. Needham, G. Anyarambhatla, G. Kong and M. W. Dewhirst, Cancer Res., 2000, 60, 1197–1201 CAS.
- D. Z. Chen, Q. S. Tang, X. D. Li, X. J. Zhou, J. Zang, W. Q. Xue, J. Y. Xiang and C. Q. Guo, Int. J. Nanomedicine, 2012, 7, 4973–4982 CrossRef CAS.
- D. Baba, Y. Seiko, T. Nakanishi, H. Zhang, A. Arakaki, T. Matsunaga and T. Osaka, Colloids Surf., B: Biointerfaces, 2012, 95, 254–257 CrossRef CAS.
- X. X. Xie, X. F. Shao, F. P. Gao, H. K. Jin, J. M. Zhou, L. H. Du, Y. Y. Zhang, W. W. Ouyang, X. W. Wang, L. Y. Zhao, X. D. Zhang and J. T. Tang, Oncol. Rep., 2011, 25, 1573–1579 CAS.
- M. Urano, M. Kuroda and Y. Nishimura, Int. J. Hyperthermia, 1999, 15, 79–107 CrossRef CAS.
- W. C. Dewey, Int. J. Hyperthermia, 1994, 10, 457–483 CrossRef CAS.
- W. Wang, H. Liang, L. Hamilton, M. Fraylich, K. Shakesheff, B. Saunders and C. Alexander, Adv. Mater., 2009, 2118, 1809–1813 CrossRef.
- A. O. Saeed, S. Dey, S. M. Howdle, K. J. Thurecht and C. Alexander, J. Mater. Chem., 2009, 19, 4529–4535 RSC.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671–678 CrossRef CAS.
- Y. Dong and S.-S. Feng, Int. J. Pharm., 2007, 342, 208–214 CrossRef CAS.
- U. Westedt, M. Kalinowski, M. Wittmar, T. Merdan, F. Unger, J. Fuchs, S. Schäller, U. Bakowsky and T. Kissel, J. Controlled Release, 2007, 119, 41–51 CrossRef CAS.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Préat, J. Controlled Release, 2009, 133, 11–17 CrossRef CAS.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- T. Betancourt, B. Brown and L. Brannon-Peppas, Nanomedicine, 2007, 2, 219–232 CrossRef CAS.
- T. Betancourt, K. Shah and L. Brannon-Peppas, J. Mater. Sci.: Mater. Med., 2009, 20, 387–395 CrossRef CAS.
- J. Kreuter, Pharm. Acta Helv., 1983, 58, 242–250 CAS.
-
J. Kreuter, in Colloidal Drug Delivery Systems, ed. J. Kreuter, M. Dekker, 1994, pp. 219–342. ISBN 0824792149 Search PubMed.
- J.-F. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2009, 12, 343–353 CrossRef CAS.
- D. J. Callahan, W. Liu, X. Li, M. R. Dreher, W. Hassouneh, M. Kim, P. Marszalek and A. Chilkoti, Nano Lett., 2012, 12, 2165–2170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2bm00184e |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a