Cell attachment and proliferation on high conductivity PEDOT–glycol composites produced by vapour phase polymerisation
Elise M.
Stewart
a,
Manrico
Fabretto
*bc,
Mischa
Mueller
c,
Paul J.
Molino
a,
Hans J.
Griesser
b,
Robert D.
Short
c and
Gordon G.
Wallace
*a
aARC Centre of Excellence for Electromaterials Science, Innovation Campus, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: gwallace@uow.edu.au; Fax: +61 2 4298 1499; Tel: +61 2 4221 3127
bIan Wark Research Institute, University of South Australia, Mawson Lakes, SA 5095, Australia. E-mail: rick.fabretto@unisa.edu.au; Fax: +61 8 8302 3683; Tel: +61 8 8302 3675
cMawson Institute, University of South Australia, Mawson Lakes, SA 5095, Australia. Fax: +61 8 8302 5639; Tel: +61 8 8302 5612
First published on 4th December 2012
Abstract
High conductivity poly(3,4-ethylene dioxythiophene) (PEDOT) was synthesised using vacuum vapour phase polymerization (VVPP). The process produces PEDOT composites which incorporate glycol within the polymer. To assess biocompatibility, a suite of analytical techniques were utilised in an effort to characterise the level of glycol present and its impact on cell attachment and proliferation. A small decrease in fibroblast cell attachment and proliferation was observed with increasing glycol content within the PEDOT composite. Keratinocyte cell attachment and proliferation by comparison showed an increase. As such, the results herein indicate that cell attachment and proliferation depends on the individual cell lines used and that the impact of glycol within the PEDOT composite is negligible. This positive outcome prompted investigation of this polymer as a platform for electro-stimulation work. Application of oxidising and reducing potentials to the PEDOT composite were utilised to examine the effect on biocompatibility. Significant effects were seen with altered protein presentation on the reduced surface, and lower mass adsorbed at the oxidised surface. Keratinocytes interacted significantly better on the reduced surface whereas fibroblasts displayed dependence on protein density, with significantly lower spreading on the oxidised surface. Understanding how proteins interact at electrically biased polymer surfaces and in turn affect cell behaviour, underpins the utilisation of such tunable surfaces in biomedical devices.
1. Introduction
The use of inherently conducting polymers (ICPs) as an alternative to traditional metallic materials for charge transport has, in recent times, seen these materials subjected to intense investigation. Of the many conducting polymers that have been synthesised and studied, poly(3,4-ethylenedioxythiophene) (PEDOT) is arguably one of the most highly conductive and environmentally stable.1–4 The facile manner in which PEDOT can be synthesised using a variety of protocols, such as wet chemical oxidation, electrochemical polymerisation or vapour phase polymerisation (VPP), has resulted in it becoming one of the most commonly used ICPs. Importantly the energetic state of the surface, the conductivity and the optical transmission of PEDOT can be altered by changing the doping level within the polymer. This can be achieved either by electrical or chemical means and exploiting this phenomenon has resulted in a plethora of potential applications such as power storage and generation,5 organic transistors (OFETs),6,7 and drug delivery devices, bioactive-platforms and biosensors.8For biological applications, such as tissue engineering, it is important to create a structure or scaffold that provides physical and/or chemical support conducive to interactions between the substrate and cells, as well as between neighbouring cells.9–11 As such, the surface properties of biomaterials are critically important in that they determine the nature of interactions with cells via the protein mediators of such interactions.12 Modification of the conducting polymer surface properties can be effected by altering the polymer composition,13 surface roughness,14 the type of dopant,15 and the doping level,16 as well as the redox state of the polymer.15 Each of these properties has been shown to affect cell behaviour.17–20 Furthermore, the application of electrical stimuli has long been known to influence the manner in which nerves,21 cartilage,22 skin tissue23 and bone24 heal. Therefore, careful manipulation of these physical, chemical or electrical cues has the potential to influence cell behaviour in some desired manner.
While numerous bio-compatibility studies have been performed on polypyrrole25–28 and to a lesser extent PEDOT,29,30 interestingly, a survey of the current literature revealed no bio-material studies with VPP PEDOT. Previous studies31,32 have shown that the conductivity of vapour phase polymerised (VPP) PEDOT can be as high as ca. 1500 S cm−1, which is only one order of magnitude smaller than some metals or alloys. Such a result opens up the possibility of using VPP PEDOT as both the bio-platform and the electrode material for use in electrically active devices. One of the most commonly used oxidants in the VPP process is Fe(III) Tosylate; however, its use is not without problems. Due to its high (apparent) reactivity, PEDOT films having poor conductivity due to conjugation defects have been reported.33 To overcome this problem, various methods have been employed to reduce the (apparent) reactivity of the oxidant. One method utilises a glycol based surfactant as a means of reducing the reactivity of the oxidant solution via a steric hindrance mechanism.34 The consequence of using glycol, however, is that a PEDOT/glycol composite is formed during polymer synthesis.32 Even though glycol on a biomaterial surface is known to inhibit the attachment of proteins and cells,35,36 and is at odds with the general requirements for most tissue engineering applications, the possibility of utilising such a high conductivity polymer for electrically stimulated cell studies would seem to be highly desirable. The effect of glycol on protein adsorption and cell adhesion is dependent on the molecular weight and distribution (i.e. surface density) present on the surface, as it is generally attributed to being a steric hindrance phenomenon.37,38 Previous studies39–41 using various glycols have either coated the entire substrate in an effort to inhibit cell attachment, or have patterned the surface in some manner in an attempt to produce directed/preferential cell attachment and proliferation. Irrespective of the desired outcome being sought, these previous studies have coated glycol onto the respective substrates. To the best of our knowledge, biological studies have not been performed with polymers containing glycol inherently within the polymer matrix, rather they have simply being adsorbed onto the surface. An interesting question is whether incorporated glycol produces, by its inherent surfactant activity, a surface concentration high enough to affect cell attachment and spreading.
One of the first steps in establishing the suitability of a new material for bio-engineering purposes is to confirm its biocompatibility using in vitro tests with a variety of cell types. As stated, previous studies with different cells have shown that PEDOT is indeed capable of supporting cell attachment and proliferation. This study outlines the synthesis of ultra-high conductivity PEDOT using the VPP technique, and its fabrication into thin films suitable for in vitro use. Furthermore, a suite of surface characterisation techniques were employed in an effort to elucidate the manner in which glycol is incorporated into the PEDOT polymer composite. Our results indicate that the incorporation of glycol has minimal impact on cell attachment and proliferation. The conducting nature of the polymer was also exploited to examine the effects of applying oxidising or reducing potentials on the adsorption of proteins and cell behaviour on this biased surface.
2. Materials and methods
2.1. Materials
Fe(III) Tosylate (Fe(Tos)3) was purchased from H. C. Starck as a 40 wt% solution in butanol (Baytron CB 40). 3,4-Ethylenedioxythiophene (EDOT) monomer and the tri-block polymer poly(ethylene glycol–propylene glycol–ethylene glycol) (PEG–PPG–PEG, referred to as ‘glycol’), Mw = 2900 Da. were obtained from Aldrich. Tris base, ethylenediaminetetraacetic acid (EDTA), Triton X-100 and phosphate buffered saline (PBS) tablets were purchased from Sigma. Dulbecco's Modified Eagles Medium (DMEM), Fetal Bovine Serum (FBS), Trypsin-EDTA, QuantIt Pico-Green Reagent, Phalloidin-Alexa 488, 4′,6-diamidino-2-phenylindole (DAPI) and human plasma fibronectin (Fn) were purchased from Invitrogen. All chemicals were used as received.2.2. Vacuum vapour phase polymerisation of PEDOT
PEDOT samples were synthesised on glass microscope slides. The slides were washed using a mild detergent, followed by an ethanol and high purity water rinse. Prior to spinning the oxidant the substrates were air plasma treated (PDC-32G, Harrick Inc.) for 2 min. The 40 wt% Fe(Tos)3 solution was further diluted with butanol to produce either a 16 or 26.6 wt% solution. Glycol in the weight range 0 to 65 wt% (with respect to Fe(Tos)3) was then added to the oxidant solution. The solution was spin-coated (400B-6NPP, Laurell Technologies Inc.) at a speed of 1900 RPM for 20 seconds and placed on a 70 °C hot plate for 1 minute to evaporate the butanol. Samples were removed from the hotplate and immediately placed into a 115 L vacuum chamber oven (Binder, Germany) set to 35 °C. The chamber was pumped down to 45 mbar before allowing EDOT monomer and water vapour ingress. Samples were removed after 25 min and placed on a 70 °C hotplate for 2 minutes to anneal the polymer. The sample was then carefully rinsed in an ethanol bath to remove the spent oxidant.2.3. X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) was performed using a Kratos AXIS Ultra DLD spectrometer, using monochromatic AlKα radiation (hν = 1486.7 eV). The unit is equipped with a magnetically confined charge compensation system. Spectra were recorded using an acceleration voltage of 15 keV at a power of 225 W.Survey spectra were collected with a pass energy of 160 eV and the analysis area was 300 × 700 μm. High-resolution spectra were obtained using a 20 eV pass energy and an analysis area of ≈300 700 μm. Data analysis was performed using CasaXPS software (Casa Software Ltd). All binding energies were referenced to the low energy C1s peak at 285.0 eV. Core level envelopes were curve-fitted with the minimum number of mixed Gaussian–Lorentzian component profiles. Curve fitting was performed with a Gaussian–Lorentzian mixing ratio (typically 30% Lorentzian and 70% Gaussian functions), and the full width at half maximum and the positions and intensities of the peaks unconstrained.
2.4. Time-of-flight secondary ion mass spectrometry
ToF-SIMS analyses were performed with a PHI TRIFT V nanoTOF instrument (PHI Electronics Ltd, USA). A 30 keV, pulsed primary 197Au+ ion beam was used to sputter and ionise species from each sample surface. Spectra were acquired for 60 seconds from an area of 100 × 100 μm. The corresponding total primary ion fluence was less than 1 × 1012 ions per cm2 (operating within the static SIMS regime1). A dual beam charge neutralization system using a combination of low energy ions (≤10 eV) and electrons (≤25 eV) was employed to provide improved charge neutralisation. Mass axis calibration was performed with CH3+, C2H5+ and C3H7+ in positive mode and CH−, C2H− and Cl− in negative mode. A mass resolution of m/Δm > 7000 at nominal m/z = 27 amu (C2H3+) was achieved.2.5. Conductivity
Resistivity, R, (Ω/□) was measured using a four-point probe (RM3, Jandel Engineering), tip radius 100 μm, 60 g preset load and measurements were taken at room temperature (22 ± 2 °C) and humidity (35% ± 5%). Results are the average of at least nine measurements. AFM images and measurements (NTEGRA, NT-MDT) were performed in tapping mode. Polymer film thickness, t, was measured by carefully scoring the surface using a soft scalpel and performing line scans across the groove. Reported thickness is the average of at least 4 line scans. PEDOT conductivity, σ, (S cm−1) is given by;| σ = 1/(R × t) | (1) |
2.6. Contact angle goniometry measurements
Contact angle measurements were performed on a custom made goniometer. Droplet shapes were recorded with a M-852 colour CCD camera attached to an Olympus SZ-PT stereomicroscope. Droplet shapes were analysed using the drop snake software.42 The advancing contact angle was recorded by slowly increasing the drop volume and ensuring that the contact line was not “pinned” by any chemical or physical defect. The advancing contact line was halted and then an image captured after 3 seconds. The receding contact angle was recorded by allowing the drop to evaporate and waiting until the contact line transitioned from an advancing state to a receding state, allowing the contact line to move freely. Care was taken to ensure that the receding contact line did not exhibit any “pinning” prior to capturing the image used to calculate the contact angle. The results are the average of at least 5 captured images and use both the right and left contact lines of the drop.2.7. Atomic force microscopy
PEDOT film roughness measurements (RMS) were performed by scanning the surface with an atomic force microscope (NTEGRA, NT-MDT). Scans were performed over two length scales (area of 100 μm2 and 1 μm2) at a scan speed of 1 Hz. The root mean square (RMS) roughness values were calculated using the NT-MDT image analysis software.2.8. Protein adsorption
2.9. Cell culture
Mouse fibroblast (3T3) and human keratinocyte (HaCaT) cell lines, were originally sourced from the ATCC (American Type Culture Collection) and were used as models for epidermal cell types. Each cell type was cultured in DMEM (Dulbecco's Modified Eagle's Medium, Invitrogen, Sydney, Australia) containing 4 mM L-glutamine and 10% Fetal Bovine Serum (FBS). Cells were cultured at 37 °C in a humidified, 5% CO2 atmosphere and were subcultured twice weekly.2.10. Cell adhesion and proliferation on VPP PEDOT
To quantify the metabolic activity of cells attached to the polymers at various times, unbound cells were removed, and polymers/cells washed twice in serum free media before cell number was quantified using the CytoTox96TM LDH Assay Kit (Promega) according to manufacturer's specifications. Cell lysate was incubated with assay reagent for 30 min at room temperature, before the reaction was stopped. Absorbance measurements (adjusted for background) were taken at 490 nm using a SpectraMax 190 (Molecular Devices).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2. Samples were placed into a humidified 37 °C incubator with 5% CO2 atmosphere for 12 h prior to application of CalceinAM (1/200 dilution in culture media) and a further 15 min incubation. Media was then removed and replaced with PBS prior to imaging using an AxioImager microscope fitted with an AxioCAM Mrm camera. Average cell area per cell body was then calculated using ImageJ software and compared between bias conditions.
000 cells per cm2. Samples were placed into a humidified 37 °C incubator with 5% CO2 atmosphere for 12 h prior to application of CalceinAM (1/200 dilution in culture media) and a further 15 min incubation. Media was then removed and replaced with PBS prior to imaging using an AxioImager microscope fitted with an AxioCAM Mrm camera. Average cell area per cell body was then calculated using ImageJ software and compared between bias conditions.
2.11. Immunocytochemistry
Cell morphology was investigated on the polymers at different time points following cell incubation on each substrate as described above. Cells were rinsed twice in PBS to remove unbound cells, then fixed using 50% (v/v) methanol:acetone for 5 min on ice, followed by two subsequent PBS washes. Samples were then incubated with the actin stain Phalloidin-Alexa 488 (diluted 1:200 in PBS/1% BSA) for 20 min at room temperature in the dark, rinsed twice in PBS, and incubated with 1 μg mL−1 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 5 min at room temperature in the dark. Solutions were removed and replaced with fresh PBS then imaged using an AxioImager microscope fitted with an AxioCAM Mrm camera and overlayed using AxioVison 4 software (Zeiss).
3. Results
3.1. Vapour phase polymerisation and PEDOT conductivity
The goal was to produce PEDOT having as high a conductivity value as possible, with the aim being to utilise this substrate in electro-stimulation cell work. With this in mind a series of VPP PEDOT films were synthesised by systematically increasing the amount of PEG-PPG-PEG (glycol surfactant) in the oxidant solution from 0 to 65 wt%. Given that the oxidant is spin-cast onto the substrate and then dried on a hot plate to remove unbound solvent, the glycol content presented in Fig. 1 is given as a weight percent of the dry-oxidant layer, rather than as a weight percent of the original solution. With no glycol addition, the conductivity of the PEDOT film was 200 S cm−1. As the glycol weight percentage was increased, a corresponding increase in conductivity was recorded, with a maximum conductivity of 1486 S cm−1 being achieved at a glycol loading of 48 wt%. Increasing the weight percentage beyond this level resulted in decreased conductivity, with 783 S cm−1 being recorded at the highest loading of 65 wt%. The rationale for pursuing high conductivity PEDOT as a bio-compatible material is that the polymer could serve as an effective electrode biomaterial for electro-stimulation cell work, a feature which has received an increased level of investigation.43,44 To ascertain whether any of the glycol that is contained within the oxidant layer becomes embedded in the synthesised PEDOT polymer, a combination of analytical techniques were used.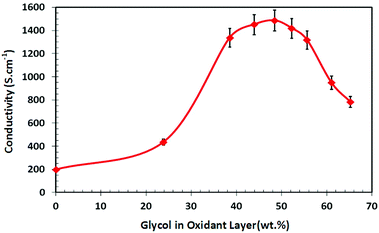 | ||
| Fig. 1 VPP PEDOT conductivity as a function of weight percentage of the glycol surfactant within the oxidant layer. (Glycol content within PEDOT capped at 16 wt%, see Fig. 2.) | ||
3.2. XPS analysis of VPP PEDOT
Surface located glycol is known to have detrimental effects on cell attachment and proliferation. Previous work45 synthesising PEDOT had indicated that using oxidant solutions with glycol additives resulted in films that contained glycol, in effect producing a PEDOT–glycol composite material. This being the case it was of paramount importance to quantify the amount of glycol present and its distribution and domain sizes. Does the glycol reside uniformly within the polymer matrix or does it become enriched at the surface? Is it a homogeneous or heterogeneous distribution? XPS spectra were collected for the various PEDOT samples and the O1s peak was used to quantify the amount of glycol by de-convoluting the peak into its constituent peaks (example given in ref. 45). Not surprisingly, peak fitting revealed no glycol peak for the sample made without glycol in the oxidant layer, but did show glycol present in increasing amounts for all other samples. Fig. 2 shows the level of glycol within the PEDOT film as a function of glycol content within the oxidant layer. As can be seen, the glycol content within the PEDOT film increased until it formed a plateau (≈16 wt%) at a corresponding glycol content within the oxidant layer of ≈48 wt%. Since XPS measures the approximate top 7–10 nm of a sample46 the question still remains; is the glycol present only on the surface, or embedded evenly throughout the PEDOT matrix, or distributed in an inhomogeneous manner.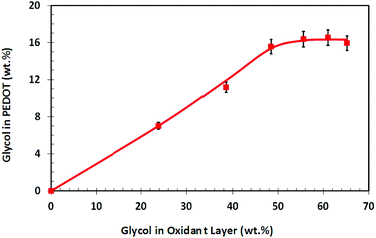 | ||
| Fig. 2 Weight percentage of glycol surfactant within the PEDOT polymer as a function of the glycol content in the oxidant layer. | ||
3.3. Contact angle measurements
Contact angle (CA) measurements are extremely sensitive to the energetic state of a surface but unambiguous quantitative analysis requires smooth homogeneous surfaces, whereas our PEDOT samples possess substantial roughness (Fig. 3). Thus, we have used (CA) measurements as a qualitative measure. The results (Table 1) for both advancing, θa, and receding, θr, contact angles show a general trend downwards with increasing glycol content originally contained within the oxidant layer. This result is at odds with the XPS data which showed that glycol within the PEDOT film reached a plateau of ≈16 wt% at a corresponding 48 wt% glycol within the oxidant layer. Based on the XPS data one would reasonably have expected the same contact angles to be recorded for both 48 and 61 wt% glycol within the oxidant layer, but this was not the case. Separating the effects of physical roughness and chemical heterogeneity is problematic. To resolve the issue, AFM imaging was performed in an effort to obtain physical topography measurements and ToF-SIMS analysis was conducted, as the latter technique provides compositional information at the uppermost surface of a sample (i.e. top 1–2 nm).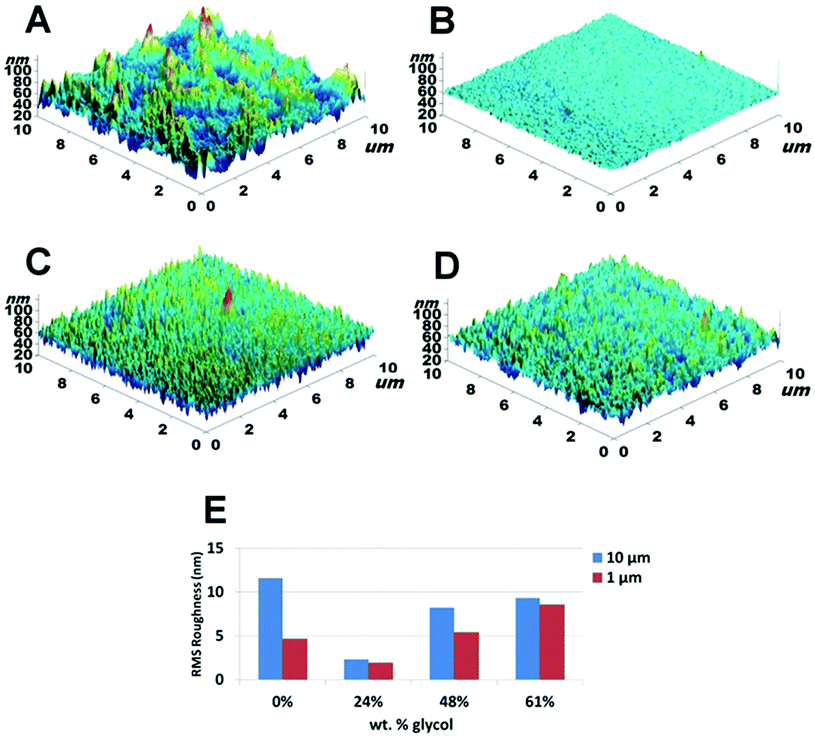 | ||
| Fig. 3 AFM topography images for samples made with: (A) 0%; (B) 24%; (C) 48% and (D) 61% glycol in oxidant layer. (E) The RMS roughness measured at length scales of 1 and 10 μm. (Note: RMSstd dev. < 10% relative for all samples and measured scan areas.) | ||
| Glycol in oxidant layer (wt%) | Contact angle (θa) | Contact angle (θr) |
|---|---|---|
| 0 | 63 ± 4 | 24 ± 2 |
| 24 | 61 ± 2 | 22 ± 4 |
| 48 | 54 ± 2 | 21 ± 4 |
| 61 | 49 ± 3 | 16 ± 3 |
3.4. AFM analysis
Fig. 3 shows 3D-images and RMS roughness for PEDOT films synthesised with varying amounts of glycol in the oxidant layers. Calculated RMS roughness values were obtained for 1 μm and 10 μm scans (length scales appropriate to cell size) and are given in Fig. 3E. Qualitatively the images show that there is a clear difference between the topography of the PEDOT film without glycol and the films synthesised with glycol. While the topography images of the PEDOT films that have glycol incorporated show a homogenous distribution of roughness over the scan areas of interest, the film without glycol (Fig. 3A) shows an inhomogeneous distribution. This can be seen by comparing the difference in roughness between the 1 μm and 10 μm scans where a marked increase in roughness between the two length scales is observed. Within the series of PEDOT films containing glycol, the roughness increased with increasing glycol content, with a maximum roughness of 9.3 nm recorded for the 61 wt% glycol sample at a scan size of 10 μm. Interestingly, phase contrast imaging showed no discernible differences at length scales down to 500 nm indicating that the incorporation of the glycol surfactant within the PEDOT film was either homogeneous and, importantly, at a length scale below that examined (i.e. <500 nm), or that the glycol resided below the top surface layer. To resolve this ToF-SIMS analyses were performed on the PEDOT films.3.5. ToF-SIMS analysis of VPP PEDOT
Positive ToF-SIMS spectra were analysed to study the extent of changes in surface chemistry with the incorporation of glycol into the PEDOT films. As ToF-SIMS has a high surface sensitivity (ca. 1–2 nm), only the top most surface layer is analysed. This aspect is important, as it is the top layer which governs the interactions of proteins and cells with the substrate.In Fig. 4 the normalised counts for major fragment ions assignable to PEG (45 amu) and PPG (59 amu) from the glycol surfactant are shown. The peak intensities were normalised using 126 peaks in each ToF-SIMS spectrum. The data show that both moieties were present on the PEDOT surface, and that their concentration increased as the amount of glycol within the oxidant layer was increased. Small counts were recorded at 45 and 59 amu for 0 wt% glycol due to PEDOT-derived fragment ions with very similar masses to the glycol-derived fragment ions. Since glycol is a high-energy material, a corresponding decrease in the contact angle should result as the concentration increases at the surface of the PEDOT film. Thus, the ToF-SIMS spectra are in qualitative agreement with the contact angle data. The XPS data on the other hand are not in agreement as they show a glycol plateau at 16 wt%, yet the contact angle decreased further as glycol within the oxidant layer was increased beyond 48 wt%. Given the fact that XPS measures the first 7–10 nm of a material, compared to ToF-SIMS which measures the top 1–2 nm, one can reconcile the two results by inferring that a glycol-rich surface is formed with a relatively less concentrated region just below, as the glycol within the oxidant layer is increased beyond 48 wt%. This surface enrichment is not measurable by XPS due to its monolayer nature and the modest additional amount on the surface.
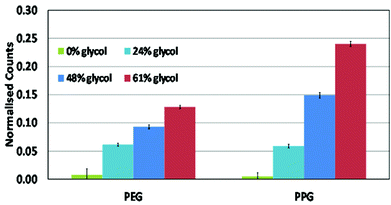 | ||
| Fig. 4 ToF-SIMS relative measurements of the PEG and PPG moieties in the glycol surfactant, which now resides within the PEDOT polymer surface (i.e. top 1–2 nm) for the different glycol weight percents originally in the oxidant layer (95% confidence level with N = 10 samples). | ||
3.6. Protein adsorption on VPP PEDOT
| Glycol in oxidant layer (wt%) | Surface area (% increase) | Mass adsorbed (ng cm−2) | Δf/ΔD |
|---|---|---|---|
| 0 | 7.6 | 1319 ± 137 | −20.7 |
| 24 | 0.7 | 1558 ± 103 | −15.5 |
| 48 | 2.8 | 1271 ± 106 | −16.1 |
| 61 | 1.5 | 1539 ± 62 | −15.8 |
Frequency versus dissipation curves indicated that there was no significant difference in the properties of the adsorbed protein layer, with a single binding event occurring.47 The viscoelastic properties of the protein layers appeared to be comparable between the three surfaces containing glycol, with the exception being the layer deposited on the PEDOT without glycol (Table 2, Δf/ΔD) which was slightly stiffer. The conformation of the protein is thus likely to be similar on each surface, which is important as alterations in surface morphology, chemistry or surface charge can cause differences in the viscoelastic properties of the protein layer. These properties indicate a similar water content of the adsorbed protein layer on each surface, allowing comparison between mass adsorbed per unit area for each surface.47,49
The mass of protein adsorbed to each surface was examined as the surface properties of the PEDOT films, particularly the contact angle and ToF-SIMS results suggested that there might be significant effects on the adsorption of protein due to the increased level of glycol present (Fig. 4). AFM indicated that no phase segregation was evident on the length scales investigated. The protein adsorption, however, did not appear to be significantly affected by the physical or chemical surface properties (Table 2), indicating that the glycol coverage on all samples was below the density required to significantly reduce protein adsorption.
The mass adsorbed to biased surfaces was determined utilising the Voigt model method as previously explained. A general trend is seen with more mass adsorbed to the reduced samples compared with the oxidised samples (Fig. 5). In order to compare the masses directly, the mechanical properties of each layer should be similar. The Δf/ΔD ratio provides an indicator of this measurement and shows that the mechanical properties of the fibronectin layers adsorbed to the biased surfaces are indeed different.
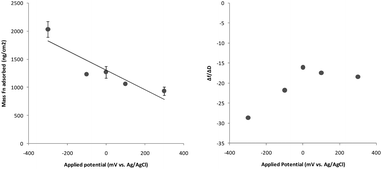 | ||
| Fig. 5 Properties of the fibronectin protein layer adsorbed to the 48 wt% PEDOT surface. (a) Representation of the trending change in adsorbed mass as a function of charge applied. Means represent the average of two samples and error bars represent the range. (b) Changes in mechanical properties as a function of charge applied represented by the Δf/ΔD ratio. | ||
Following the application of ±100 mV or +300 mV to the PEDOT substrate, the adsorbed layer of fibronectin did not appear to be significantly mechanically different to the adsorbed layer on the unbiased surface. However the application of higher reducing potential significantly altered the mechanical properties of the fibronectin layer. The −300 mV reducing potential caused the adsorbing layer to form a denser, less hydrated protein film, as indicated by the significantly higher Δf/ΔD ratio. Whereas the application of an oxidising potential did not alter the Δf/ΔD ratio significantly from the unbiased protein layer properties. These results might suggest a significant difference in protein presentation on biased surfaces,50 particularly at reducing potentials.
3.7. Cell adhesion and proliferation on VPP PEDOT
The following PEDOT samples were selected for cell work: 0, 24, 48 and 61 wt% glycol addition. The adhesion of both fibroblast and keratinocyte cell lines was examined following a 2 h incubation on the surface of the polymers (Fig. 6). Unbound cells were removed by washing and cell numbers quantified. The highest number of fibroblast cells were retained on TCP substrate after 2 h incubation (Fig. 6a). This number was significantly higher (p < 0.05) than adhesion to any of the PEDOT surfaces. Comparing between the PEDOT surfaces however, there was only a significant difference between the number of cells bound after 2 h on 0 and 61 wt%. This indicated that at the extremes of glycol incorporation there is a significant effect on cell adhesion; however, mid-range glycol incorporation does not significantly alter fibroblast cell adhesion. A similar response was seen with keratinocytes, where a significantly higher number of cells (p < 0.05) adhered to the TCP surface than to any of the PEDOT surfaces at 2 h (Fig. 6b).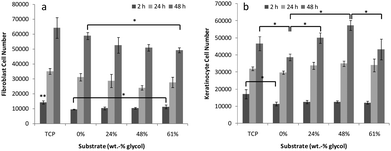 | ||
| Fig. 6 Cell adhesion and proliferation data of (a) 3T3 fibroblasts and (b) HaCaT keratinocytes on PEDOT substrates over 48 h. Means are averages of 4 replicates and error bars are standard error. * Indicates significant difference between indicated measurements, ** indicates difference from all other measurements at the same time point. | ||
Following 48 h incubation on the surfaces, the fibroblast population remained at a higher number on the TCP substrate compared to the PEDOT substrates, however, good proliferation was maintained on all surfaces tested. The PEDOT substrates supported the proliferation of fibroblasts regardless of the amount of surface exposed glycol (i.e. surface energy) or the physical roughness. The initial difference in cell adhesion observed at 2 h did not impact on the proliferation of cells on the PEDOT surface with 0 wt% glycol, with this surface supporting a higher growth rate than the other PEDOT substrates. At 48 h a significantly higher cell number was reached on the 0 wt% glycol than on 61 wt% glycol surface. The keratinocyte cell line proliferated best on the 48 wt% substrate, with a significantly higher cell number reached than on 0 or 61 wt% after 48 h. The 0 wt% PEDOT material supported the lowest proliferation rate, significantly below that on TCP, 24 and 48 wt%.
Taken together, these results suggest that the cell behaviour on these surfaces is cell line dependent, with 0 wt% supporting the lowest adhesion but highest proliferation rate of fibroblasts, and the lowest adhesion and proliferation rate of keratinocytes. The 0 wt% glycol substrate supported the lowest cell adhesion after 2 h compared to the glycol composite PEDOT surfaces. This was contrary to speculation that the surface presence of glycol would inhibit this binding event. Based on these results, it is likely that there are other physicochemical factors acting on the cell adhesion and proliferation kinetics.
3.8. Cell morphology on VPP PEDOT
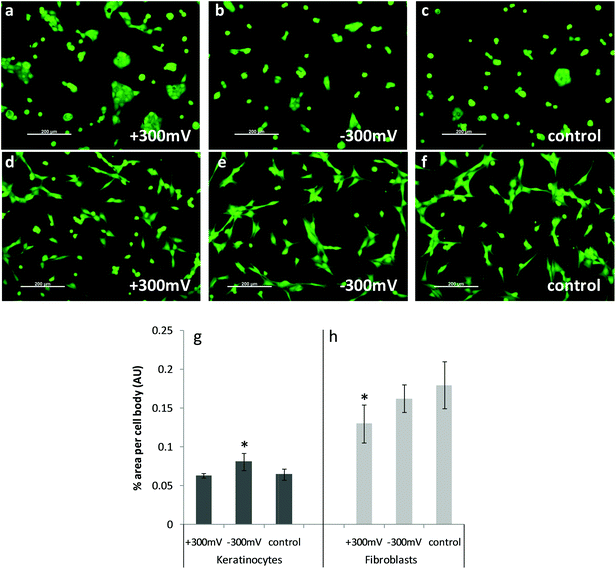 | ||
| Fig. 7 Calcein stained images of live cells on PEDOT 48 wt% glycol which were biased prior to incubation with complete media, followed by cell seeding. Keratinocytes on variously charged surfaces (as indicated on image) are shown in (a) to (c); with fibroblasts shown in (d) to (f). Graphs shown in (g) and (h) show relative % area per cell body for each cell type. Means are averages of >6 analysed images with error bars indicating the standard error of the mean. * Indicates significant difference to other measurements within the group. | ||
4. Discussion
4.1. Physical aspects of VPP PEDOT polymers
Ex vivo tissue engineering offers a long term adjunct to conventional cell replacement methodologies, such as donor transplants or grafting, for the repair or replacement of damaged tissue. Within the context of materials engineering, the ability to successfully create an artificial environment conducive to desired cell proliferation will depend on the characteristics that can be tailored into the candidate host materials. Of the many possibilities, conducting polymers have shown considerable potential and, as such, have received significant attention in the bio-materials literature. The ability to alter the physical, chemical and electrical properties of these polymers opens the door to a vast array of possibilities. The focus of this study was to investigate whether VPP PEDOT could be used as a host substrate that would support in vitro studies, with the longer term goal of also using the polymer as the electrode in electro-stimulation studies.The synthesis of high conductivity PEDOT can be accomplished using several facile methods, of which one is VPP. One of the easiest and most reproducible methodologies employs the surfactant glycol to moderate the polymerisation rate, resulting in high conductivity PEDOT. This comes at a cost, however, as the use of glycol results in the formation of a PEDOT–glycol composite material. There are numerous studies reporting that the addition of glycol onto substrate surfaces has an adverse or inhibiting effect on the adhesion and proliferation of different cell lines. To this end a systematic study was conducted to ascertain the presence of glycol on the surface of VPP PEDOT, and whether it was at a surface density that would be problematic.
The highest conductivity PEDOT obtained in this study was ca. 1500 S cm−1. This was achieved using an oxidant layer loading of 48 wt% glycol, which produced a PEDOT polymer composite with 16 wt% (refer Fig. 2) glycol. Increasing the glycol loading within the oxidant layer did not result in any additional glycol being present in the PEDOT according to XPS data. Interestingly, however, the advancing and receding contact angles decreased further when the glycol loading in the oxidant layer was increased to 61 wt%, even though XPS data indicated that the level of glycol had reached a plateau. XPS is able to measure down to a depth of approximately 7–10 nm, and so cannot be considered a true surface sensitive technique. To elucidate this apparent conflict ToF-SIMS was utilised, as the penetration depth is only of the order of 1–2 nm. The results (Fig. 4) showed that the topmost layer did indeed contain more glycol, even though XPS had indicated that the total glycol content had reached a plateau of 16 wt%. The XPS and ToF-SIMS results can be reconciled by suggesting that there is surface enrichment of glycol after the PEDOT matrix becomes saturated with glycol. Such a conclusion is supported by the contact angle data which showed a monotonic decrease in both advancing and receding angles. Given that there is a significant level of glycol present on the surface of the PEDOT film, AFM phase contrast imaging was employed to examine the glycol presentation at the surface. The technique however was unable to detect any phase changes at the length scales measured (i.e. 1 and 10 μm). The result indicates that the incorporation of glycol within the PEDOT composite is at a length scale well below the typical dimensions of the cell lines used in this study. Such a result suggests that the impact of glycol within the PEDOT polymer may be minimal in nature, and this aspect is discussed in the following section.
4.2. Biological aspects of VPP PEDOT substrates
Glycol is well known for its antifouling capability, and for its non-toxic and non-immunogenic behaviour. Its inclusion in biomaterial formulations is generally to inhibit interaction with proteins though it is also exploited for its mechanical properties. The PEDOT glycol composites examined in this study were prepared to exploit the high conductivity of the material. This desirable property as well as the good biocompatibility of PEDOT deems the material a good candidate for applying stimulus to cells and tissues. Protein adsorption is a good indicator of cell interactions at a surface, as cell adhesion is mediated by certain proteins. Fibronectin was examined as a model serum protein involved in the adhesion of many cell types. The interactions of fibronectin at the surface of each material was examined by QCM and indicated that there was a single binding event occurring to form the adsorbed protein layer. Similar properties observed on each PEDOT glycol composite surface despite the significantly different amounts of exposed glycol moieties at the material surfaces. The 0 wt% PEDOT surface was significantly rougher and displayed a slightly stiffer protein layer. The mass of protein adsorbed on the surfaces per unit area showed slight variations with 24 and 61 wt% adsorbing slightly higher amounts of protein than 0 and 48 wt% PEDOT, however these differences were not significant. These observations did not correlate with surface energy or exposed glycol, indicating that there may be other contributing physicochemical factors which impact on protein adsorption.The properties of the cell interactions with the PEDOT materials were then examined to determine any correlations with observed glycol content or protein behaviour. Initial adhesion of both cell types examined was lowest on the rough 0 wt% PEDOT surface, though this surface showed slightly higher adsorption of protein mass, indicating a possible role of surface topography on cell adhesion. Following initial adhesion however, each cell type behaved slightly differently on the surfaces, with fibroblasts proliferating best on 0 wt% PEDOT, even though initial adhesion was low, and keratinocytes proliferating best on 48 wt% PEDOT. Effects of glycol at the surface on the cell morphology also appeared to be insignificant with cells on PEDOT retaining a similar morphology to those on TCP at comparable time points.
Taken together, these high conductivity PEDOT surfaces with different amounts of surface exposed glycol appear to provide good support for cell adhesion and proliferation. These results indicate their suitability for further investigation into the delivery of electrical stimulus via the materials in the development of biomedical devices.
Examination of the effect of the application of charge to a material on protein interactions occurring at its surface provides insights into how a material will interact with cells and tissues in vivo. This study has shown that highly conductive PEDOT glycol composites are certainly compatible with different cell types. Charge application altered the surfaces in such a way that they interacted with the model protein tested here, fibronectin, in a manner dependent on the level of potential applied, and whether the material was pushed toward a reducing or oxidising potential. Oxidised PEDOT adsorbed less protein onto its surface, however the mechanical properties of the protein layer were maintained similar to unbiased PEDOT. This correlated with significantly less area covered by fibroblast cells, indicating lower adhesive properties of this protein coated surface. Reduced PEDOT however, while it demonstrated higher mass adsorption to its surface, also displayed altered mechanical properties of this adsorbed layer. This alteration towards a denser, less hydrated protein layer with increased reducing potential in turn altered the adhesion of keratinocytes, with these cells displaying more area covered per cell on this surface. The difference in cell spreading of the cell types tested indicate a difference in dependence on protein presentation vs. protein density at material surfaces between the two cell types.
5. Conclusions
High conductivity PEDOT glycol composites display conductivities only one order of magnitude lower than some metals and alloys. This desirable property combined with its demonstrated ability to support the adhesion and proliferation of a number of cell types indicate this material to be a useful alternative to current metal technologies for implantable devices. An examination into the effect of biasing the PEDOT surface by the application of oxidising or reducing potentials revealed significant effects on the protein layer adsorbed at the surface, and hence cell-type dependent behaviour at the surface. This observed difference opens up the possibility of tuning PEDOT surfaces by the application of potential to alter cell behaviour at the material interface by influencing the mediating protein layer.Acknowledgements
The authors acknowledge Miss Amy Gelmi for assistance with AFM imaging. The authors gratefully acknowledge the Australian Research Council for funding.Notes and references
- A. Nardes, et al. , Org. Electron, 2008, 9, 727–734 CrossRef CAS.
- J. Ouyang, et al. , Adv. Funct. Mater., 2005, 15(2), 203–208 CrossRef CAS.
- B. Winther-Jensen and F. Krebs, Sol. Energy Mater. Sol. Cells, 2006, 90, 123–132 CrossRef CAS.
- B. Winther-Jensen and K. West, React. Funct. Polym., 2006, 66, 479–483 CrossRef CAS.
- F. Zhang, et al. , Adv. Mater., 2002, 14(9), 662–665 CrossRef CAS.
- W. Wu, et al. , Adv. Funct. Mater., 2008, 18, 810–815 CrossRef CAS.
- N. Senthilkumar, et al. , J. Macromol. Sci., Part A: Pure Appl. Chem., 2010, 47, 484–490 CrossRef CAS.
- D. Bernards, et al. , J. Mater. Chem., 2007, 18, 116–120 RSC.
- M. M. Stevens and J. H. George, Science, 2005, 310(5751), 1135–1138 CrossRef CAS.
- D. E. Discher, P. Janmey and Y.-l. Wang, Science, 2005, 310(5751), 1139–1143 CrossRef CAS.
- L. G. Griffith, Ann. N. Y. Acad. Sci., 2002, 961(1), 83–95 CrossRef CAS.
- J. Huang, et al. , Adv. Funct. Mater., 2005, 15(2), 290–296 CrossRef CAS.
- A. Gürsel, et al. , React. Funct. Polym., 2003, 57(1), 57–65 CrossRef.
- L. Niu, et al. , Synth. Met., 2001, 122, 425–429 CrossRef CAS.
- A. Gelmi, M. Higgins and G. Wallace, Biomaterials, 2010, 31, 1974–1983 CrossRef CAS.
- H. Bai, et al. , Polymer, 2007, 48, 5259–5267 CrossRef CAS.
- K. Svennersten, et al. , Biomaterials, 2009, 30(31), 6257–6264 CrossRef CAS.
- C. Salto, et al. , Langmuir, 2008, 24(24), 14133–14138 CrossRef CAS.
- K. Anselme, et al. , Biomaterials, 2000, 21(15), 1567–1577 CrossRef CAS.
- R. A. Green, et al. , Biomaterials, 2008, 29(24–25), 3393–3399 CrossRef CAS.
- A. Quigley, et al. , Adv. Mater., 2009, 21, 4393–4397 CrossRef CAS.
- R. Aaron and D. Ciombor, J. Cell. Biochem., 1993, 52(1), 42–46 CrossRef CAS.
- J. Wong, R. Langer and D. Ingber, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(8), 3201–3204 CrossRef CAS.
- D. Ciombor and R. Aaron, J. Cell. Biochem., 1993, 52(1), 37–41 CrossRef CAS.
- A. Rowlands and J. Cooper-White, Biomaterials, 2008, 29, 4510–4520 CrossRef CAS.
- K. Gilmore, et al. , Biomaterials, 2009, 30, 5292–5304 CrossRef CAS.
- S. Meng, et al. , J. Biomed. Mater. Res., Part A, 2008, 87A(2), 332–344 CrossRef CAS.
- J. Razal, et al. , Adv. Funct. Mater., 2009, 19, 3381–3388 CrossRef CAS.
- S. Richardson-Burns, et al. , Biomaterials, 2007, 28, 1539–1552 CrossRef CAS.
- R. Green, N. Lovell and L. Poole-Warren, Acta Biomater., 2010, 6, 63–71 CrossRef CAS.
- M. Fabretto, et al. , Thin Solid Films, 2011, 519, 2544–2549 CrossRef CAS.
- D. Evans, et al. , J. Mater. Chem., 2012, 22(30), 14889–14895 RSC.
- B. Winther-Jensen and K. West, Macromolecules, 2004, 37, 4538–4543 CrossRef CAS.
- M. Fabretto, et al. , J. Mater. Chem., 2009, 19, 7871–7878 RSC.
- E. Wischerhoff, et al. , Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS.
- E. Tziampazis, J. Kohm and P. Moghe, Biomaterials, 2000, 21, 511–520 CrossRef CAS.
- T. Satomi, et al. , J. Nanosci. Nanotechnol., 2007, 7(7), 2394–2399 CrossRef CAS.
- S. Jeon, et al. , J. Colloid Interface Sci., 1991, 142(1), 149–158 CrossRef CAS.
- R. Michel, et al. , Langmuir, 2005, 21, 12327–12332 CrossRef CAS.
- T. Ekblad, et al. , Adv. Funct. Mater., 2010, 20, 2396–2403 CrossRef CAS.
- K. Uhlig, et al. , Soft Matter, 2010, 6, 4262–4267 RSC.
- A. Stalder, et al. , Colloids Surf., A, 2006, 286(1), 92–103 CrossRef CAS.
- S. F. Cogan, Annu. Rev. Biomed. Eng., 2008, 10, 275–309 CrossRef CAS.
- L. He, et al. , Colloids Surf., B, 2011, 87(2), 273–279 CrossRef CAS.
- M. Fabretto, et al. , Polymer, 2011, 52, 1725–1730 CrossRef CAS.
- D. Briggs, Cambridge University Press, Cambridge, NY, 1998.
- F. Höök, et al. , Langmuir, 1998, 14(4), 729–734 CrossRef.
- A. J. Garcia, M. D. Vega and D. Boettiger, Mol. Biol. Cell, 1999, 10(3), 785–798 CAS.
- M. B. Hovgaard, et al. , J. Phys. Chem. B, 2008, 112, 8241–8249 CrossRef CAS.
- P. J. Molino, et al. , Langmuir, 2012, 28(22), 8433–8445 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |